
Abstract
Investigation of the association between microbial communities in different compartments and the health status of coral hosts will support an in-depth understanding of the relationship between the microbial symbionts and coral health. In this study, next-generation sequencing of 16S ribosomal RNA genes (rDNA) and ribosomal RNA (rRNA) was used to characterize bacterial communities from the mucus and tissues of healthy and bleached Pocillopora damicornis. The cDNA (active bacterial members) and DNA (the total bacterial community) libraries of the same coral sample were similar, suggesting the fact that the bacteria inhabiting P. damicornis were active. Upon coral bleaching, the bacterial communities changed considerably in the tissue, but remained stable in the coral mucus layer. The relative abundance of Endozoicomonas decreased sharply in the bleached coral tissue. Presumably, microbial function based on the taxonomic compositions was accordingly changed in coral tissue, but not in the mucus layer, when the coral bleached. This study suggests that both rRNA- and rDNA-based methods for bacterial community analysis are fit for evaluating P. damicornis health implications. Furthermore, the results of this study demonstrate the differential responses of mucus- and tissue-associated bacterial communities to coral bleaching.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The coral-associated bacteria community includes endosymbiotic, symbiotic, and loosely associated partners (Ainsworth et al. 2015). They are distributed among several anatomical compartments, including the skeleton, polyp tissue, and external surface mucopolysaccharide layer (Bourne and Munn 2005; Brown and Bythell 2005; Sweet et al. 2011; Krediet et al. 2013; Ainsworth et al. 2015). These three compartments within a coral colony (i.e., mucus, tissue, and skeleton) are colonized by distinct bacterial communities, as shown by using 16S rRNA gene profiling (Rohwer et al. 2002; Sweet et al. 2011; Bourne et al. 2013; Li et al. 2014). Coral-associated bacteria have important functions in carbon (Brown and Bythell 2005; Littman et al. 2009; Thurber et al. 2009; Kimes et al. 2010; Webster and Thomas 2016), nitrogen (Lema et al. 2012, 2014; Sharp et al. 2012), and sulfur (Raina et al. 2010, 2016; Garren et al. 2014) cycling.
Coral bleaching, the disruption of the symbiotic relationship between corals and their photosynthetic algae, can lead to coral morbidity and mortality and has substantially decimated coral reefs worldwide (Heron et al. 2016; Oliver et al. 2018). In addition to the loss of symbiotic Symbiodiniaceae, the overall composition of coral-associated bacterial communities changes when coral bleaches (Ainsworth and Hoegh-Guldberg 2009; Lee et al. 2015; Hadaidi et al. 2017). The abundance of potential pathogens was found to increase in bleached coral mucus (Nguyen-Kim et al. 2015) and tissue (Li et al. 2021). Several bacterial taxa, such as Sphingomonadaceae, Flavobacteriaceae, and Rhodobacteraceae, were more abundant in bleached coral (Goyen et al. 2019; Botté et al. 2022; Li et al. 2021). There is a consensus that the bacterial communities in coral tissues change during bleaching (Pantos et al. 2003; Bourne and Munn 2005; Bourne et al. 2008; Gardner et al. 2019; Li et al. 2021), although there is some argument about changes in coral mucus communities (Lee et al. 2015; Nguyen-Kim et al. 2015; Hadaidi et al. 2017).
The microniche-specific bacterial community might be attributed to the unique set of physicochemical properties of these microhabitats (Sweet et al. 2011; Engelen et al. 2018). Owing to the extreme diurnal variation of oxygen saturation levels (from supersaturated to nearly anoxic) (Shashar et al. 1996) and exuded organic carbon in dissolved form which creates a nutrient-rich medium, Proteobacteria (mostly Alpha- and Gamma proteobacteria) and Bacteroidetes were found to be enriched in coral mucus (Sweet et al. 2011; Lee et al. 2015; Marchioro et al. 2020). These mucus-associated bacterial communities showed host specificity (Sweet et al. 2011; Li et al. 2014; Marchioro et al. 2020). In contrast to those in the mucus layer, bacterial communities in the coral tissue are embedded within a relatively stable microenvironment (Bourne and Munn 2005; Sweet et al. 2011; Lee et al. 2015). Previous studies have revealed that bacterial communities associated with coral tissues, such as Porites astreoides, Acropora muricata, and Pocillopora damicornis, were mostly comprised of members belonging to the phyla Proteobacteria and Actinobacteria (Klaus et al. 2005; Sweet et al. 2011; Krediet et al. 2013; Ainsworth et al. 2015). Additionally, Endozoicomonas (Gammaproteobacteria) is abundant and often considered a potential endosymbiont in corals for example, Acropora hemprichii and Pocillopora damicornis (Neave et al. 2016, 2017; Li et al. 2021). Similar to those in the coral mucus, the tissue-associated bacterial communities also show host specificity (Rohwer et al. 2002; Daniels et al. 2011; Li et al. 2014; Lee et al. 2015; Neave et al. 2017). Owing to the apparent differences in bacterial communities associated with different coral compartments, it is important to consider compartments when investigating the relationship between bacterial symbionts and coral health (Rosenberg et al. 2007; Bourne et al. 2008; Ainsworth et al. 2010; Garren et al. 2014). However, the bleaching responses of microbial communities associated with different coral compartments have seldom been directly compared (Lee et al. 2015).
