
Abstract
The recently developed CRISPR/Cas9 system is a promising technology for targeted genome editing in a variety of species including plants. However, the first generation systems were designed to target one or two gene loci at a time. We designed a new multiplex CRISPR/Cas9 system that allows the co-expression of six sgRNA modules in one binary vector using a simple (three steps) cloning strategy in Arabidopsis. The transcription of the sgRNA modules is under the control of three different RNA Polymerase III-dependent promoters. We tested the efficiency of the new multiplex system by targeting six of the fourteen PYL families of ABA receptor genes in a single transformation experiment. One line with mutations in all six targeted PYLs was identified from 15 T1 plants. The mutagenesis frequency for the six individual PYL targets in the T1 lines ranged from 13 to 93 %. In the presence of ABA, the transgenic line identified as containing mutations in all six PYL genes produced the highest germination rate in the T2 progeny (37 %). Among these germinated seedlings, half of the analyzed plants (15/30) were homozygous mutants for at least four targeted genes and two plants (6.7 %) contained homozygous mutations in five of the targeted PYLs and the other targeted PYL had biallelic mutations. Homozygous sextuple mutants were identified in the T3 progeny and characterized together with previously described triple and sextuple PYL mutants. We anticipate that the application of this multiplex CRISPR/Cas9 system will strongly facilitate functional analysis of genes pathways and families.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reverse genetics is the most popular approach for gene functional studies in plants (Alonso and Ecker 2006). Targeted gene silencing by RNA interference (RNAi) has been widely employed for loss-of-function gene studies, and for Arabidopsis thaliana, a vast collection of T-DNA mutant lines with known insertion sites has been available for some time (Alonso and Ecker 2006; Waterhouse et al. 1998). However, both approaches have important practical limitations. Silencing levels achieved by RNAi in individual lines are variable and the stability of silencing over several generations is not guaranteed. In addition the production of mutant collections with characterized insertion sites similar to the one existing in Arabidopsis is not practical in most plant species. The emergence of the Streptococcus pyogenes derived CRISPR/Cas9 system as a powerful tool for genome editing can facilitate gene functional studies in a variety of organisms, including plants (Doudna and Charpentier 2014). The engineered CRISPR/Cas system is composed of a single-stranded guide RNA (sgRNA) for target DNA recognition via base pairing, and a CRISPR-associated protein (Cas9) for DNA cleavage ~3 bp upstream of the protospacer adjacent motif (PAM = ‘NGG’) (Jinek et al. 2012). In the absence of homologous DNA templates, the CRISPR/Cas9-triggered double-stranded breaks will be repaired via the error-prone non-homologous end joining (NHEJ) pathway, resulting in the generation of frame-shift mutations in the targeted genes (Cong et al. 2013; Mali et al. 2013).
Successful application of the CRISPR/Cas9 gene editing system has been reported in a variety of plant species (Brooks et al. 2014; Fauser et al. 2014; Feng et al. 2013; Jia and Wang 2014; Jiang et al. 2013; Li et al. 2013a; Mao et al. 2013; Miao et al. 2013; Nekrasov et al. 2013; Shan et al. 2013; Upadhyay et al. 2013; Xie and Yang 2013; Xing et al. 2014; Zhang et al. 2014). However, most of the first generation CRISPR/Cas9 systems used for plant gene editing usually provided only one or two docking sites for sgRNA modules, thus limiting the gene-targeting capacity of the system. New CRISPR/Cas9 systems have been designed for multiplex gene editing in plants thus allowing systematic study of gene families or metabolic pathways (Xie et al. 2015; Xing et al. 2014).
In animals, multiplex gene editing can be achieved by co-injection of in vitro transcribed sgRNAs and Cas9 mRNA into the one-cell embryo (Li et al. 2013b, c; Wang et al. 2013). However, in plants, this method is not practical due to the physical barrier provided by the cell wall. Instead, the Agrobacterium-mediated gene transformation method is used for delivering the CRISPR/Cas9 system into germline cells (Katavic et al. 1994). To perform multiplex gene targeting in plants, the crux of the problem is how to construct multiple sgRNA modules into one binary vector along with the expression cassette for Cas9. One solution is to mimic the native composition of type II or type III CRISPR arrays (Cong et al. 2013; Nissim et al. 2014). For such RNA precursors, the tandem repeated crRNAs or sgRNAs units are first transcribed from a RNA Pol III (Pol III) promoter and then subjected to the excision of an engineered endonuclease system to release the individual RNA components. Recently, a similar design hijacking the plant endogenous tRNA processing system for sgRNA maturation was reported in rice (Xie et al. 2015). However, a potential risk of applying these strategies is that transformation of highly repetitive DNA sequences might cause transgenic gene silencing in a variety of species (Ma and Mitra 2002).
To reduce the repetitiveness of the sgRNA cassette, we adopted a relatively conservative strategy by engineering three sgRNAs modules with different Poll III-dependent promoters. These three sgRNA modules were designed with adaptive restriction sites to allow one step assembly of multiple fragments. Using a previously developed CRISPR/Cas9 system with two separated sgRNA docking sites (Mao et al. 2013), we generated a sextuple CRISPR/Cas9 vector containing six sgRNAs. Using this upgraded CRISPR/Cas9 system, we successfully targeted six of the 14 PYR/PYL gene family members (PYR1, PYL1, PYL2, PYL4, PYL5 and PYL8) in a single transformation event. These six PYR/PYL genes encode a branch of the ABA receptors in Arabidopsis (Park et al. 2009). In the absence of these ABA receptors, the newly obtained pyl sextuple mutant exhibited an ABA-insensitive phenotype similar to the previously described sextuple mutant, which has a mixed background of both Col-0 and Ler (Gonzalez-Guzman et al. 2012). The targeting efficiency of the three sgRNA modules was studied using YFP single-stranded annealing assay (SSA) in protoplasts. Our results show that the AtU6 and At7SL promoters had relatively higher activity than the AtU3b promoter for targeted gene modification in protoplasts. The sequential order of these sgRNA modules in the expression cassettes had no effect on their gene targeting efficiency. Thus, we propose that this upgraded CRISPR/Cas9 system can be used for efficient multiplex gene editing in Arabidopsis with up to six sgRNAs. Future application of this system will facilitate the generation of high order mutants for the functional analysis of gene families in Arabidopsis and other plants.
