
Abstract
Phylogenetic analysis of cloned 16S rRNA gene fragments from Cornitermes cumulans (T) and its termite mound soil (TM) from the Brazilian Cerrado (Brazilian Savannah) revealed a great diversity of sequences. The bacteria detected in T and TM samples were associated with the following major lineages: Spirochaetes (T), Firmicutes (T), Synergistetes (T), Cyanobacteria (T), Fibrobacteres (T), Elusimicrobia (T), Chloroflexi (TM), Verrucomicrobia (TM), Gemmatimonadetes (TM), Armatimonas (TM), Proteobacteria (T and TM), Actinobacteria (T and TM), Bacteroidetes (T and TM), Planctomycetes (T and TM), and Acidobacteria (T and TM). All archaeal sequences only obtained from TM sample were associated with uncharacterized Crenarchaeota. The high values that were obtained for the diversity indices and evenness are indicative of high bacterial diversity from T and TM libraries, whereas the TM archaeal library exhibited low diversity. Therefore, our findings revealed differences between the bacterial communities from termite mounds and those from C. cumulans, the latter of which represents a specific bacterial composition when compared to other termite species.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Soil contains a diverse and vast biota, and the microorganisms that make up this biota perform an essential role within the environment by participating in the cycling and flux of various nutrients and by influencing structure formation and the maintenance of soil quality (Martins 2010). In addition to microorganisms, termites also have a large influence on soil characteristics and are known as “soil engineers”.
Termites are a group of insects that have an ecological impact on carbon turnover through cellulose degradation and the cycling of nutrients, which is of the utmost importance to the environment. Different species of termites build unique types of nests, such as galleries and simple chambers, underground nests, arboreal nests, or mounds. The mounds built by these insects are usually made of a mineral matrix that is mixed with feces or saliva, depending on the species of termite. These structures are the leading cause of soil physical changes, including the ability to hold water, density and structural stability, soil chemical modifications, and organic matter content (Fall et al. 2007). These changes can affect the activity and diversity of bacteria, which in agricultural systems are directly influenced by changes in soil (Jangid et al. 2008).
Cornitermes cumulans (Kollar 1932) is a termite species that belongs to the family Termitidae, and only some members construct a nest mound, known as “mound termites,” because of the huge nest they build and because of their high prevalence in grazing land. This species is unique to the Neotropics, and their nests are the most abundant in Brazil, especially in the Cerrado (Brazilian Savannah); thus, this termite is an important species for the ecosystem. Using a mineral matrix and saliva, C. cumulans can construct mounds that range in height from 50 to 100 cm. The feeding habits of this termite species are based on litter that includes mostly leaves and the dead roots of grasses.
Despite of the known impact of termite activity on the soil affecting the diversity and microbial rhizosphere communities, the prokaryotic composition of these organisms and of their nests has been scarcely explored. Therefore, in this study, we sought to illuminate the prokaryotic community diversity associated with C. cumulans and its termite mound soil by means of 16S rRNA clone libraries.
Material and methods
Sample collection
C. cumulans workers were collected under aseptic conditions from a nest at the Ecological Station of the Universidade Federal de Minas Gerais (19º52′36″ S - 43°58′16″ W), a region that has been characterized as dry grassland (a type of Brazilian Cerrado), in November 2009. Their termite mound soil (height 1.3 m, TM) and neighbor soil (NS), which was 60 cm from the mound, were also sampled. The opening created in the wall of the mound to collect the soil was 40 cm deep. After 48 h, this gap was refilled, indicating intense termite activity.
Soil samples, abiotic parameters, and the taxonomic identification of the termites
The quantitative mineral composition of TM and NS samples were determined through the quantification of P, Fe, Zn, Mn, and Cu using Mehlich 1 as an extractant; the quantification of Ca, Mg, and Al using KCl (1 mol/L) as an extractant; and the quantification of H + Al using calcium acetate (0.5 mol/L) as an extractant (EMBRAPA 1997). pH and granulometry analyses of the TM and NS samples were also performed (Ruiz 2005). The termite taxonomy (Constantino 1999) was performed at Unidade Laboratorial de Referência em Pragas Urbanas, Instituto Biológico, São Paulo.