DNA-based high-throughput sequencing is the most widely used method for investigating microbial communities (Ward et al. 1990; Herlemann et al. 2011; Bourne et al. 2013; Li et al. 2014) that provides information regarding the total microbial community, including active, moribund, encysted, and metabolically inactive members, as well as nonliving genetic material (Hu et al. 2016). As RNA is more susceptible to degradation than DNA (Egge et al. 2015), RNA-based analysis (mainly using a complementary DNA sequence library) provides information regarding active members (Hu et al. 2016; Tian and Li 2017; Ziegler et al. 2017). From this perspective, RNA-based sequencing may be better than DNA-based methods for evaluating the responses of a microbial community to environmental perturbations (Terrado et al. 2011; Egge et al. 2015). Previous studies targeting both 16S rRNA and rDNA have shown significant differences between the active and the total bacterial communities in various habitats, including seawater (Wang et al. 2019), sea ice (Stecher et al. 2016), atmosphere (Klein et al. 2016), benthic biofilms in streams (Wilhelm et al. 2014), and mosses (Tian and Li 2017). Until now, coral-associated bacterial communities have mainly been characterized using DNA-based sequencing (Sweet et al. 2011; Li et al. 2014, 2021; Hadaidi et al. 2017). Although RNA-based 16S rRNA gene sequencing has been used by Ziegler et al. (2017), there have been few direct comparisons between coral-associated bacterial communities revealed by DNA- versus RNA-based sequencing. Therefore, we do not know whether the results of these two distinct methods must be interpreted differently.
In this study, we investigated the compositions and potential functions of bacterial communities in the tissues and mucus of healthy and bleached corals via DNA- and RNA-based 16S rRNA gene sequencing. Our aims were to (1) determine whether the bacterial communities revealed by DNA- and RNA-based 16S rRNA gene sequencing are different, (2) characterize the bacterial communities in different compartments of healthy and bleached corals, and (3) determine whether the bacterial community changes differently in the coral tissue and mucus layer under different health conditions.
Materials and methods
Coral sample collection
Coral Pocillopora damicornis samples were collected at a depth of 3–5 m in October 2014 in the Luhuitou Reef (109°47′ E, 18°19′ N) area, Sanya. Hammers and chisels were used to collect fragments (approximately 10 cm3) from three healthy and three partially bleached coral colonies in the same area with a distance of 0.5 m between colonies. One fragment was cut from each healthy coral colony, and one bleached fragment was cut from each partially bleached colony. These fragments were separately placed in sterile sampling bags, kept in a low-temperature storage box, and then sent to the laboratory within 20 min. In the laboratory, corals were rinsed with 0.2-μm-filtered (Merck Millipore, Burlington, MA, USA) autoclaved seawater to clean surfaces and remove loosely bound bacteria. A 700-μL mucus sample was extracted from each fragment with a 1-mL sterile syringe and then diluted with 100 mL sterile seawater before being filtered through a 0.2-μm membrane. The filter membrane was stored in a 2-mL frozen storage tube (Corning, Corning, NY, USA), immediately frozen with liquid nitrogen, and stored in a freezer at − 80 °C. The coral fragment was cut into small sections, placed in a sterilized centrifuge tube, and then immediately frozen with liquid nitrogen and stored at − 80 °C.
DNA and RNA extraction
DNA and RNA were extracted simultaneously from each sample using an All Prep DNA/RNA Universal Kit (Qiagen, Dusseldorf, Germany). For the mucus sample, the filter membrane was cut into pieces and used for DNA and RNA extraction (Frias-Lopez et al. 2002; Weinbauer et al. 2002). Coral tissue was removed from the fragment using a sterile syringe with sterile deionized water; the tissue suspension was then centrifuged at 13,000 × g for 5 min, and the cell pellet was subsequently collected. The membranes and cell pellets were transferred into 1.5 mL centrifuge tubes containing 600 μL Buffer RLT Plus. The tubes were placed on the Vortex Genie2 (Mo Bio, Carlsbad, CA, USA) and vortexed at maximum speed for 10 min, before being centrifuged at 18,000 × g for 3 min. After transferring the supernatant into the filter column provided by the kit, the DNA and RNA were extracted stepwise according to the manufacturer’s instructions. All RNA samples were purified using RNase-Free DNase Set (Qiagen) according to the manufacturer’s instructions. After purification, all samples were investigated using PCR with the primers 27F and 1492R (Lane 1991) targeting the bacterial 16S rRNA genes. Fifty microliter of PCR reaction mix was run in triplicate per sample using multiplex PCR master mix (Takara, Kyoto, Japan), with a 200 ng template and final primer concentration of 0.2 μM. PCR cycling conditions were 95 °C for 5 min, followed by 30 cycles of 95 °C for 30 s, 54 °C for 45 s and 72 °C for 90 s, with a final extension time of 5 min. PCR products were run on a 1% ultra -pure agarose gel, and negative amplification indicated that the DNA was removed in the template, i.e., there was no DNA in the RNA samples.
PCR amplification and sequencing
Reverse transcription of the total RNA was carried out according to the protocol of the Promega GoScript Reverse Transcription System (Promega, Madison, WI, USA). All cDNA and DNA were amplified using the primers 341F and 805R (Herlemann et al. 2011) targeting the V3-V4 hypervariable regions. MiSeq indexing adaptors were added via PCR according to the Illumina 16S metagenomic sequencing library preparation protocol. 50 μL of the PCR reaction mix was run in triplicate per sample using multiplex PCR master mix, with 200 ng DNA or cDNA template and a final primer concentration of 0.2 μM. PCR cycling conditions were 95 °C for 5 min, followed by 35 cycles of 95 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s, with a final extension time of 5 min. Triplicate PCR products were pooled, run on a 2% ultra-pure agarose gel, and purified using a QIAquick Gel Extraction Kit (Qiagen). Sequencing was performed on an Illumina MiSeq platform with 2 × 300 bp paired-end reads.
Sequence processing
The 16S rRNA gene sequences were processed using the QIIME 2 Pipeline version 2020.02 (Bolyen et al. 2019). The Demux plugin (Bolyen et al. 2019) was used to visualize the interactive quality plots and to proofread quality. The DADA2 plugin (Callahan et al. 2016) was then applied to remove primers, truncate poor-quality fragments, dereplicate, identify chimeras, and merge paired-end reads. Taxonomy was assigned to Amplicon Sequence Variants (ASVs) using the q2-feature-classifier (Bokulich et al. 2018) which employs the classify-sklearn naïve Bayes taxonomy classifier against the SILVA 132 99% OTUs reference sequences (Quast et al. 2012). After clustering the ASVs, the metagenomic functional content was predicted using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) (Langille et al. 2013) for each DNA and cDNA library. The “Picrust2_pipeline.py” command was used to predict functions and annotate KO abundances. The command “add_comm.py” was used to annotate KO information. The Kyoto Encyclopedia of Genes and Genomes (KEGG) results were generated using the commands “pathway_pipeline.py” and “add_merchant.py”. For quality control, the Nearest Sequenced Index (weighted NSTI) was calculated for each sample. The results were in a satisfactory range for metagenomic predictions (mean NSTI = 0.17 ± 0.12 s.d.) (Langille et al. 2013).