Results
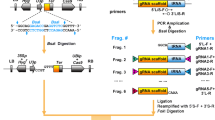
Workflow of vector construction for sextuple gene targeting in Arabidopsis
The workflow to produce the sextuple CRISPR/Cas9 vector for targeted gene modification is shown in Fig. 1. To construct multiple sgRNA modules for gene targeting in Arabidopsis, three different Pol III-dependent gene promoters (AtU6-26, AtU3b and At7SL-2) were cloned from the Arabidopsis genome to drive the expression of the sgRNAs. For target recognition, 20-nt guide oligo-nucleotides were synthesized with appropriate adaptors for seamless ligation with the three sgRNA modules separately. Note that the 5′ adaptor sequences of those synthesized oligo-nucleotides were actually determined by the designated sgRNA promoters (Table 1). Subsequently, the three customized sgRNA modules were digested with the corresponding restriction enzymes for cloning into the Cas9 expression vector in tandem. The resulting CRISPR/Cas9 vectors were used directly for plant transformation (for 3-gene targeting) or as an intermediate vector for further construction of the sextuple construct. In the later case, two intermediate CRISPR/Cas9 vectors with three sgRNAs each were constructed in parallel from the beginning. To clone the second sgRNA cassette, it was first amplified from its own intermediate vector by PCR, and then cloned into the construct already containing the first three sgRNAs to generate the final CRISPR/Cas9 sextuple vector.

Pipeline for the construction of a sextuple CRISPR/Cas9 system for multiplex gene editing in Arabidopsis. The assembly of a sextuple CRISPR/Cas9 expression vector can be accomplished in three steps. (1) Clone synthesized guide oligo-nucleotides into the sgRNA modules. For ligation with the three sgRNA scaffolds containing different Pol III promoters, a specific 5-nt adapter should be added to the 5′ end of the sense oligo-nucleotides. The sequences of the adaptors for the sense oligos are determined by their promoters. (2) Assemble the first three sgRNA modules (sgRNAs 1, 2 and 3) into the expression vector containing Cas9. The three customized sgRNA modules can be digested directly from their hosting vectors with the indicated restriction enzymes or after PCR amplification by M13F/M13R. The digested fragments are subsequently cloned into the HindIII and XmaI sites of the binary vector containing Cas9 in tandem to produce an intermediate vector with three sgRNAs. To construct the sextuple vector, a second triple intermediate vector containing sgRNAs 4, 5 and 6 is generated as described above (cloning sgRNAs 4, 5, and 6 into the HindIII and XmaI sites of the binary vector containing Cas9). (3) To clone the second sgRNA cassette into the intermediate vector, the fragment containing sgRNAs 4, 5 and 6 is amplified by PCR with primers containing KpnI and EcoRI. The amplicon is then digested with KpnI and EcoRI and cloned into the intermediate vector to generate the final CRISPR/Cas9 binary vector with six sgRNAs. The three Pol III-dependent promoters are labeled with different color. Red arrowheads indicate the M13F and M13R sites in plasmids. Blue rectangles indicate the left and right border sites of T-DNA. HygR hygromycin resistance gene, pVS1-Rep bacterial origin of replication, pAtUBQ promoter of AtUBQ1 gene, tUBQ terminator of AtUBQ1 gene, hspCas9 human-codon optimized Cas9 gene
Assemblage of a sextuple CRISPR/Cas9 vector for PYL gene targeting
In Arabidopsis, perception of the phytohormone abscisic acid (ABA) is mediated by proteins in the 14-member PYR/PYL family (Park et al. 2009). A PYR/PYL receptor sextuple mutant, namely pyr1pyl1pyl2pyl4pyl5pyl8 (pyl112458) (all mutations except pyl2 were in the Col-0 background) was reported to be insensitive to high ABA concentrations (Gonzalez-Guzman et al. 2012). To facilitate the screening of sextuple mutants as well as to test the gene editing efficiency of the multiplex CRISPR/Cas9 system, these six PYR/PYL genes were selected as targets for editing. For most of the PYL genes, the sgRNA target sites were chosen from the CDS region, while for PYL8, the sgRNA target site was located within the 5′ untranslated (UTR) region (Fig. 2a). Phylogenetic analysis of PYL/PYR genes, reveals that the six targeted PYL/PYR genes can be classified into three clades and each gene shows limited sequence similarity with other members (Table S1). To predict the specificity of the sgRNAs, potential off-target sites were investigated using the Blastn algorithm with the full length guide sequences (including 3nt PAMs) as queries. In total, 24 putative off-targets were identified in the Arabidopsis genome (Exp <3.0) with all but one containing at least two mismatches. The most significant putative off-target sequence belonged to PYL1 with a single mismatch compared to the target site for sgR-PYR1 (Table S2). The arrangement of these PYL targeting sgRNA modules in the CRISPR/Cas9 sextuple construct is shown in Fig. 2b. This binary vector was used for Agrobacterium-meditated transformation of Arabidopsis. In subsequent analysis, the six PYL genes were always presented in the order of PYL1 > PYR1 > PYL4 > PYL2 > PYL8 > PYL5 following the order of sgRNAs that target these genes.