DNA extraction
Total DNA from the TM sample was extracted using the UltraClean Mega Soil DNA Isolation® kit (MoBio Laboratories, USA) according to the manufacturer’s instructions. Isolated DNA was stored at −20 °C until further processing. The extraction of total DNA from termites was performed according to Corby-Harris et al. (2007). Prior to DNA extraction, the exterior of termites was washed with the following (in order): sterile distilled water (five times), 70 % ethanol (five times), 6 % sodium hypochlorite (five times), and sterile distilled water (five times). The termites were then macerated in liquid nitrogen.
PCR amplification of the 16S rRNA gene, cloning, and sequencing
Fragments of the 16S rRNA gene were amplified from C. cumulans (T) and TM genomic DNA for the construction of bacterial and archaeal clone libraries. The bacterial 16S rRNA gene fragment was amplified by touchdown PCR according to Freitas et al. (2008), using the conserved primer set 8f (5′AGAGTTTGATCMTGGCTCAG 3′) and 907r (5′ACGGHTACCTTGTTACGACTT 3′) (Lane 1991). The sequences of the archaeal primers were 16SAf (5′TTATTGGGCCTAAAGCRTC3′) and 1400Ar (5′CGGCGAATTCGTGCAAGGAGCAGGGAC3′) (Clementino et al. 2007). The 16S rRNA gene was amplified by PCR according to Clementino et al. (2007). The amplicons were confirmed using agarose gel electrophoresis. Five separate PCR amplifications were pooled before cloning. The PCR products were purified using a DNA extraction kit (Fermentas, Canada) for cloning.
Clone libraries were constructed for bacterial and archaeal communities present in the TM and bacteria present in the T by cloning the PCR products that were obtained with the different primer sets. These purified PCR products were cloned into the pJET1.2/blunt cloning vector using the CloneJET ™ PCR Cloning Kit (Fermentas) according to the manufacturer’s instructions. Electrocompetent Escherichia coli XL1 Blue were transformed with the ligations according to the manufacturer’s instructions. Plasmid DNA was extracted using the GeneJETTM Plasmid Miniprep Kit (Fermentas).
DNA sequencing reactions were performed with the DYEnamic ET Dye Terminator Cycle Sequencing Kit, and the products were run on a MegaBACE 1000 capillary sequencer (GE Healthcare, UK) according to the manufacturers’ instructions. Partial 16S rRNA gene sequences were checked for quality, aligned, and edited to produce a consensus using the programs Phred v. 0.020425 (Erwing and Green 1998), Phrap v. 0.990319 (Green 1994), and Consed 12.0 (Gordon et al. 1998). Chimeras were checked using Bellerophon software (http://comp-bio.anu.edu.au/bellerophon/bellerophon.pl). The sequences were compared with the Silva database (release 108) (Pruesse et al. 2007) and the approximate phylogenetic affiliations were conducted with ARB software (Ludwig et al. 2004) using the neighbor-joining method (Saitou and Nei 1987).
Operational taxonomic units (OTUs) were determined by using DOTUR software (Schloss and Handelsman 2005); similarity levels between sequences of at least 97 % have been proposed for classifying a microorganism at the species level (Drancourt et al. 2000). Coverage of the clone libraries was calculated using the equation \( \mathrm{C}=1-\left( {{n \left/ {N} \right.}} \right)\times 100 \), where n is the number of unique OTUs and N is the number of sequences analyzed in the library (Good 1953). The nucleotide sequences from OTUs were deposited in the GenBank database with accession numbers JN830961-JN831088.