Statistical analyses
ASVs with relative abundances less than 0.005% were removed (Bokulich et al. 2013). The communities were compared based on the Aitchison distances among the samples. Briefly, the ASV and KEGG pathway abundance matrices were represented by centered log-ratio (CLR)-transformed (zero value abundance with plus one processing), and the R language (version 3.6.1) “robcomposition” package (version 2.2.1) (Templ et al. 2011) was used to calculate the Aitchison distance between each pair of samples. The similarity between the DNA and cDNA libraries of the same sample was defined as 1 minus the Aitchison distance between them. According to the distance matrix, principal coordinates analysis (PCoA) was performed to show the differences among the samples, and non parametric multivariate analysis of variance (PERMANOVA) was used to analyze the significance of the differences. PCoA and PERMANOVA were performed using the vegan package (version 2.5–6) in R. As the bacterial communities revealed by DNA and cDNA libraries within each sample were highly similar, the samples were grouped by compartment and health status (without considering the factor DNA versus cDNA library) for PCoA and PERMANOVA.
The ALDeX2 package (version 1.18.0) (Fernandes et al. 2013) in R language was used to identify the differential ASVs and KEGG pathways based on the abundance matrix of ASVs and the functional genes, respectively. The differential ASVs and KEGG pathways between the DNA and cDNA libraries within each category of coral samples were identified. Differentials were also identified among coral sample categories to address the factors of coral compartment and health status (combining DNA and cDNA libraries within each coral sample). After CLR-transformation of the abundance matrix with the “aldex.clr” function, the significance of each factor was tested using the “aldex.kw” function. Regression fitting and the Benjamini–Hochberg corrected p-value variables were used by default in this analysis.
Results
Compositions of the coral-associated bacterial communities
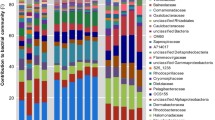
After quality control and removal of contaminated sequences, 435,603 sequences were retained from 24 libraries (12 cDNA and 12 DNA libraries) and 946 ASVs were yielded. All sequences were classified into 22 bacterial phyla or classes (Fig. 1), and 1.27% of all ASVs were unclassified. Gammaproteobacteria were predominant in healthy coral mucus (relative abundances 94–99%) and tissue (62–99%), except for one cDNA library of a tissue sample (2C), in which Alphaproteobacteria were dominant (54%). Gammaproteobacteria remained dominant in bleached coral mucus (83–95%), whereas Chloroflexi (30–46%), Alphaproteobacteria (4–13%), Acidobacteria (4–6%), and Actinobacteria (3–7%) were abundant in bleached coral tissue, except in the DNA (B2) and cDNA (B2C) libraries of the same tissue sample in which Gammaproteobacteria was a major group (98% and 96%, respectively).

Bacterial community composition in the coral samples. 1, 2, and 3 represent DNA libraries; 1C, 2C, and 3C represent cDNA libraries
The similarities (defined as one minus the Aitchison distance) between bacterial communities of the same coral sample revealed by the cDNA and DNA libraries were ˃89%, with 89–96% in healthy coral tissue, 93–95% in healthy coral mucus, 92–93% in bleached coral tissue, and 92–93% in bleached coral mucus (Figs. 1 and 2a). Additionally, the results of differential ASV analysis showed that only three ASVs were detected in the DNA and cDNA libraries of healthy coral mucus: a Gammaproteobacteria was enriched in the DNA libraries, whereas a Marinobacterium sp. and Alteromonas sp. were enriched in the cDNA libraries. No differential ASVs were observed in other comparisons between DNA and cDNA libraries. Moreover, the potential functional compositions predicted from the taxonomic compositions did not show a significant difference between the DNA and cDNA libraries of the same coral samples.

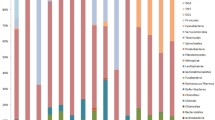
PCoA results based on ASVs (a) and KEGG pathways (b). HT and BT represent healthy and bleached coral tissue, respectively; HM and BM represent healthy and bleached coral mucus, respectively
Distinct bacterial communities associated with coral mucus and tissue
Bacterial communities were distinct in mucus and tissue, both for the healthy (PERMANOVA, R2 = 0.550, F = 12.247, p < 0.01) and bleached (PERMANOVA, R2 = 0.492, F = 9.684, p < 0.01) corals (Fig. 2a). Additionally, bacterial community functions were significantly different between the tissue and mucus of the healthy (PERMANOVA, R2 = 0.350, F = 5.380, p < 0.01) and bleached (PERMANOVA, R2 = 0.361, F = 5.660, p < 0.01) corals (Fig. 2b).
Furthermore, we defined significantly enriched (i.e., more abundant) or reduced (i.e., less abundant) ASVs using a corrected threshold of significance (p < 0.05; Fig. 3). In comparisons between healthy coral tissue and mucus, ASVs belonging to Marinobacterium, Alteromonadaceae, Rhodobacteraceae, Saccharospirillaceae, Bermanella, Litoricola, and Vibrio were enriched in the mucus layer, whereas three Endozoicomonas ASVs were enriched in the tissue. In bleached coral, ASVs affiliated to Marinobacterium, Alteromonadaceae, Bermanella, Saccharospirillaceae, Litoricola and Vibrio were mainly enriched in the mucus compared with that in the tissues (Fig. 3).

