
Abstract
The CRISPR/Cas9 genome-editing system has emerged as a popular powerful tool for biological research. However, the process of selecting efficiently edited Cas9-free plants is usually laborious and time consuming. Here, we demonstrated P2A to be the most efficient self-cleaving peptide for fusing Cas9 and GFP in Arabidopsis and then used Cas9-P2A-GFP to develop a novel CRISPR/Cas9 system. Additionally, a pair of isocaudomer restriction enzymes were selected to conveniently assemble multiple sgRNAs. In this system, the GFP fluorescence intensity in T1 transgenic plants indicates the expression level of the Cas9 protein, which correlates well with the editing efficiency. Furthermore, Cas9-free plants can be easily selected by examining GFP fluorescence in T2 transgenic plants. The efficient knockout of BRI1, BZR1 and BES1 demonstrated the robustness of our new system. Thus, we designed a novel CRISPR/Cas9 system that can generate Cas9-free multiplex mutants efficiently in Arabidopsis and possibly in other plant species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) genome-editing systems, derived from the prokaryotic type II adaptive immune system, have been widely used to exploit genetic variation in a variety of organisms, and many CRISPR systems have been generated for gene editing in plants (Horvath and Barrangou 2010; Mao et al. 2019). The conventional CRISPR/Cas9 system includes two components, a single-guide RNA (sgRNA) for recognizing and matching the target DNA and a Cas9 endonuclease for cleaving the target DNA, which can be driven by different promoters. In the model plant Arabidopsis, constitutive promoters such as the Cauliflower mosaic virus (CaMV) 35S or maize ubiquitin promoter have been extensively used to drive Cas9 for gene editing in several plant species (Feng et al. 2013; Li et al. 2013; Mao et al. 2013; Ma et al. 2015). Furthermore, egg cell-specific promoters have been used to control Cas9 to generate homozygous mutants in a single generation (Wang et al. 2015b). The promoter of the YAO gene, which is highly expressed in active dividing cells, has been used to drive Cas9 to improve editing efficiency (Yan et al. 2015). Germline-specific promoters have been used to overcome the defects of growth or fertility in T1 plants caused by noninheritable somatic mutations (Mao et al. 2016).
Recently, the selection of Cas9-free plants has received increasing attention. If Cas9 and sgRNA exist in edited plants, it is difficult to distinguish whether a mutation is inherited from the last generation or newly produced in the current generation. This is important, because the new mutations produced in the current generation may be not heritable as somatic mutations. In addition, the constitutive existence of Cas9 and sgRNA might increase the risk of off-target effects. Moreover, obtaining Cas9-free edited plants makes complementation experiments possible, as the complemented wild-type gene would otherwise be mutated as well. In agriculture, edited plants exhibiting CRISPR/Cas9 expression cassettes are regarded as transgenic plants, and it is usually more difficult to obtain approval for their commercial applications. Thus, it is critical to identify Cas9-free genome editing mutants. Conventional methods for identifying Cas9-free plants depend on genotyping, which is time consuming and laborious. A Cas9-free system based on mCherry fluorescence that is specifically expressed in Arabidopsis seeds under the control of the At2S3 promoter has been reported, and transgene-free seeds in the T2 generation can be selected via visual isolation (Gao et al. 2016; Yu and Zhao 2019). This system can make the selection of Cas9-free plants convenient.
To generate multiple mutations simultaneously, several systems for multiplex genome editing have been developed. One method is to use different Pol III promoters to drive sgRNAs, which are assembled via the use of multiple restriction enzymes (Zhang et al. 2016). In addition, isocaudomer enzymes have been widely used to assemble multiple sgRNA cassettes. Spe I/Nhe I, BamH I/Bgl II, Xba I/Nhe I, and Xho I/Sal I are frequently used isocaudomer enzyme pairs (Wang et al. 2015a; Yan et al. 2015). Moreover, the method-based Golden Gate ligation, or Gibson Assembly, technique has been used to produce tandem sgRNA cassettes (Ma et al. 2015).