Results and discussion
Physicochemical properties of the samples
The TM and NS samples had different physicochemical compositions (Table 1). All minerals, except for Al, were enriched in the TM compared to the NS sample. The lower concentration of Al found in the TM can be explained by the complexes formed by this ion with soil organic matter (Sarcinelli et al. 2009) because it is known that termite mounds contain more organic matter than soil. The granulometry analysis revealed that the TM and NS samples contained 50 and 41 % clay content, respectively; a characteristic property of the Cerrado soil. Therefore, the data are consistent with studies from other research groups that have revealed an increased richness in clay and minerals from termite mounds with respect to the neighboring soil (Holt and Lepage 2000; Ndiaye et al. 2004). According to Holt and Lepage (2000), this enrichment occurs because the termites select neighboring soil particles that are rich in minerals to build their nests. Previous study showed that lower clay and organic matter content of soils around mounds suggest that termite activity can reduce the bioavailability of soil nutrients for plants (Adekayode and Ogunkoya 2009).
The physicochemical differences between TM and NS may influence the composition of their microbial communities. Indeed, the dendrogram generated from the ARDRA fingerprint revealed that the TM and NS environments harbored distinct bacterial communities (data not shown). It is well known that the plant species are the dominant factors influencing the composition of the rhizosphere microbial communities (Barret et al. 2011). However, it should be noted that the termite activity leads to nutrient depletion of soil possibly disturbing this microbial community, which has a plant growth-promoting effect.
General features of the libraries
To obtain information on the composition and structure of the TM and T prokaryotic communities, three clone libraries of 16S rRNA genes were constructed, and 328 randomly selected clones from the three libraries were sequenced (Table 2). Of the 328 sequences, 286 were used for further analyses after quality control and the removal of chimeric sequences.
The clone libraries constructed from the T and TM samples were evaluated by rarefaction analysis, when the OTUs were defined at ≥97 % sequence identity (data not shown). The rarefaction curves of bacteria were far from saturation, and the coverage values of clone libraries were 63 and 68 %, for T and TM, respectively, indicating that further sequencing of more clone libraries would have revealed additional diversity. In contrast, the rarefaction curve of archaea (TM library) reached an asymptote, indicating that most of the diversity of archaea was detected in the sample (Good’s coverage 95 %).
Diversity indices are a valuable tool with which to quantify diversity in a community and describe its numerical structure (Hill et al. 2003). They reflect the richness and the relative abundance of OTUs in each environment. Values from the Shannon and Simpson indices demonstrated a high diversity of bacteria in the TM and T libraries compared to the archaeal TM library (Table 2). These data indicated a relatively even OTU distribution, whereas the low Shannon and high Simpson diversity indices in the archaea from the TM library are due to the presence of a few dominant OTUs. Additionally, both the Chao1 and ACE values indicated a greater diversity of bacterial species in the TM and T libraries.
Phylogenetic composition of bacterial clone libraries
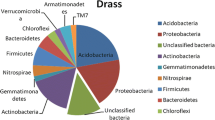
Soil habitats contain a great diversity of macro- and microorganisms, and bacteria are the largest group with respect to species richness and community size (Neher 1999). The phylogenetic analysis of 16S rRNA gene sequences constitutes an important tool for the assessment of complex microbial diversity. The phylogenetic diversity of the 16S rRNA gene clones from the TM and T libraries is shown in Fig. 1. The two libraries showed that OTUs belong to several bacterial phyla. Furthermore, five (one OTU from T library and four OTUs from TM library) of the 127 bacterial OTUs detected were considered to be unclassified at the phylum level and, thus, may represent new bacterial taxa.