Bacterial ASVs with significant alterations in relative abundance. Each cell represents the log10-transformed relative abundance of each ASV. The relative abundance was transformed by log10(X + 1). 1, 2, and 3 represent DNA libraries; 1C, 2C, and 3C represent cDNA libraries. *ASV was enriched in healthy coral tissue compared with that in healthy coral mucus and in bleached coral tissue. #ASV was enriched in healthy coral mucus compared with that in healthy coral tissue. §ASV was enriched in bleached coral mucus compared with that in bleached coral tissue. ASVs without symbols appeared to be enriched in both the healthy and bleached coral mucus
Analysis of the relative abundances of functional genes involved in different KEGG pathways revealed significantly altered pathways. These were identified using ALDeX2 with a corrected threshold of significance (p < 0.05; Supplementary Fig. 1). Between healthy coral tissues and mucus, a total of 19 pathways were significantly different. Among them, functional genes involved in chlorocyclohexane and chlorobenzene degradation, bacterial chemotaxis, flagellar assembly, and D-arginine and D-ornithine metabolism were enriched in mucus, with > twofold higher relative abundances compared with tissue (Supplementary Fig. 1a). Between bleached coral tissue and mucus, a total of 34 pathways differed significantly. Among them, functional genes involved in glycosaminoglycan degradation, and chlorocyclonhexane and chlorobenzene degradation were enriched in mucus, with > threefold higher relative abundances in comparison with that in tissue (Supplementary Fig. 1b).
Differential responses of mucus- and tissue-associated bacterial communities to coral bleaching
The bacterial communities were significantly different between healthy and bleached coral tissues (PERMANOVA, R2 = 0.446, F = 8.039, p < 0.01), whereas they were similar in the mucus (PERMANOVA, R2 = 0.126, F = 1.444, p > 0.1; Fig. 2a). In addition, bacterial functions were significantly different between the healthy and bleached tissues (PERMANOVA, R2 = 0.351, F = 5.407, p < 0.01), whereas they were similar between the healthy and bleached mucus (PERMANOVA, R2 = 0.090, F = 0.991, p > 0.1) (Fig. 2b).
Three Endozoicomonas ASVs were enriched in healthy coral tissue, whereas their relative abundances decreased by more than 90% in bleached coral tissue (Fig. 3). In healthy coral tissue, the bacterial functions were mainly enriched in five pathways—notch signaling, primary and secondary bile acid biosynthesis, steroid hormone biosynthesis, and glycosaminoglycan degradation with 10 fold higher relative abundances than that in bleached tissue. In the bleached coral tissue, bacterial function was mainly enriched in proteasome, biosynthesis of type II polyketide backbone, N-glycan biosynthesis, D-arginine and D-ornithine metabolism, tetracycline biosynthesis, biosynthesis of ansamycins, and carotenoid biosynthesis (Supplementary Fig. 1c).
Discussion
Although DNA- and RNA-based sequencing has been used to reveal the total bacterial community and its active part, respectively, in various habitats (Hu et al. 2016; Stecher et al. 2016; Tian and Li 2017), these methods have not been applied simultaneously in coral holobionts. In this study, we analyzed the bacterial community structures in the tissue and mucus of healthy and bleached corals by using both DNA- and RNA-based 16S rRNA gene sequencing. The results show that coral-associated microbial communities revealed by investigating DNA and RNA/cDNA libraries were similar, in contrast to previous results obtained in seawater (Wang et al. 2019), sea ice (Stecher et al. 2016), and seagrass (Ling et al. 2018; Zhang et al. 2018; He and Guodong 2020). Given the consistent results obtained in this study by using these two methods, we suggest that both DNA- and RNA-based 16S rRNA gene sequencing are efficient and sensitive methods for active bacteria detection in P. damicornis. Considering that only one coral species was investigated in this study, the comparison is worth extending and validating in other coral species.
In this study, the functions of the bacterial community were predicted using PICRUSt. It has been suggested that there are two main limitations of amplicon-based functional prediction: (1) rare environment-specific functions are less likely to be identified, and (2) amplicon-based predictions cannot provide sufficient resolution to distinguish strain-specific functionality (Douglas et al. 2020). Therefore, the predicted functions revealed in this study should be interpreted with caution.
Investigation of the spatial organization of bacterial communities within the coral holobiont is crucial for understanding the relationship between coral and bacterial assemblages (Li et al. 2014). Consistent with the results of previous studies (Rohwer et al. 2002; Sweet et al. 2011; Bourne et al. 2013; Li et al. 2014), we found that the coral P. damicornis tissue and mucus layer were colonized by distinct bacterial communities. These distinct bacterial communities might be shaped by different microhabitats within the coral host (Sweet et al. 2011; Engelen et al. 2018). Compared with the variable environment in the mucus layer, coral tissue provided a more stable and host-controlled environment (Bourne and Munn 2005; Lee et al. 2015).
Coral tissue-associated bacterial communities altered when bleaching occurred. The dynamic nature of the coral-associated bacterial community is considered to be a mechanism that allows for the rapid adaptation of corals in a changing environment (Reshef et al. 2006). Our results suggest the potential for functions, such as tetracycline and ansamycins biosynthesis, to be enriched in bleached coral tissue, which might support this hypothesis, considering their contribution to the inhibition of opportunistic pathogens. Thus, we suggest that the shifting bacterial communities in the tissue of bleached P. damicornis might be an acclimatation to perturbation.