Targeted gene modification of PYL genes using the sextuple CRISPR/Cas9 system. a Schematics of the PYL gene loci. CDS regions are shown in yellow and the mRNA transcripts are depicted in black lines. Location and the sequences of the sgRNA targets are indicated with PAM and GC % content in red. b Schematic of the sextuple CRISPR/Cas9 vector used to modify PYL genes. Red arrowheads indicate the M13F and M13R sites in plasmids. Blue rectangles indicate the left and right border sites of T-DNA. HygR hygromycin resistant gene, pVS1-Rep bacterial origin of replication, pAtUBQ promoter of AtUBQ1 gene, tUBQ terminator of AtUBQ1 gene, hspCas9 human-codon optimized Cas9 gene
Fifteen individual T1 transformants were analyzed by sequencing their PYL target regions. Triple mutants (six plants) and quintuple mutants (three plants) were identified as the two dominant multiple mutations in the population, while only one sextuple mutant (line #11) was detected (Fig. 3a). The mutation efficiencies for the different loci were highly variable with PYR1, PYL1 and PYL2 showing high efficiency (>70 % of T1 plants), while for the remaining three loci the efficiency was low (13–26 %) (Fig. 3b). The combination patterns of the PYL mutations observed in the 15 T1 plants is summarized in Fig. 3c.

Characterization of the CRISPR/Cas9-induced PYL gene mutations in the T1 generation. a Frequency distribution of the chimeric pyl multiple mutants. b Mutagenesis frequency at the six individual PYL gene targets. c Summary of the different PYL mutation combinations in T1 plants. PCR products of the six PYL target loci in each T1 plant were sequenced for mutation detection. Samples with detected gene mutations are shown in green. The T1 line containing sextuple gene mutations is highlighted in red
Moreover, we investigated the putative off-target site of sgR-PYR1 in the PYL1 gene but no mutations were detected among the 15 T1 plants (Fig. S1).
Identification of pyl sextuple mutants in the T2 generation
To facilitate the screening of heritable pyl sextuple mutants, T2 seeds originating from the 15 individual T1 plants were germinated on MS medium supplemented with 1 µM ABA. The ratios of seedlings growing in the presence of 1 µM ABA for lines #11 and #12 were much higher than those observed for WT seeds, suggesting that the targeted PYLs were extensively mutated in plants derived from these two lines (Fig. 4a). To identify pyl sextuple mutants, 122 T2 plants (previously germinated on 1 µM ABA) were randomly selected for analysis of targeted gene mutations by sequencing, among which 30 were from line #11. Overall, the abundance of homozygous multiple mutants were in inverse proportion to the number of simultaneously mutated gene loci. Line #11 was an exception to this rule with most of its T2 homozygotes being quadruple mutants (37 %). No sextuple homozygotes were identified (Fig. 4b). The segregation ratios of homozygous mutants for each of the six PYL loci in line #11 were above the population average (Fig. 4c). Analysis of the mutation patterns for the 122 analyzed T2 plants, shows that all of the 11 quadruple and the 4 quintuple homozygotes were derived from line #11 (Fig. 4d; Table S3). The zygosity type of all 30 individuals from line #11 was determined by amplifying the corresponding loci by PCR, cloning the products into plasmids and sequencing of the inserts. Analysis of the four quintuple mutants shows that two of the non-homozygous PYL loci were biallelic (PYL4 locus in #11-3 and PYL2 locus in #11-4) while the remaining two were heterozygous (PYL8 locus in #11-24 and #11-28) (Fig. 4d).

Characterization of the CRISPR/Cas9 induced PYL gene mutations in the T2 generation. a Screening for ABA-insensitive T2 mutants with 1 μM ABA. Survival ratios of the screened T2 populations are shown at the bottom. b Frequency distribution of homozygous pyl multiple mutants in the T2 population. Averages for all lines are shown in red while individual values for line #11 are shown in blue. c Segregation ratios of homozygous mutants for the six PYL genes in the T2 population. Averages for all lines are shown in red while individual values for line #11 are shown in blue. d Summary of the zygosity and genotype of PYL mutations in the T2 population of line #11. PCR products of the six PYL target loci in each T2 plant were sequenced for mutation detection. The four zygosity types are shown in different colors and the four homozygous quintuple mutants are highlighted in red. Mutation types of the homozygous target sites are shown. Plus symbol denotes insertion, minus symbol denotes deletion, R symbol represents replacement, A/T/C/G nucleotide type, Number nucleotide number
Segregation of homozygous sextuple mutants in T3 generation
To obtain homozygous pyl sextuple mutants, two T2 quintuple mutant lines (#11-4 and #11-24) were grown to maturity, allowed to self-pollinate and used for further genotyping in the T3 generation. Twelve individuals were analyzed for each line. The segregation ratio of each PYL locus is summarized in Table 2. It was confirmed that the PYL2 locus in #11-4 and the PYL5 locus in #11-24 were biallelic and heterozygous, respectively, while all the other targeted gene loci contained homozygous mutations as expected. In the T3 generation for both lines the presence of T-DNA insertions was detectable by PCR (data not shown) but the amplified PYL5 wild type alleles in #11-24 were remained uncleaved, suggesting that the integrated CRISPR/Cas9 genes might be silenced after several generations.