Phylogenetic distribution of bacterial 16S rDNA sequences generated from T and TM samples
Actinobacteria and Acidobacteria comprised the largest fractions of the TM clone library, whereas Verrucomicrobia, Gemmatimonadetes, and Armatimonadetes were minor members. Members of Proteobacteria, Actinobacteria, and Acidobacteria have been reported to constitute the largest groups of bacterial communities derived from a variety of soils, including Cerrado soil. Thus, for the most part, our findings corresponded to the results from the studies of Hugenholtz et al. (1998), Kent and Triplett (2002), Zhang and Xu (2008), and Quirino et al. (2009).
Previous studies on bacterial diversity in termite nests have been reported (Fall et al. 2007, Long et al. 2010). In our sample, the bacteria found in TM library were associated with nine phyla (Fig. 1). In other studies, using DGGE (Fall et al. 2007) to analyze the variation of the internal transcribed spacer region, coupled with the construction of 16S rRNA gene clone libraries and subsequent sequencing (Long et al. 2010), the bacteria were associated with six and four phyla, respectively. Verrucomicrobia, Planctomycetes, and Gemmatimonadetes were detected in our analysis, although they were not observed in the analyses by Long et al. (2010) and Fall et al. (2007). In contrast to our work, the phylum Firmicutes was dominant in the study by Long et al. (2010).
Thirty percent of all classified sequences from the T clone library corresponded to Spirochaetes (which were represented exclusively by Treponema genus), Firmicutes, and Actinobacteria. Other important minor bacterial phyla included Planctomycetes, Cyanobacteria, Fibrobacteres, Acidobacteria, and Elusimicrobia, the latter being a typical phylum from termites gut (Ikeda-Ohtsubo et al. 2010). Most OTUs (67.2 %) from the T clone library were associated with other sequences that had previously been obtained from other termite species, and the remaining OTUs from soil and gut of other insects. Furthermore, all bacterial phyla found in T library were previously reported as symbionts of other species of termites (Hongoh 2010). The phyla Spirochaetes and Fibrobacteres are known to act in the hydrolysis of lignocellulose and are considered essential symbionts of the termites gut (Hongoh 2011).
Previous cultivation-independent studies have been performed on the bacterial diversity of the lower and higher termite gut with different feeding habits. Among these, Macrotermes michaelseni (Mackenzie et al. 2007) and Macrotermes gilvus (Hongoh et al. 2006) have been reported to feed on litter and are found in savannah regions, similar to C. cumulans. Despite eating habits and habitat similarities between these two species and C. cumulans, the bacterial communities among them were very distinct. The bacteria found in the gut of M. michaelseni were less diverse, and Bacteroidetes (formally referred to as Cytophaga-Flexibacter-Bacteroides) was the dominant group (Mackenzie et al. 2007), whereas Bacteroidetes and Firmicutes were the phyla predominant in M. gilvus (Hongoh et al. 2006).
Most OTUs affiliated with the phyla outlined in Figs. 2 and 3 were phylogenetically related to uncultured bacteria from other termite species and from the soil. Nevertheless, some OTUs related to the Actinobacteria, Proteobacteria, Bacteroidetes, and Acidobacteria could be classified at the genus level, i.e., Marmoricola, Nocardioides, Planosporangium, Conexibacter, Microlunatus, and Actinomadura (Actinobacteria of the TM clone library); Rhodococcus, Luteococcus, and Propionibacterium (Actinobacteria of the T clone library); Caulobacter, Phenylobacterium, Altererythrobacter, Pedomicrobium, and Burkholderia (Proteobacteria of the TM clone library); Chromobacterium, Paracoccus, Pantoea, and Novosphingobium (Proteobacteria of the T clone library); Flavisolibacter (Bacteroidetes of the TM clone library); and Edaphorobacter (Acidobacteria of the TM library). Some of these genera, such as Burkholderia and Bradyrhizobium, are important for soil nitrogen fixation and the stimulation of plant growth, respectively (Hennecke 1990; Preisig et al. 1996; Pandey et al. 2005).