Endozoicomonas was a dominant group in the healthy coral tissue, whereas its relative abundance decreased markedly in bleached coral tissue; these findings are consistent with the results of previous reports (Bourne et al. 2008; Gardner et al. 2019; Shiu and Tang 2019; Li et al. 2021). The dominance of Endozoicomonas in healthy coral tissue implies that it may play an important role in coral health (Rosenberg et al. 2007; Galand et al. 2018; Pogoreutz et al. 2018). Endozoicomonas has been hypothesized to play a role in providing nutrients, constructing the host microbiome, and synthesizing amino acids and antibacterial substances (Shiu and Tang 2019). The difference in the relative abundance of Endozoicomonas between healthy and bleached corals further supports the hypothesis that this group could be an indicator of the health status of P. damicornis (Rosenberg et al. 2007; Neave et al. 2017; Galand et al. 2018; Pogoreutz et al. 2018). Both the results of this study and a previous report (Li et al. 2021) showed that diverse bacteria, including Acidobacteria, Chloroflexi, Dadabacteria, and Nitrospirae, were enriched in bleached coral tissue, whereas the abundance of Endozoicomonas was reduced. The reduced abundance of Endozoicomonas might diminish its regulatory effect on the coral-associated bacterial community (Rua et al. 2014; Neave et al. 2016, 2017; Li et al. 2021).
In this study, the bacterial communities associated with P. damicornis mucus were found to be stable when bleaching occurred. This result is consistent with a previous report, in which a stable mucus-associated microbiome was observed in healthy and bleached Porites lobata (Hadaidi et al. 2017). However, the coral Acropora muricata showed a successive shift in the microbial community associated with its mucus layer under thermal stress (Lee et al. 2015). These inconsistent observations might be attributed to the different coral species. We speculate that a stable microbial community structure could help mucus maintain its protective function (Brown and Bythell 2005; Ritchie 2006; Bythell and Wild 2011; Hadaidi et al. 2017) after coral bleaching. Additionally, Bermanella and Alteromonadaceae were abundant in healthy and bleached coral mucus, this finding could be attributed to their metabolism of polysaccharides (Lenihan and Edmunds 2010), which are the main components of coral mucus (Brown and Bythell 2005; Sweet et al. 2011; Krediet et al. 2013). The enrichment of Vibrio in mucus might be because of its chemotaxis toward the mucus layer (Garren et al. 2014).
We noticed that there were two libraries (2 and 2C) of a healthy tissue and two (B2 and B2C) of a bleached tissue that showed differential bacterial community structures in comparison with others in the same group (i.e., healthy or bleached tissue). These results might be related to the different genotypes of the coral host (Pantos et al. 2015; Ziegler et al. 2017; Miller et al. 2020). This result suggests that the genotype of coral samples should be assessed in future studies.
This study provided the first comparison of bacterial communities revealed by using both DNA- and RNA-based 16S rRNA gene sequencing in coral holobionts, and we found that they were similar between cDNA (active bacterial members) and DNA (the total bacterial community) libraries of the same coral sample. The results of this study show that bacterial assemblages tended to be dynamic in the tissue of P. damicornis when bleaching occurs, whereas they were rigid in the mucus (Fig. 4). The dominant genus in healthy coral tissue, namely Endozoicomonas, showed a dramatic decrease in relative abundance upon coral bleaching, suggesting that this group might be an indicator of coral health. These results will advance our understanding of the relationship between coral health and bacterial symbionts.

Differential responses of the microbial communities in coral tissue and mucus to bleaching. Samples were collected from three healthy and three partially bleached coral colonies. The metagenomic functional content was predicted using PICRUSt based on the results of 16S rRNA gene sequencing
Data availability
The datasets generated during the current study have been deposited and are publicly available in the Sequence Read Archive repository under BioProject ID PRJNA732546.
References
Ainsworth TD, Hoegh-Guldberg O (2009) Bacterial communities closely associated with coral tissues vary under experimental and natural reef conditions and thermal stress. Aquat Biol 4:289–296
Ainsworth TD, Thurber RV, Gates RD (2010) The future of coral reefs: a microbial perspective. Trends Ecol Evol 25:233–240. https://doi.org/10.1016/j.tree.2009.11.001
Ainsworth TD, Krause L, Bridge T, Torda G, Raina J-B, Zakrzewski M, Gates RD, Padilla-Gamiño JL, Spalding HL, Smith C (2015) The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J 9:2261. https://doi.org/10.1038/ismej.2015.39
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG (2013) Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10:57–59. https://doi.org/10.1038/nmeth.2276
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Caporaso JG (2018) Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:1–17. https://doi.org/10.1186/s40168-018-0470-z