Phenotypic analysis of CRISPR/Cas9-generated PYL sextuple mutants
The homozygous pyl sextuple mutants from line #11-4 (112458-C) were subjected to phenotypic analysis of root elongation and plant development in response to ABA. The previously reported pyr1pyl1pyl4 triple mutant (114-T) and pyr1pyl1pyl2pyl4pyl5pyl8 (112458-T) sextuple mutant were used for comparison (Gonzalez-Guzman et al. 2012). Seedlings were grown on vertically oriented MS plates for 5 days before being transplanted to new MS plates with or without ABA supplementation at the indicated concentrations. All three pyl mutants were highly resistant to ABA-mediated inhibition of root growth at several ABA concentrations (5–50 µM), but there were slight differences among them (Fig. 5a, b). For example, the 112458-T mutant had shorter roots than the 112458-C or 114-T mutant in the absence of ABA, but in the presence of high concentrations of ABA (20–50 µM) their roots were the longest among the three mutants, suggesting that this mutant is more resistant to ABA than the other two (Fig. 5c). In addition, we noticed that 5–10 μM ABA stimulated plant growth for pyl mutants, especially for the two sextuple mutants (Fig. 5c, d). We also found that the fresh weight of the 112458-C mutant was lower than that of the 112458-T mutant under all of the conditions, but their responses to ABA treatment were similar (Fig. 5d). Our results demonstrate the efficiency of the upgraded CRISPR/Cas9 system to generate multiple inheritable mutations within three generations in Arabidopsis.

Phenotype analysis of pyl mutants. (a, b) 14-day-old seedlings grown on MS0 medium without (a) or with the addition of 50 μM ABA 5 days after germination (b). c Quantification of root length in 10-day-old seedlings grown on MS0 medium supplemented with different concentrations of ABA. Data are averages ± SE (n = 20). Asterisks indicate P < 0.05 (Student’s t test) when comparing data of 112458-T mutant with 112458-C and 114-T mutants under 0 or 50 μM ABA treatment. d Fresh weight of 10-days-old seedlings grown on MS0 medium supplemented with different concentrations of ABA. Data are averages ± SE of four seedlings as a group (n = 5)
In vivo evaluation of the gene targeting efficiency for the sgRNA modules
We observed that the mutagenesis frequencies for the PYL4, PYL5 and PYL8 loci were quite low; two of the PYLs were targeted by sgRNAs driven by the At7SLpromoter (Fig. 3b). Previous studies in rice suggested that a low GC content in the sgRNA sequence might affect gene targeting efficiency (Zhang et al. 2014). However in this case, the GC content of the guide sequences was comparable (Fig. 2a), thus we hypothesize that the transcriptional activity of the At7SL promoter might be lower than the AtU6 and AtU3b promoters. To compare the gene targeting efficiencies of the three sgRNA modules, they were customized to target the multiple recognition sites (MRS) in the YFFP reporter gene (Mao et al. 2013). In protoplasts, the CRISPR/Cas9-induced DSBs in the YFFP reporter gene can be restored via the SSA DNA repair pathway so that the gene targeting efficiency of the three sgRNA modules can be evaluated by counting the number of YFP positive cells (Mao et al. 2013; Zhang et al. 2012) (Fig. 6a). Two different guide sequences were used for each sgRNA module. Our results show comparable ratios of YFP positive cells in transfected protoplasts for the three sgRNA modules and not much difference was observed between the two guide sequences (Fig. 6b). However, the average fluorescence intensities of YFP positive protoplasts in protoplasts transfected with the two AtU3b-driven sgRNAs were lower than those driven by the AtU6 or At7SL promoter (Fig. 6c). These results suggest that the AtU3b promoter was less efficient than the AtU6 and At7SL promoter for triggering DSB.

Gene editing efficiency of the sgRNA modules using the YFP single-stranded anneal assay. a A schematic showing the mechanism of CRISPR/Cas9-mediated SSA repair of the YFP gene in protoplasts. The sgRNA transcripts derived from the three sgRNA modules are assembled into the Cas9 protein to target the MRS region in YFFP gene. The CRISPR/Cas9 induced DSBs can be repaired via single-stranded anneal to restore a functional YFP gene for detection. b Fluorescence activated cell sorting (FASC) quantification of YFP positive cells in transfected protoplasts. The plasmids used for transfection are shown at the top. The ratios of YFP positive cells in each transfected population are shown at the bottom. c Fluorescence microscope images of the transfect protoplasts. The plasmids used for transfection are shown at the top. The fluorescence intensity of each transfected population were evaluated by FASC and labeled as average ± SD (n > 2000)
Moreover, two of the three poorly performing sgRNAs were placed in the last position of the sgRNA cassettes. We thus hypothesize that the relative position of the sgRNA module might affect its mutagenesis activity. To test this possibility, we reversed the order of the AtU6 and the At7SL modules (Fig. 7a). The PYR1, PYL1 and PYL4 gene loci were targeted in protoplast assays but no differences in mutagenesis efficiency was observed (Fig. 7b).

Influence of the sgRNA module position on mutagenesis efficiency. a Schematics of two different sgRNA cassettes containing the sgRNA modules in reverse order. Both cassettes were designed to target three PYL genes in protoplasts. b Surveyor assay evaluating the mutagenesis efficiency of the three PYL genes targeted by the two CRISPR/Cas9 cassettes described in a. The cleavage efficiencies of the PCR products were calculated using the Bio-rad image lab software and shown at the bottom
Discussion
Designing an easy-to-assemble multiplex CRISPR/Cas9 system for gene editing in Arabidopsis
In this study we developed a CRISPR/Cas9 platform that allows sextuple gene editing in Arabidopsis with a single binary vector. To increase the capacity of the CRISPR/Cas9 system originally described (Mao et al. 2013), we designed a strategy to insert three sgRNA modules into a single sgRNA docking site so that the assembly of a sextuple gene editing system can be achieved in three steps using conventional molecular cloning.
It has been reported that transgenes containing three or four direct repeats can induce post-transcriptional gene silencing (PTGS) in tobacco at high frequency (80–100 %), regardless of the relative strength of the promoter used. To reduce the repetitiveness of the inserted sgRNA modules, we used three different Arabidopsis Pol III-dependent promoters to drive the expression of the individual sgRNAs. In addition, the two cloning sites for the sgRNA modules were separated by the Cas9 expression cassette. As we have shown, this multiplex CRISPR/Cas9 system was able to generate homozygous sextuple mutations in a single transformation experiment, although its efficiency might be further increased by using stronger RNA polymerase III promoters.