Phylogenetic tree showing the affiliation of bacterial OTUs from TM library. Phylogenetic relationships were inferred with ARB software with Silva database (release 108) using the neighbor-joining method

Phylogenetic tree showing the affiliation of bacterial OTUs from T library. Phylogenetic relationships were inferred with ARB software with Silva database (release 108) using the neighbor-joining method
Archaeal community composition in termite mounds
The construction of the archaeal 16S rRNA gene clone library was successfully obtained only for the sample TM, despite 10 attempts to do with the sample T. In an endeavor to obtain the amplification of the 16S RNA gene fragment (T sample), different archaeal primers set were used namely: 16SAf and 1400Ar (Clementino et al. 2007); 25 F and 1492R (Lane 1991); 21 F and 958R; (DeLong 1992); and mcrA-F and mcrA -R (Luton et al. 2002). According to Hongoh (2010), the abundance of archaea in the gut of termite species that have already been studied ranged from 0 to 10 %. Therefore, our results can be explained by the absence or low abundance of archaea in the gut of C. cumulans. Moreover, according to Brune (2006), the abundance of archaea in the gut of termites is apparently related to diet. In termites that feed on litter, such as C. cumulans, the abundance of archaea is lower (0.1–1.7 %) than in termites feeding soil (1.4–3.1 % archaea).
The phylogenetic analysis of the TM archaeal 16S rRNA gene revealed a total of two OTUs, indicating that the archaeal community was poorly diverse. Both OTUs were affiliated with the phylum Crenarchaeota. In addition, one OTU gathered 37 clones, whereas another OTU contained only one clone. It should be mentioned that all OTUs were uncharacterized archaea.
The identification of archaea from non-extreme environments is important for understanding their ecological role in different environments. The incidence of archaea inhabiting the soil is extremely low, ranging from 0.5 to 3.8 % of all prokaryotes that inhabit this environment (Ochsenreiter et al. 2003; Timonen and Bomberg 2009). Although members of the phylum Crenarchaeota have previously been isolated from extreme environments, they are often found in many less-extreme environments, including different soil types, such as that of the Cerrado (Bintrim et al. 1997; Ochsenreiter et al. 2003; Clementino et al. 2007; Timonen and Bomberg 2009). Our results are in agreement with the work of Tagliaferro (2005) which only detected clones belonging to the phylum Crenarchaeota.
Similar to previous studies of termite species, this survey detected a wide variety of phylogenetic taxa. The analysis of 16S rRNA gene sequences revealed that a specific microbiota is associated with the termite species and not with the feeding habits or habitats of the termites. These data support the hypothesis of Hongoh et al. (2006) that microorganisms in the intestinal tracts of termites have coevolved with their hosts. The survey also detected a broader phylogenetic breadth of taxa than any previous study of termite mounds, including several taxa that had not been previously detected in termite mounds. The results of this study provide the first insights into the bacterial diversity in the gut of a member of the mandibulate nasute grass-feeding termite, a group unique to the Neotropics and its nest.