Bolyen E, Rideout J, Dillon M, Bokulich N, Abnet C, Al-Ghalith G, Alexander H, Alm E, Arumugam M, Asnicar F, Bai Y, Bisanz J, Bittinger K, Brejnrod A, Brislawn C, Brown C, Callahan B, Caraballo-Rodríguez A, Chase J, Cope E, Da Silva R, Diener C, Dorrestein P, Douglas G, Durall D, Duvallet C, Edwardson C, Ernst M, Estaki M, Fouquier J, Gauglitz J, Gibbons S, Gibson D, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley G, Janssen S, Jarmusch A, Jiang L, Kaehler B, Kang K, Keefe C, Keim P, Kelley S, Knights D, Koester I, Kosciolek T, Kreps J, Langille M, Lee J, Ley R, Liu Y, Loftfield E, Lozupone C, Maher M, Marotz C, Martin B, McDonald D, McIver L, Melnik A, Metcalf J, Morgan S, Morton J, Naimey A, Navas-Molina J, Nothias L, Orchanian S, Pearson T, Peoples S, Petras D, Preuss M, Pruesse E, Rasmussen L, Rivers A, Robeson M, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song S, Spear J, Swafford A, Thompson L, Torres P, Trinh P, Tripathi A, Turnbaugh P, Ul-Hasan S, van der Hooft J, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber K, Williamson C, Willis A, Xu Z, Zaneveld J, Zhang Y, Zhu Q, Knight R, Caporaso J (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. https://doi.org/10.1038/s41587-019-0209-9
Botté ES, Cantin NE, Mocellin VJ, O’Brien PA, Rocker MM, Frade PR, Webster NS (2022) Reef location has a greater impact than coral bleaching severity on the microbiome of Pocillopora acuta. Coral Reefs 41:63–79. https://doi.org/10.1007/s00338-021-02201-y
Bourne DG, Munn CB (2005) Diversity of bacteria associated with the coral Pocillopora damicornis from the Great Barrier Reef. Environ Microbiol 7:1162–1174. https://doi.org/10.1111/j.1462-2920.2005.00793.x
Bourne D, Iida Y, Uthicke S, Smith-Keune C (2008) Changes in coral-associated microbial communities during a bleaching event. ISME J 2:350–363. https://doi.org/10.1038/ismej.2007.112
Bourne DG, Dennis PG, Uthicke S, Soo RM, Tyson GW, Webster N (2013) Coral reef invertebrate microbiomes correlate with the presence of photosymbionts. ISME J 7:1452–1458. https://doi.org/10.1038/ismej.2012.172
Brown B, Bythell J (2005) Perspectives on mucus secretion in reef corals. Mar Ecol Prog Ser 296:291–309. https://doi.org/10.3354/meps296291
Bythell JC, Wild C (2011) Biology and ecology of coral mucus release. J Exp Mar Biol Ecol 408:88–93. https://doi.org/10.1016/j.jembe.2011.07.028
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Daniels CA, Zeifman A, Heym K, Ritchie KB, Watson CA, Berzins I, Breitbart M (2011) Spatial heterogeneity of bacterial communities in the mucus of Montastraea annularis. Mar Ecol Prog Ser 426:29–40. https://doi.org/10.3354/meps09024
Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, Huttenhower C, Langille MG (2020) PICRUSt2 for prediction of metagenome functions. Nat Biotechnol 38:685–688. https://doi.org/10.1038/s41587-020-0548-6
Egge ES, Johannessen TV, Andersen T, Eikrem W, Bittner L, Larsen A, Sandaa RA, Edvardsen B (2015) Seasonal diversity and dynamics of haptophytes in the Skagerrak, Norway, explored by high-throughput sequencing. Mol Ecol 24:3026–3042. https://doi.org/10.1111/mec.13160
Engelen AH, Aires T, Vermeij MJ, Herndl GJ, Serrao EA, Frade PR (2018) Host differentiation and compartmentalization of microbial communities in the azooxanthellate cupcorals Tubastrea coccinea and Rhizopsammia goesi in the Caribbean. Front Mar Sci 5:391. https://doi.org/10.3389/fmars.2018.00391
Fernandes AD, Macklaim JM, Linn TG, Reid G, Gloor GB (2013) ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-Seq. PLoS ONE 8:e67019. https://doi.org/10.1371/journal.pone.0067019
Frias-Lopez J, Zerkle AL, Bonheyo GT, Fouke BW (2002) Partitioning of bacterial communities between seawater and healthy, black band diseased, and dead coral surfaces. Appl Environ Microbiol 68:2214–2228. https://doi.org/10.1128/AEM.68.5.2214-2228.2002
Galand PE, Chapron L, Meistertzheim A-L, Peru E, Lartaud F (2018) The effect of captivity on the dynamics of active bacterial communities differs between two deep-sea coral species. Front Microbiol 9:2565. https://doi.org/10.3389/fmicb.2018.02565
Gardner SG, Camp EF, Smith DJ, Kahlke T, Osman EO, Gendron G, Hume BC, Pogoreutz C, Voolstra CR, Suggett DJ (2019) Coral microbiome diversity reflects mass coral bleaching susceptibility during the 2016 El Niño heat wave. Ecol Evol 9:938–956. https://doi.org/10.1002/ece3.4662
Garren M, Son K, Raina J-B, Rusconi R, Menolascina F, Shapiro OH, Tout J, Bourne DG, Seymour JR, Stocker R (2014) A bacterial pathogen uses dimethylsulfoniopropionate as a cue to target heat-stressed corals. ISME J 8:999–1007. https://doi.org/10.1038/ismej.2013.210
Goyen S, Camp EF, Fujise L, Lloyd A, Nitschke MR, LaJeunensse T, Kahlke T, Ralph PJ, Suggett D (2019) Mass coral bleaching of P. versipora in Sydney Harbour driven by the 2015–2016 heatwave. Coral Reefs 38:815–830. https://doi.org/10.1007/s00338-019-01797-6
Hadaidi G, Röthig T, Yum LK, Ziegler M, Arif C, Roder C, Burt J, Voolstra CR (2017) Stable mucus-associated bacterial communities in bleached and healthy corals of Porites lobata from the Arabian Seas. Sci Rep 7:1–11. https://doi.org/10.1038/srep45362
He X, Guodong J (2020) Responses of AOA and AOB activity and DNA/cDNA community structure to allylthiourea exposure in the water level fluctuation zone soil. Environ Sci Pollut Res 27:15233–15244. https://doi.org/10.1007/s11356-020-07952-9
Herlemann DP, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ, Andersson AF (2011) Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5:1571–1579. https://doi.org/10.1038/ismej.2011.41
Heron SF, Maynard JA, Van Hooidonk R, Eakin CM (2016) Warming trends and bleaching stress of the world’s coral reefs 1985–2012. Sci Rep 6:1–14. https://doi.org/10.1038/srep38402
Hu SK, Campbell V, Connell P, Gellene AG, Liu Z, Terrado R, Caron DA (2016) Protistan diversity and activity inferred from RNA and DNA at a coastal ocean site in the eastern North Pacific. FEMS Microbiol Ecol 92:fiw050. https://doi.org/10.1093/femsec/fiw050