For all the currently available CRISPR/Cas9 systems in plants, it is laborious to genotype many transgenic plants to select the lines with target mutations. The available Cas9-free system in Arabidopsis described above also presents this disadvantage (Gao et al. 2016; Yu and Zhao 2019); in this system, Cas9 and mCherry are separated in two expression cassettes driven by different promoters, and the fluorescence intensity is, therefore, not correlated with the expression level of Cas9 and cannot be used to predict the efficiency of gene editing. To improve the previous Cas9-free system in Arabidopsis, we referred to the design applied in mammalian cells (Platt et al. 2014) and used the self-cleaving P2A peptide to fuse Cas9 and GFP. Thus, the GFP fluorescence intensity can be used to predict the expression level of Cas9 and editing efficiency, while the activity of Cas9 is not affected, since the GFP protein can be cleaved out automatically. By combining Cas9-P2A-GFP with multiple sgRNA assembly using a pair of isocaudomer restriction enzymes, we developed a novel CRISPR system for efficiently generating Cas9-free multiplex mutants. This system was demonstrated in Arabidopsis and could also be used in other plant species.
Results
The P2A peptide shows the highest cleavage efficiency in Arabidopsis protoplasts
To generate a CRISPR system in plants that in which the abundance of functional Cas9 is reported visually, similar to the strategy in mammalian cells (Platt et al. 2014), we searched for self-cleaving peptides suitable for plants. The 2A peptides, which are belong to the picornaviruses, have been demonstrated to exhibit self-cleavage properties in various animal cells and tissues (Kim et al. 2011). To test whether 2A peptides also function in the plant system, five different 2A peptides derived from the foot-and-mouth disease virus (F2A-1, F2A-2), equine rhinitis A virus (E2A), Thosea asigna virus (T2A) or porcine teschovirus-1 (P2A), were used to link Cas9 and GFP (Kim et al. 2011; Buren et al. 2012; Supplemental Table 1). Two nuclear localization signal (NLS) peptides were fused with Cas9 at its C- and N-termini. The cleavage efficiency of these 2A peptides was evaluated in protoplasts through confocal microscopy and western blotting analyses. In contrast to the nuclear localization of Cas9-CP-GFP, some of the GFP fluorescence in Cas9-2A-GFP-transformed protoplasts was obviously localized in the cytoplasm, indicating that the GFP protein was cleaved from Cas9 (Fig. 1A). P2A displayed the highest cleavage efficiency among all these 2A peptides (Fig. 1B, C). Due to the equal expression of Cas9 and GFP followed by highly efficient self-cleavage by P2A, the intensity of GFP fluorescence reflects the abundance of Cas9 well, and the GFP protein does not interrupt the function of Cas9. Thus, P2A was selected for the fusion of Cas9 and GFP in our novel CRISPR/Cas9 system in Arabidopsis.