References
Adekayode FO, Ogunkoya MO (2009) Comparative study of clay and organic matter content of termite mounds and the surrounding soils. Afr Crop Sci Conf Proc 9:379–384
Barret M, Morrissey JP, O’Gara F (2011) Functional genomics analysis of plant growth-promoting rhizobacterial traits involved in rhizosphere competence. Biol Fertil Soils 47:729–743
Bintrim SB, Donohue TJ, Handelsman J, Roberts GP, Goodman RM (1997) Molecular phylogeny of Archaea from soil. PNAS 94:277–282
Brune A (2006) Symbiotic associations between termites and prokaryotes. Springer, Berlin
Clementino MM, Fernandes CC, Vieira RP, Cardoso AM, Polycarpo CR, Martins OB (2007) Archaeal diversity in naturally occurring and impacted environments from a tropical region. J Appl Microbiol 103:141–151
Constantino R (1999) Chave ilustrada para identificação dos gêneros de cupins (Insecta: Isoptera) que ocorrem no Brasil. Pap Avulsos de Zool 40:387–448
Corby-Harris V, Pontaroli AC, Shimkets LJ, Bennetzen JL, Habel KE, Promislow DEL (2007) Geographical distribution and diversity of bacteria associated with natural populations of Drosophila melanogaster. Appl Environ Microbiol 73:3470–3479
Delong EF (1992) Archaea in coastal marine environments. Proc Natl Acad Sci U S A 89:5685–5689
Drancourt M, Bollet C, Carlioz A, Martelin R, Gayral J-P, Raoult D (2000) 16S Ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol 34:3623–3630
EMPRESA BRASILEIRA DE PESQUISA AGROPECUÁRIA - EMBRAPA (1997) Manual de métodos de análise de solo, 2nd edn. Rio de Janeiro, Centro Nacional de Pesquisa de Solos, p 212
Erwing B, Green P (1998) Base- calling of automated sequencer traces using Phred II, error probabilities. Genome Res 8:186–194
Fall S, Hamelin J, Ndiaye F, Assigbetse K, Aragno M, Chotte JL, Brauman A (2007) Differences between bacterial communities in the gut of a soil-feeding termite (Cubitermes niokoloensis) and its mounds. Appl Environ Microbiol 7:5199–5208
Freitas DB, Lima-Bittencourt CI, Reis MP, Costa PS, Assis PS, Chartone-Souza E, Nascimento AMA (2008) Molecular characterization of early colonizer bacteria from wastes in a steel plant. Lett Appl Microbiol 47:241–249
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40:237–262
Gordon D, Abajian C, Green P (1998) Consed: a graphical tool for sequence finishing. Genome Res 8:195–202
Green P (1994) PHRAP documentation. URL: http://www.phrap.org. Accessed 17 Apr 2011
Hennecke H (1990) Nitrogen fixation genes involved in Bradyrhizobium japonicum soybean symbiosis. FEBS 268:422–426
Hill TCJ, Walsh KA, Harris JA, Moffett BF (2003) Using ecological diversity measures with bacterial communities. FEMS Microbiol Ecol 43:1–11
Holt JA, Lepage M (2000) Termites and soil properties. Kluwer Academic, Norwell
Hongoh Y (2010) Diversity and genomes of uncultured microbial symbionts in the termite gut. Biosci Biotechnol Biochem 74:1145–1151
Hongoh Y (2011) Toward the functional analysis of uncultivable, symbiotic microorganisms in the termite gut. Cell Mol Life Sci 68:1311–1325
Hongoh Y, Ekpornprasit L, Inoue T, Moriya S, Trakulnaleamsai S, Ohkuma M, Noparatnaraporn N, Kudo T (2006) Intracolony variation of bacterial gut microbiota among castes and ages in the fungus-growing termite Macrotermes gilvus. Mol Ecol 15:505–516
Hugenholtz P, Pitulle C, Hershberger KL, Pace N (1998) Novel division level bacterial diversity in a yellowstone hot spring. J Bacteriol 180:366–376
Ikeda-Ohtsubo W, Desai M, Stingl U, Brune A (2010) Phylogenetic diversity of ‘Endomicrobia’ and their specific affiliation with termite gut flagellates. Microbiology 153:3458–3465
Jangid K, Williams MA, Franzluebbers AJ, Sanderlin JS, Reeves JH, Jenkins MB, Endale DM, Coleman DC, Whitman WB (2008) Relative impacts of land-use, management intensity and fertilization upon soil microbial community structure in agricultural systems. Soil Biol Biochem 40:2843–2853