Factors influencing the gene targeting efficiency of the tandem sgRNA modules
Analysis of the mutagenesis efficiency for the six PYL loci revealed that PYL4, PYL5 and PYL8 showed relatively low efficiencies compared with PYR1, PYL1 and PYL2. In vivo experiments in Arabidopsis protoplasts showed that, although the AtU3b promoter had slightly reduced activity, all three promoters had comparable strength (Fig. 6b, c). Unlike the AtU6 and At7SL promoters, which use a guanine “G” as the transcriptional start site, the transcriptional start site of the AtU3b promoter was an adenosine “A”. To validate the effect of this transcriptional start site in sgRNA expression, additional experiments with very strict controls will be required.
Since no obvious transcriptional differences were observed among the three RNA polymerase III promoters, we hypothesized that the sgRNA mutagenesis activity could be influenced by its relative position in the expression cassette. However, this hypothesis was also disproved since swapping the positions of the AtU6 and At7SL promoters in the sgRNA cassette did not influence their gene targeting efficiencies (Fig. 7a, b). It is therefore obvious that, in addition to the GC content and transcriptional level, the targeting efficiency of sgRNAs may be affected by other factors such as chromosome structure and the transcriptional level of the targeted genes.
Generation of sextuple mutants with a single transformation experiment
To evaluate the gene targeting efficiency of the multiplex CRISPR/Cas9 system, six of the 14 PYR/PYL gene family members were selected as targets for mutagenesis. Previous studies have shown that these six PYR/PYL genes have non-redundant functions in ABA perception and their sextuple mutants exhibited a dramatic ABA-insensitive phenotype (Gonzalez-Guzman et al. 2012). Even though we obtained only 15 transgenic plants in the T1 generation, one of them was identified as containing sextuple mutations. Compared to the other lines, this sextuple mutation line produced more ABA-insensitive seedlings in the T2 generation, with all of the 11 homozygous quadruple mutants and the four quintuple mutants identified in the T2 population belonging to this line. Our results show that the selection of T1 mutated lines can be critical to increase the rate of homozygous mutants in the T2 population.
Phenotype analysis of the CRISPR/Cas9-generated T3 homozygous pyl sextuple mutant (112458-C) performed along with two previously described pyl mutants (114-T and 112458-T) (Gonzalez-Guzman et al. 2012) revealed very similar, although not identical, ABA-insensitive phenotypes. Slight differences were nevertheless observed between the newly generated 112458-C mutant and the sextuple 112458-T mutant such as the reduced fresh weight displayed by 112458-C in the absence or presence of ABA at different concentrations. These differences were most probably caused by the fact that the mutation in PYL8 was in the 5′ UTR in the CRISPR/Cas9-generated mutant. We targeted the 5′ UTR of PYL8 in an attempt to study the regulation of PYL8 by the untranslated region.
The multiplex CRISPR/Cas9 system can facilitate functional studies targeting multiple genes and gene families
Conventional methods used to generate multiple mutants are laborious and time consuming. While crossing individual mutants to obtain double mutants is relatively easy, the difficulty increases exponentially when trying to produce higher order mutants given the normal Mendelian segregation ratios. In addition, duplication events have sometimes resulted in the presence of tandemly arranged gene repeats in close proximity within the chromosome. In these cases the affected genes have a strong genetic linkage making the production of multiple mutants by crossing practically impossible. Although non-homologous multiple mutations can be generated frequently by CRISPR/Cas9, the advantage of this gene editing tool is that researchers can screen for transgenic lines showing the highest mutation frequency at most of the targeted positions. Thus, the chance of obtaining homozygous multiple mutants will be largely increased. Our easy-to-use CRISPR/Cas9 system for multiplex gene editing in Arabidopsis can prove invaluable in these situations. We expect that by co-transforming two sextuple CRISPR/Cas9 vectors it will be possible to generate duodecuple mutants.
Methods
Vector construction
To construct the pEx-ptAtUBQ-Cas9 vector, the ptAtUBQ-Cas9 module from psgR-Cas9-At was cloned into the XmaI-KpnI site of pCAMBIA1300 binary vector. This ptAtUBQ-Cas9 module contains a 680 bp AtUBQ1 promoter, a human code optimized Cas9 gene and a 208 bp AtUBQ1 terminator as described before (Mao et al. 2013). The three sgRNA modules were synthesized using overlapping PCR. The AtU6-26, AtU3b, At7SL-2 promoter were amplified from the Arabidopsis genome by PCR using primer pairs: AtU6-F-HindIII/AtU6-sgR-R, AtU3b-XhoI-F/AtU3b-sgR-R and At7SL-XbaI-F/At7SL-sgR-R, respectively. And their corresponding sgRNA scaffolds with overlapping sequences was also amplified by PCR using primer pairs: sgR-U6-F/sgR-XhoI-R, sgR-U3b-F/sgR-XbaI-R and sgR-7SL-F/sgR-R-XmaI, respectively. The sequences of these primers are listed in Table S4. For each sgRNA module, overlap PCR was performed using primer pairs at the remote end. The obtained products were then cloned into the HindIII-XmaI site of pMD18T vector (Takara, Japan) to generate pAtU6-sgR, pAtU3b-sgR and pAt7SL-sgR vector, respectively.
To construct a pEX-6XsgR-Cas9 vector for targeting PYR/PYL genes, two complementary 24-bp oligonucleotides with a 20-bp target sequence were synthesized for each PYR/PYL targets. These oligo pairs were annealed to generate a double-strand DNA with 4-bp overhangs on both ends and cloned into BbsI sites of the three sgRNA modules, respectively following a standard procedure (Liu et al. 2015). The sequences of the synthesized DNA oligos are shown in Table S4. In total, six sgRNA modules were generated to target the six PYR/PYL genes. They were named pAtU6-sgR-PYL1, pAtU6-sgR-PYL2, pAtU3b-sgR-PYR1, pAtU3b-sgR-PYL8, pAt7SL-sgR-PYL4 and pAt7SL-sgR-PYL8.