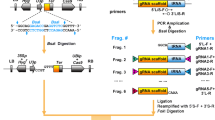
CRISPR/Cas9 system for efficiently generating Cas9-free multiplex mutants in Arabidopsis. A–C Cleavage efficiency of several 2A peptides. A Expression of GFP and Cas9-Linker-GFP in Arabidopsis mesophyll protoplasts. Left to right: GFP channel, bright field, and merged images. F2A-1, E2A, F2A-2, P2A and T2A are various 2A peptides, and CP is the abbreviation for control peptide. B Immunoblot analysis of the cleavage efficiency of the indicated 2A peptides. Total proteins were extracted from the Arabidopsis mesophyll protoplasts shown in panel A and analyzed by western blotting using a GFP antibody. C Quantitation of the cleavage efficiency from panel B using ImageJ. Five independent experiments were performed, and the data are shown as the mean ± SEM. D Schematic diagram of the construction of multiple sgRNA expression cassettes using isocaudomer restriction enzymes. The primers for the sgRNAs were annealed and inserted into the Bbs I restriction enzyme site between AtU6-26 and the scaffold to generate sgRNA cassettes. Two sgRNA cassettes were digested with Spe I/Sal I and Nhe I/Sal I for the sgRNA fragment and plasmid skeleton, respectively, and then ligated together. Similarly, unlimited multiple tandem sgRNA cassettes could be ligated together. E The physical map of the CRISPR/Cas9 system for efficiently generating Cas9-free multiplex mutants. Cas9-P2A-GFP was driven by the double CaMV 35S promoter or the UBQ10 promoter and terminated with rbcS-E9 terminator. Multiple sgRNA cassettes were assembled and then inserted into the binary vector with Cas9-P2A-GFP using Kpn I and Sal I. HygR hygromycin resistance gene, GmR gentamicin sulfate resistance gene, KanR kanamycin resistance gene, SmR spectinomycin resistance gene. HygR and KanR belong to the binary vector of pCambia1300-Cas9-P2A-GFP, while GmR and SmR belong to pJim19-Cas9-P2A-GFP
A construct for multiplex genome editing and Cas9-free mutant selection
In functional genomics studies, multiple genes often need to be edited simultaneously. To conveniently perform multiplex gene editing, a method based on isocaudomer restriction enzymes was used (Fig. 1D; Yan et al. 2015; Wang et al. 2015a). The primers of the sgRNAs were annealed and inserted between AtU6-26 and the scaffold using Bbs I (Supplemental Figs. 1 and 2). Spe I and Nhe I are a pair of isocaudomers, and multiple sgRNA cassettes can be assembled continuously via digestion using Spe I/Sal I and Nhe I/Sal I followed by ligation (Fig. 1D). Furthermore, the tandem sgRNA cassettes can be digested and inserted into the binary vector containing Cas9-P2A-GFP driven by the double Cauliflower mosaic virus (CaMV) 35S or Arabidopsis ubiquitin 10 gene promoter (UBQ10 Pro) using Kpn I and Sal I (Fig. 1E; Supplemental Figs. 1 and 2). Using this construct, the protein level of Cas9 can be detected easily by checking the fluorescence intensity in transgenic plants, and multiple genes can be edited simultaneously. The general principle is the selection of T1 plants with high GFP fluorescence and T2 plants without fluorescence to obtain Cas9-free multiplex mutants.
Selection of Cas9-free multiplex mutants based on simple fluorescence detection
To use the Cas9-P2A-GFP module to generate multiplex genome-edited Cas9-free plants, we designed a pipeline. First, multiple sgRNA expression elements can be assembled using isocaudomer restriction enzymes and inserted into a binary vector containing Cas9-P2A-GFP using Kpn I and Sal I (Supplemental Fig. 1). Second, transformed seeds are grown on MS plates with antibiotics, and then seedlings with high GFP expression are selected by fluorescence stereoscopy or microscopy. T1 transgenic seedlings with strong GFP fluorescence are transferred to the soil to produce offspring. Third, in the T2 transgenic plants, individuals without GFP expression are selected and genotyped to obtain Cas9-free multiplex gene-edited mutants (Fig. 2A).