Kent AD, Triplett EW (2002) Microbial communities and their interactions in soil and rhizosphere ecosystems. Annu Rev Microbiol 56:211–236
Lane DJ (1991) 16S/23S rRNA sequencing. John Wiley and Sons, New York
Long YH, Xie L, Liu N, Yan X, Li MH, Fan MZ, Wang Q (2010) Comparison of gut-associated and nest-associated microbial communities of a fungus growing termite (Odontotermes yunnanensis). Insect Sci 17:265–276
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar BA, Lai T et al (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371
Luton PE, Wayne JM, Sharp RJ, Riley PW (2002) The mcrA gene as an alternative to 16S rRNA in the phylogenetic analyses of methanogen populations in landfill. Microbiology 148:3521–3530
Mackenzie LM, Muigai AT, Osir EO, Lwand W, Keller M, Toledo G, Boga HI (2007) Bacterial diversity in the intestinal tract of the fungus-cultivating termite Macrotermes michaelseni (Sjöstedt). Afr J Biotechnol 6:658–667
Martins MA (2010) Microbiologia do solo. URL: http://www.uenf.br/Uenf/Downloads/LSOL_345_1113400965.pdf. Accessed 20 Jan 2011
Ndiaye RLD, Lepage M, Sall CE, Brauman A (2004) Nitrogen transformations associated with termite biogenic structure in a dry savanna ecosystem. Plant Soil 265:189–196
Neher DA (1999) Soil community composition and ecosystem processes: comparing agricultural ecosystems with natural ecosystems. Agrofor Syst 45:159–185
Ochsenreiter T, Selezi D, Quaiser A, Bonch-Osmolovskaya L, Schleper C (2003) Diversity and abundance of Crenarchaeota in terrestrial habitats studied by 16S RNA surveys and real time PCR. Environ Microbiol 5:787–797
Pandey P, Kang SC, Maheshwari DK (2005) Isolation of endophytic plant growth promoting Burkholderia sp. MSSP from root nodules of Mimosa pudica. Curr Sci India 89:177–180
Preisig O, Zufferey R, Hennecke H (1996) The Bradyrhizobium japonicum fixGHIS genes are required for the formation of the high-affinity cbb3-type cytochrome oxidase. Arch Microbiol 165:297–305
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196
Quirino BF, Pappas GJ, Tagliaferro AC, Collevatti RG, Neto EL, Silva MRSS, Bustamante MMC, Krüger RH (2009) Molecular phylogenetic diversity of bacteria associated with soil of the savanna-like Cerrado vegetation. Microbiol Res 164:59–70
Ruiz HA (2005) Incremento da exatidão da análise granulométrica do solo por meio da coleta da suspensão (silte + argila). R Bras Ci Solo 29:297–300
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sarcinelli TS, Schaefer CEGR, Lynch LS, Arato HD, Viana JHM, Filho MRA, Gonçalves TT (2009) Chemical, physical and micromorphological properties of termite mounds and adjacent soils along a toposequence in Zona da Mata, Minas Gerais State, Brazil. Catena 76:107–113
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Tagliaferro AC (2005) Análise molecular da diversidade bacteriana de solos do Cerrado utilizando bibliotecas de rDNA 16S—Uma perspectiva biotecnológica. 2005. Dissertação de Mestrado. Universidade Católica de Brasília.
Timonen S, Bomberg M (2009) Archaea in dry soil environments. Phytochem Rev 8:505–518
Zhang L, Xu Z (2008) Assessing bacterial diversity in soil. J Soil Sediment 8:379–388
Acknowledgments
We thank Fundação de Amparo à Pesquisa do Estado de Minas Gerais, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Pró-Reitoria de Pesquisa da Universidade Federal de Minas Gerais, and Conselho Nacional de Desenvolvimento Científico e Tecnológico for providing financial support. We thank Francisco José Zorzenon from Biological Institute, São Paulo, for termite identification.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Costa, P.S., Oliveira, P.L., Chartone-Souza, E. et al. Phylogenetic diversity of prokaryotes associated with the mandibulate nasute termite Cornitermes cumulans and its mound. Biol Fertil Soils 49, 567–574 (2013). https://doi.org/10.1007/s00374-012-0742-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00374-012-0742-x