To assemble these sgRNA modules into the pEx-ptAtUBQ-Cas9 vector, the three sgRNA modules were first amplified from their host vectors by PCR using primer pair M13F/M13F. The reaction mix was set up as follows:
0.4 μL | Forward primer (M13F) (10 µM) |
0.4 μL | Reverse primer (M13R) (10 µM) |
2 μL | 10X Ex Taq buffer (Mg2+ plus) |
1.6 μL | dNTP mix (2.5 mM) |
0.1 μL | Ex Taq polymerase (5 units/μL) (Takara, Japan) |
Up to 20 μL | ddH2O |
Perform PCR using the following cycling conditions:
94 °C 2 min, (94 °C 10 s, 58 °C 15 s, 68 °C 20 s) X 35 cycles, 68 °C 5 min, keep at 12 °C.
The amplified fragments were then digested by HindIII and XhoI (for pAtU6-sgRNA), XhoI and XbaI (for pAtU3b-sgRNA) and XbaI and XmaI (for pAt7SL-sgRNA) in separate reactions at 37 °C for 2 h:
X μL | Amplified fragments (100 ng) |
5 μL | 10X NEB CutSmart Buffer |
1 μL | Restriction enzyme 1 (20 units/μL) (New England Biolab, USA) |
1 μL | Restriction enzyme 2 (20 units/μL) (New England Biolab, USA) |
Up to 50 μL | ddH2O |
And the binary vector pEx-ptAtUBQ-Cas9 was digested by HindIII and XmaI accordingly:
X μL | PEx-ptAtUBQ-Cas9 (1 μg) |
5 μL | 10X NEB CutSmart Buffer |
1 μL | HindIII (20 units/μL) (New England Biolab, USA) |
1 μL | XmaI (20 units/μL) (New England Biolab, USA) |
Up to 50 μL | ddH2O |
The three digested sgRNA fragments were then mixed at equal volume ratio and purified together by column before assembly, while the Phenol–Chloroform extraction method was recommended for purifying the digested binary vector.
Two triplicated CRISPR/Cas9 vectors named pEx-3XsgR-PYL114-Cas9 and pEx-3XsgR-PYL285-Cas9 was assembled, respectively as follows and incubated at 16 °C for 2 h:
X μL | Purified sgRNA mix (>50 ng) |
Y μL | pEx-ptAtUBQ-Cas9 (HindIII and XmaI digested) (>10 ng) |
1 μL | 10XT4 ligase buffer |
0.5 μL | T4 DNA ligase (400 units/µL) (New England Biolab, USA) |
Up to 10 μL | ddH2O |
Transform the ligation reaction into DH5 alpha competent cells and plate cells on LB plates containing 50 µg/mL kanamycin for selection. Positive clones were identified by colony PCR as follows:
0.4 μL | Forward primer (AtU6-F-Kpn) (10 µM) |
0.4 μL | Reverse primer (sgR-R-EcoRI) (10 µM) |
2 μL | 10X Ex Taq buffer (Mg2+ plus) |
1.6 μL | dNTP mix (2.5 mM) |
0.1 μL | Ex Taq polymerase (5 units/μL) (Takara, Japan) |
Up to 20 μL | ddH2O |
Perform PCR using the following cycling conditions:
94 °C 5 min, (94 °C 15 s, 58 °C 20 s, 68 °C 50 s) X 35cycles, 68 °C 5 min, keep at 12 °C. The positive PCR product is about 1.5 Kb in length.
To generate the final sextuple CRISPR/Cas9 construct, the three sgRNA modules in the pEx-3XsgR-PYL285-Cas9 vector was amplified by PCR using primer pair AtU6-F-KpnI/sgR-R-EcoRI. The reaction mix was set up as follows:
0.4 μL | Forward primer (AtU6-F-Kpn) (10 µM) |
0.4 μL | Reverse primer (sgR-R-EcoRI) (10 µM) |
2 μL | 10X Ex Taq buffer (Mg2+ plus) |
1.6 μL | dNTP mix (2.5 mM) |
0.1 μL | Ex Taq polymerase (5 units/μL) (Takara, Japan) |
Up to 20 μL | ddH2O |
Perform PCR using the following cycling conditions:
94 °C 2 min, (94 °C 15 s, 58 °C 20 s, 68 °C 50 s) X 35 cycles, 68 °C 5 min, keep at 12 °C.
The amplified sgR-PYL285 fragments were then digested by KpnI and EcoRI at 37 °C for 2 h:
X μL | Amplified sgR-PYL285 fragments (100 ng) |
5 μL | 10X NEB CutSmart Buffer |
1 μL | KpnI (20 units/μL) (New England Biolab, USA) |
1 μL | EcoRI (20 units/μL) (New England Biolab, USA) |
Up to 50 μL | ddH2O |
And the pEx-3XsgR-PYL114-Cas9 binary vector was digested by KpnI and EcoRI accordingly.