Kimes NE, Van Nostrand JD, Weil E, Zhou J, Morris PJ (2010) Microbial functional structure of Montastraea faveolata, an important Caribbean reef-building coral, differs between healthy and yellow-band diseased colonies. Environ Microbiol 12:541–556. https://doi.org/10.1111/j.1462-2920.2009.02113.x
Klaus JS, Frias-Lopez J, Bonheyo GT, Heikoop JM, Fouke BW (2005) Bacterial communities inhabiting the healthy tissues of two Caribbean reef corals: interspecific and spatial variation. Coral Reefs 24:129–137. https://doi.org/10.1007/s00338-004-0447-1
Klein AM, Bohannan BJ, Jaffe DA, Levin DA, Green JL (2016) Molecular evidence for metabolically active bacteria in the atmosphere. Front Microbiol 7:772. https://doi.org/10.3389/fmicb.2016.00772
Krediet CJ, Ritchie KB, Paul VJ, Teplitski M (2013) Coral-associated micro-organisms and their roles in promoting coral health and thwarting diseases. Proc R Soc B 280:20122328. https://doi.org/10.1098/rspb.2012.2328
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Chichester, UK, pp 115–175
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Thurber RLV, Knight R (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821. https://doi.org/10.1038/nbt.2676
Lee ST, Davy SK, Tang S-L, Fan T-Y, Kench PS (2015) Successive shifts in the microbial community of the surface mucus layer and tissues of the coral Acropora muricata under thermal stress. FEMS Microbiol Ecol 91:fiv142. https://doi.org/10.1093/femsec/fiv142
Lema KA, Willis BL, Bourne DG (2012) Corals form characteristic associations with symbiotic nitrogen-fixing bacteria. Appl Environ Microbiol 78:3136–3144. https://doi.org/10.1128/AEM.07800-11
Lema KA, Bourne DG, Willis BL (2014) Onset and establishment of diazotrophs and other bacterial associates in the early life history stages of the coral Acropora millepora. Mol Ecol 23:4682–4695. https://doi.org/10.1111/mec.12899
Lenihan HS, Edmunds PJ (2010) Response of Pocillopora verrucosa to corallivory varies with environmental conditions. Mar Ecol Prog Ser 409:51–63. https://doi.org/10.3354/meps08595
Li J, Chen Q, Long L-J, Dong J-D, Yang J, Zhang S (2014) Bacterial dynamics within the mucus, tissue and skeleton of the coral Porites lutea during different seasons. Sci Rep 4:1–8. https://doi.org/10.1038/srep07320
Li J, Long L, Zou Y, Zhang S (2021) Microbial community and transcriptional responses to increased temperatures in coral Pocillopora damicornis holobiont. Environ Microbiol 23:826–843. https://doi.org/10.1111/1462-2920.15168
Ling J, Lin X, Zhang Y, Zhou W, Yang Q, Lin L, Zeng S, Zhang Y, Wang C, Ahmad M (2018) Community composition and transcriptional activity of ammonia-oxidizing prokaryotes of seagrass Thalassia hemprichii in coral reef ecosystems. Front Microbiol 9:7. https://doi.org/10.3389/fmicb.2018.00007
Littman RA, Willis BL, Pfeffer C, Bourne DG (2009) Diversities of coral-associated bacteria differ with location, but not species, for three acroporid corals on the Great Barrier Reef. FEMS Microbiol Ecol 68:152–163. https://doi.org/10.1111/j.1574-6941.2009.00666.x
Marchioro GM, Glasl B, Engelen AH, Serrão EA, Bourne DG, Webster NS, Frade PR (2020) Microbiome dynamics in the tissue and mucus of acroporid corals differ in relation to host and environmental parameters. PeerJ 8:e9644. https://doi.org/10.7717/peerj.9644
Miller N, Maneval P, Manfrino C, Frazer TK, Meyer JL (2020) Spatial distribution of microbial communities among colonies and genotypes in nursery-reared Acropora cervicornis. PeerJ 8:e9635. https://doi.org/10.7717/peerj.9635
Neave MJ, Apprill A, Ferrier-Pagès C, Voolstra CR (2016) Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas. Appl Microbiol Biotechnol 100:8315–8324. https://doi.org/10.1007/s00253-016-7777-0
Neave MJ, Michell CT, Apprill A, Voolstra CR (2017) Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci Rep 7:1–12. https://doi.org/10.1038/srep40579
Nguyen-Kim H, Bettarel Y, Bouvier T, Bouvier C, Doan-Nhu H, Nguyen-Ngoc L, Nguyen-Thanh T, Tran-Quang H, Brune J (2015) Coral mucus is a hot spot for viral infections. Appl Environ Microbiol 81:5773–5783. https://doi.org/10.1128/AEM.00542-15
Oliver JK, Berkelmans R, Eakin CM (2018) Coral bleaching in space and time. In: van Oppen MJH, Lough JM (eds) Coral bleaching. Springer, Berlin, pp 27–49
Pantos O, Cooney RP, Le Tissier MD, Barer MR, O’Donnell AG, Bythell JC (2003) The bacterial ecology of a plague-like disease affecting the Caribbean coral Montastrea annularis. Environ Microbiol 5:370–382. https://doi.org/10.1046/j.1462-2920.2003.00427.x
Pantos O, Bongaerts P, Dennis PG, Tyson GW, Hoegh-Guldberg O (2015) Habitat-specific environmental conditions primarily control the microbiomes of the coral Seriatopora hystrix. ISME J 9:1916–1927. https://doi.org/10.1038/ismej.2015.3
Pogoreutz C, Rädecker N, Cárdenas A, Gärdes A, Wild C, Voolstra CR (2018) Dominance of Endozoicomonas bacteria throughout coral bleaching and mortality suggests structural inflexibility of the Pocillopora verrucosa microbiome. Ecol Evol 8:2240–2252. https://doi.org/10.1002/ece3.3830
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2012) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. https://doi.org/10.1093/nar/gks1219
Raina J-B, Dinsdale EA, Willis BL, Bourne DG (2010) Do the organic sulfur compounds DMSP and DMS drive coral microbial associations? Trends Microbiol 18:101–108. https://doi.org/10.1016/j.tim.2009.12.002