X μL | pEx-3XsgR-PYL114-Cas9 (1 μg) |
5 μL | 10X NEB CutSmart Buffer |
1 μL | KpnI (20 units/μL) (New England Biolab, USA) |
1 μL | EcoRI (20 units/μL) (New England Biolab, USA) |
Up to 50 μL | ddH2O |
To assemble the final pEx-6XsgR-PYL114285-Cas9 binary vector, the digested sgR-PYL285 fragments were purified by column and the pEx-3XsgR-PYL114-Cas9 binary vector was purified using the Phenol–Chloroform extraction method. The ligation reaction was set up as follows and incubated at 16 °C for 2 h:
X μL | Purified sgR-PYL285 fragments (>50 ng) |
Y μL | pEx-3XsgR-PYL114-Cas9 (KpnI and EcoRI digested) (>10 ng) |
1 μL | 10XT4 ligase buffer |
0.5 μL | T4 DNA ligase (400 units/µL) (New England Biolab, USA) |
Up to 10 μL | ddH2O |
Transform the ligation reaction into DH5 alpha competent cells and plate cells on LB plates containing 50 µg/mL kanamycin for selection. Positive clones were identified by colony PCR as follows:
0.4 μL | Forward primer (tUBQ-F) (10 µM) |
0.4 μL | Reverse primer (M13R) (10 µM) |
2 μL | 10X Ex Taq buffer (Mg2+ plus) |
1.6 μL | dNTP mix (2.5 mM) |
0.1 μL | Ex Taq polymerase (5 units/μL) (Takara, Japan) |
Up to 20 μL | ddH2O |
Perform PCR using the following cycling conditions:
94 °C 5 min, (94 °C 15 s, 58 °C 20 s, 68 °C 50 s) X 35 cycles, 68 °C 5 min, keep at 12 °C. The positive PCR product is about 1.7 Kb in length.
To generate plasmids used for YFP SSA assay in protoplasts, eight sgRNA guide oligos were synthesized to form six DNA-duplexes for cloning into the three sgRNA-modules, respectively. The sequences of these DNA oligos were listed in Table S4. The obtained six sgRNA modules were then cloned into the HindIII-XmaI sites of psgR-Cas9-At to yield six transiently expressed vectors. They were named pAtU6-MRS1-Cas9, pAtU6-MRS2-Cas9, pAtU3b-MRS1-Cas9, pAtU3b-MRS1-Cas9, pAt7SL-MRS1-Cas9, pAt7SL-MRS2-Cas9. The YFFP vector used for this study has been described before (Mao et al. 2013).
To generate the expressional vector used for targeting PYR1, PYL1 and PYL4 genes in protoplasts, the three sgRNA modules in pEx-3XsgR-PYL114-Cas9 was cloned into the HindIII-XmaI site of psgR-Cas9-At to yield the psgR-114-Cas9 vector. To exchange the position of pAtU6-sgR-PYR1 and pAt7SL-sgR-PYL4 in this vector, these two sgRNA modules were first amplified by PCR using primer pair At7SL-HindIII-F/sgR-R-XhoI and AtU6-XbaI-F/sgR-R-XmaI and then cloned into the same restriction sites of psgR-Cas9-At together with the XhoI and XmaI digested AtU3b-sgR-PYL1 module to produce the psgR-411-Cas9 vector.
Plant transformation and growth conditions
Agrobacterium-mediated transformation of Arabidopsis thaliana Columbia-0 with the binary vectors was performed using the floral dipping method as previously described (Weigel and Glazebrook 2006). Seed collected from the Agrobacterium infected plants was sterilized with 2 % sodium hypochlorite for 15 min and plated on Murashige and Skoog (MS0) medium containing 30 mg/L hygromycin plus 50 mg/L Carbenicillin to inhibit Agrobacterium growth. The resulting T1 plants were transplanted to soil after growing under long-day conditions (16 h light/8 h dark) at 22 °C for 2 weeks.
Detection of targeted gene mutations
DNA was extracted from transgenic plants by the CTAB method. Genomic regions surrounding the PYR/PYL target sites were amplified by PCR using primer pairs listed in Table S4. Targeted gene mutations were detected by aligning the sequencing chromatograms of these PCR products with the wild type controls. Genotyping for the heterozygous or biallelic PYL mutations in quintuple mutant were performed by cloning the PCR products of those target loci into the pMD18T vector (Takara, Japan) for DNA sequencing. Segregation ratios of these PYL mutations were calculated by sequencing at least 24 clones.
Phenotype analysis for the pyl mutants
The pyr1pyl1pyl4 triple mutant and the pyr1pyl1pyl2pyl4pyl5pyl8 sextuple mutant have been described previously (Gonzalez-Guzman et al. 2012). Seedlings were grown on vertically oriented MS0 plates with 3 % sucrose for 5 days. Subsequently, 20 plants were transferred to new MS0 plates lacking or supplemented with ABA at a concentration of 5, 10, 20, 50 µM. Root lengths were measured by vernier caliper and the fresh weights were measured using four seedlings as a group by precision electronic autobalance 8 days after the treatment. Pictures were taken by high resolution camera 10 days after the treatment.
Transient expression of the CRISPR/Cas9 system in Arabidopsis protoplasts
Isolation of Arabidopsis mesophyll protoplasts were performed by the ‘Tape-Arabidopsis Sandwich’ method (Wu et al. 2009). Plasmids of were purified using the QIAGEN Plasmid Mini Kit (Qiagen, USA) according to manufacturers’ instructions. Transfection of the Arabidopsis protoplast was performed using the PEG-calcium method (Yoo et al. 2007). For each reaction, 10 μg total plasmids were used. The transfected Arabidopsis mesophyll protoplasts were incubated in the dark at room temperature for 12–24 h.
To analyze the gene targeting activities of different sgRNA modules, YFP positive cells were measured by flow cytometry (BECKMAN COULTER MoFloTM XDP, USA). Photographs were taken by a fluorescence microscope (IX71, Olympus, Japan) under the RFP and YFP channels. To evaluate the mutagenesis efficiency of endogenous pyl genes, protoplasts were transferred to a 1.5 mL Eppendorf tube and collected by centrifugation. Genomic DNA was extracted for mutation detection using the CTAB method (Springer 2010).
Author contribution statement
Y. M., Z. Z., and J. -K. Z. designed research; Z. Z., Y. M., S. H, W. L, performed research; Z. Z., Y. M., J.B., and J. -K. Z. analyzed data; Y. M., J. B., and J. -K. Z. wrote the paper.
References
Alonso JM, Ecker JR (2006) Moving forward in reverse: genetic technologies to enable genome-wide phenomic screens in Arabidopsis. Nat Rev Genet 7:524–536
Brooks C, Nekrasov V, Lippman ZB, Van Eck J (2014) Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-associated9 system. Plant Physiol 166:1292–1297
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096
Fauser F, Schiml S, Puchta H (2014) Both CRISPR/Cas-based nucleases and nickases can be used efficiently for genome engineering in Arabidopsis thaliana. Plant J 79:348–359
Feng Z, Zhang B, Ding W, Liu X, Yang DL, Wei P, Cao F, Zhu S, Zhang F, Mao Y, Zhu JK (2013) Efficient genome editing in plants using a CRISPR/Cas system. Cell Res 23:1229–1232
Gonzalez-Guzman M, Pizzio GA, Antoni R, Vera-Sirera F, Merilo E, Bassel GW, Fernandez MA, Holdsworth MJ, Perez-Amador MA, Kollist H, Rodriguez PL (2012) Arabidopsis PYR/PYL/RCAR receptors play a major role in quantitative regulation of stomatal aperture and transcriptional response to abscisic acid. Plant Cell 24:2483–2496
Jia H, Wang N (2014) Targeted genome editing of sweet orange using Cas9/sgRNA. PLoS One 9:e93806
Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA (2013) RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Katavic V, Haughn GW, Reed D, Martin M, Kunst L (1994) In planta transformation of Arabidopsis thaliana. Mol Gen Genet 245:363–370
Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, Zhao Y (2013a) Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol 31:681–683
Li JF, Norville JE, Aach J, McCormack M, Zhang D, Bush J, Church GM, Sheen J (2013b) Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31:688–691
Li W, Teng F, Li T, Zhou Q (2013c) Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol 31:684–686
Liu W, Zhu X, Lei M, Xia Q, Botella J, Zhu J-K, Mao Y (2015) A detailed procedure for CRISPR/Cas9-mediated gene editing in Arabidopsis thaliana. Sci Bull 60:1332–1347
Ma C, Mitra A (2002) Intrinsic direct repeats generate consistent post-transcriptional gene silencing in tobacco. Plant J 31:37–49
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826
Mao Y, Zhang H, Xu N, Zhang B, Gou F, Zhu JK (2013) Application of the CRISPR-Cas system for efficient genome engineering in plants. Mol Plant 6:2008–2011
Miao J, Guo D, Zhang J, Huang Q, Qin G, Zhang X, Wan J, Gu H, Qu LJ (2013) Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res 23:1233–1236
Nekrasov V, Staskawicz B, Weigel D, Jones JD, Kamoun S (2013) Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotechnol 31:691–693
Nissim L, Perli SD, Fridkin A, Perez-Pinera P, Lu TK (2014) Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol Cell 54:698–710
Park SY, Fung P, Nishimura N, Jensen DR, Fujii H, Zhao Y, Lumba S, Santiago J, Rodrigues A, Chow TF, Alfred SE, Bonetta D, Finkelstein R, Provart NJ, Desveaux D, Rodriguez PL, McCourt P, Zhu JK, Schroeder JI, Volkman BF, Cutler SR (2009) Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 324:1068–1071
Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, Gao C (2013) Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol 31:686–688
Springer NM (2010) Isolation of plant DNA for PCR and genotyping using organic extraction and CTAB. CSH Protoc. doi:10.1101/pdb.prot5515
Upadhyay SK, Kumar J, Alok A, Tuli R (2013) RNA-guided genome editing for target gene mutations in wheat. G3 Bethesda 3:2233–2238
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153:910–918
Waterhouse PM, Graham MW, Wang MB (1998) Virus resistance and gene silencing in plants can be induced by simultaneous expression of sense and antisense RNA. Proc Natl Acad Sci USA 95:13959–13964
Weigel D, Glazebrook J (2006) In planta transformation of Arabidopsis. CSH Protoc. doi:10.1101/pdb.prot4668
Wu FH, Shen SC, Lee LY, Lee SH, Chan MT, Lin CS (2009) Tape-Arabidopsis Sandwich—a simpler Arabidopsis protoplast isolation method. Plant Methods 5:16
Xie K, Yang Y (2013) RNA-guided genome editing in plants using a CRISPR-Cas system. Mol Plant 6:1975–1983
Xie K, Minkenberg B, Yang Y (2015) Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci USA 112:3570–3575
Xing HL, Dong L, Wang ZP, Zhang HY, Han CY, Liu B, Wang XC, Chen QJ (2014) A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol 14:327
Yoo SD, Cho YH, Sheen J (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2:1565–1572
Zhang Y, Zhang F, Li X, Baller JA, Qi Y, Starker CG, Bogdanove AJ, Voytas DF (2012) Transcription activator-like effector nucleases enable efficient plant genome engineering. Plant Physiol 161:20–27
Zhang H, Zhang J, Wei P, Zhang B, Gou F, Feng Z, Mao Y, Yang L, Xu N, Zhu JK (2014) The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol J 12:797–807
Acknowledgments
We thank Dr Pedro L. Rodriguez for kindly providing the pyr1pyl1pyl4 and pyr1pyl1pyl2pyl4pyl5pyl8 mutants. The work was supported by the Chinese Academy of Sciences. The authors declare that they have no conflicts of interest with respect to this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Mahfouz.
Y. Mao and Z. Zhang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, Z., Mao, Y., Ha, S. et al. A multiplex CRISPR/Cas9 platform for fast and efficient editing of multiple genes in Arabidopsis . Plant Cell Rep 35, 1519–1533 (2016). https://doi.org/10.1007/s00299-015-1900-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-015-1900-z