Raina J-B, Tapiolas D, Motti CA, Foret S, Seemann T, Tebben J, Willis BL, Bourne DG (2016) Isolation of an antimicrobial compound produced by bacteria associated with reef-building corals. PeerJ 4:e2275. https://doi.org/10.7717/peerj.2275
Reshef L, Koren O, Loya Y, Zilber-Rosenberg I, Rosenberg E (2006) The coral probiotic hypothesis. Environ Microbiol 8:2068–2073. https://doi.org/10.1111/j.1462-2920.2006.01148.x
Ritchie KB (2006) Regulation of microbial populations by coral surface mucus and mucus-associated bacteria. Mar Ecol Prog Ser 322:1–14. https://doi.org/10.3354/meps322001
Rohwer F, Seguritan V, Azam F, Knowlton N (2002) Diversity and distribution of coral-associated bacteria. Mar Ecol Prog Ser 243:1–10. https://doi.org/10.3354/meps243001
Rosenberg E, Koren O, Reshef L, Efrony R, Zilber-Rosenberg I (2007) The role of microorganisms in coral health, disease and evolution. Nat Rev Microbiol 5:355–362. https://doi.org/10.1038/nrmicro1635
Rua CP, Trindade-Silva AE, Appolinario LR, Venas TM, Garcia GD, Carvalho LS, Lima A, Kruger R, Pereira RC, Berlinck RG (2014) Diversity and antimicrobial potential of culturable heterotrophic bacteria associated with the endemic marine sponge Arenosclera brasiliensis. PeerJ 2:e419. https://doi.org/10.7717/peerj.419
Sharp KH, Distel D, Paul VJ (2012) Diversity and dynamics of bacterial communities in early life stages of the Caribbean coral Porites astreoides. ISME J 6:790–801. https://doi.org/10.1038/ismej.2011.144
Shashar N, Kinane S, Jokiel P, Patterson M (1996) Hydromechanical boundary layers over a coral reef. J Exp Mar Biol Ecol 199:17–28. https://doi.org/10.1016/0022-0981(95)00156-5
Shiu J-H, Tang S-L (2019) The bacteria Endozoicomonas: community dynamics, diversity, genomes, and potential impacts on corals. In: Li Z (ed) Symbiotic microbiomes of coral reefs sponges and corals. Springer, Berlin, pp 55–67
Stecher A, Neuhaus S, Lange B, Frickenhaus S, Beszteri B, Kroth PG, Valentin K (2016) rRNA and rDNA based assessment of sea ice protist biodiversity from the central Arctic Ocean. Eur J Phycol 51:31–46. https://doi.org/10.1080/09670262.2015.1077395
Sweet M, Croquer A, Bythell J (2011) Bacterial assemblages differ between compartments within the coral holobiont. Coral Reefs 30:39–52. https://doi.org/10.1007/s00338-010-0695-1
Templ M, Hron K, Filzmoser P (2011) robCompositions: an R-package for robust statistical analysis of compositional data. In: Pawlowsky-Glahn V, Buccianti A (eds) Compositional data analysis: theory and applications. John Wiley & Sons, New Jersey, pp 341–354
Terrado R, Medrinal E, Dasilva C, Thaler M, Vincent WF, Lovejoy C (2011) Protist community composition during spring in an Arctic flaw lead polynya. Polar Biol 34:1901–1914. https://doi.org/10.1007/s00300-011-1039-5
Thurber RV, Willner-Hall D, Rodriguez-Mueller B, Desnues C, Edwards RA, Angly F, Dinsdale E, Kelly L, Rohwer F (2009) Metagenomic analysis of stressed coral holobionts. Environ Microbiol 11:2148–2163. https://doi.org/10.1111/j.1462-2920.2009.01935.x
Tian Y, Li YH (2017) Comparative analysis of bacteria associated with different mosses by 16S rRNA and 16S rDNA sequencing. J Basic Microbiol 57:57–67. https://doi.org/10.1002/jobm.201600358
Wang F, Xie Y, Wu W, Sun P, Wang L, Huang B (2019) Picoeukaryotic diversity and activity in the northwestern Pacific Ocean based on rDNA and rRNA high-throughput sequencing. Front Microbiol 9:3259. https://doi.org/10.3389/fmicb.2018.03259
Ward DM, Weller R, Bateson MM (1990) 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nat 345:63–65. https://doi.org/10.1038/345063a0
Webster NS, Thomas T (2016) The sponge hologenome. Mbio 7:e00135-e1116. https://doi.org/10.1128/mBio.00135-16
Weinbauer MG, Fritz I, Wenderoth DF, Höfle MG (2002) Simultaneous extraction from bacterioplankton of total RNA and DNA suitable for quantitative structure and function analyses. Appl Environ Microbiol 68:1082–1087. https://doi.org/10.1128/AEM.68.3.1082-1087.2002
Wilhelm L, Besemer K, Fasching C, Urich T, Singer GA, Quince C, Battin TJ (2014) Rare but active taxa contribute to community dynamics of benthic biofilms in glacier-fed streams. Environ Microbiol 16:2514–2524. https://doi.org/10.1111/1462-2920.12392
Zhang LM, Duff AM, Smith CJ (2018) Community and functional shifts in ammonia oxidizers across terrestrial and marine (soil/sediment) boundaries in two coastal Bay ecosystems. Environ Microbiol 20:2834–2853. https://doi.org/10.1111/1462-2920.14238
Ziegler M, Seneca FO, Yum LK, Palumbi SR, Voolstra CR (2017) Bacterial community dynamics are linked to patterns of coral heat tolerance. Nat Commun 8:1–8. https://doi.org/10.1038/ncomms14213
Acknowledgements
This work was supported by the National Natural Science Foundation of China (41676155, 42122045, and 41890853), Guangdong Natural Science Funds for Distinguished Young Scholars (2017A030306025), and K. C. Wong Education Foundation (GJTD-2020-12).
Author information
Authors and Affiliations
Contributions
J.L. and S.Z. conceived this study. Y.Z. and J.L. performed the analyses. Y.Z., J.L., Y.C., and L.W. drafted the manuscript, and all authors commented on the manuscript and made suggestions.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that no competing interests exist.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Topic Editor Steve Vollmer
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zou, Y., Chen, Y., Wang, L. et al. Differential responses of bacterial communities in coral tissue and mucus to bleaching. Coral Reefs 41, 951–960 (2022). https://doi.org/10.1007/s00338-022-02261-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00338-022-02261-8