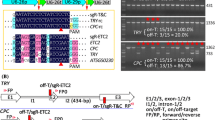
Procedure of selecting Cas9-free gene editing plants. A Workflow diagram of the transgene-free genome-editing system. The construct with target sgRNA cassettes and Cas9-P2A-GFP was delivered into Arabidopsis through Agrobacterium-mediated transformation. Seeds from transformed plants were sown in MS plates with antibiotics (hygromycin or gentamicin sulfate). T1 transgenic plants with high GFP expression were selected, whereas T2 transgenic plants without GFP expression (Cas9-free plants) were selected. The target genes were genotyped in T1 plants and Cas9-free T2 plants. B Segregation of T2 transgenic plants with or without GFP fluorescence. Four sgRNAs, two targeting BZR1 and two targeting BES1, were assembled using the method described in Fig. 1D and cloned into the binary vector p35S:Cas9-P2A-GFP (Gent). The construct was delivered to the wild-type to edit BZR1 and BES1. 3-day-old light-grown seedlings were photographed using confocal microscopy. The numbers in the lower right-hand corner of each panel are the numbers of observed plants. C Agarose gel analysis of the Cas9-P2A-GFP cassette. Three T2 plants without GFP fluorescence and nine plants with GFP fluorescence were randomly selected from the plants in panel B. PCR products were amplified using primers for Cas9 and analyzed in agarose gels to confirm the accuracy of selection based on fluorescence. D DNA sequences of the BZR1 and BES1 target sites in a representative T2 transgenic plant without GFP expression (line #10–19). BZR1 locus 1 and BZR1 locus 2 are two genomic loci containing BZR1 sgRNAs, whereas BES1 locus1 and BES1 locus 2 are two genomic loci containing BES1 sgRNAs. The PAM sequences are indicated in red, and sgRNA sequences are underlined. Plus indicates insertion, while minus indicates deletion. E Statistical results of GFP fluorescence in T1 transgenic Arabidopsis and mutations at four loci in the lines with GFP fluorescence. F Summary of the segregation of T2 plants and the genotypes of Cas9-free T2 Arabidopsis
To test the efficiency of our CRISPR/Cas9 system, BZR1 and BES1 were chosen as targets for generating Cas9-free mutants. Four sgRNAs were designed (two for BZR1 and two for BES1) and inserted into the p35S:Cas9-P2A-GFP (Hyg) binary vector. Wild-type (Col-0) plants were subjected to transformation. Four seedlings with high GFP expression were selected from 62 T1 plants in the wild-type background, and the fluorescence of two representative seedlings is shown (Supplemental Fig. 3A; Fig. 2E). The four target sites were not well edited in a representative seedling without obvious GFP expression (Supplemental Fig. 3B) but were efficiently edited in the plants with high GFP expression (Supplemental Fig. 3C, D). Among the T2 progenies from these two representative T1 plants with high GFP expression, the segregation ratio of GFP and non-GFP plants was close to 3:1, indicating a single transgenic insertion (Fig. 2B, F). The majority of the Cas9-P2A-GFP protein in T2 plants with fluorescence was self-cleaved (Supplemental Fig. 4A), indicating that P2A functions well in Arabidopsis plants, which is consistent with the results in Arabidopsis protoplasts (Fig. 1A–C). The T2 plants were further genotyped using Cas9-specific primers, which confirmed the perfect correlation between GFP fluorescence and Cas9 presence (Fig. 2C; Supplemental Fig. 4B). Sanger sequencing showed that the four sgRNA loci were edited efficiently in T2 Cas9-free plants (Fig. 2D, F). Excitingly, 8/40 (20%) T2 plants of line #10 harbored mutations at all four loci, and 3/40 (7.5%) T2 plants were homozygous at all four loci (Supplemental Table 2). The sequencing results of a representative homozygous line are shown in Fig. 2D. In another unpublished study, we constructed 12 sgRNA expression cassettes together using the method shown in Fig. 1D to target 8 SAUR genes, and at least 10 of the sgRNAs functioned well, further suggesting that this isocaudomer-based method could be used to target multiple sites simultaneously.
Efficiency of Cas9-P2A-GFP driven by different promoters
Due to the low efficiency of the 35S promoter in generating transgenic plants with obvious expression of Cas9-P2A-GFP (Fig. 2E), the Arabidopsis ubiquitin 10 (UBQ10) gene promoter was included for comparison. The Arabidopsis BRI1 gene was selected as a representative target, since the mutation of BRI1 resulted in an obvious phenotype (Li and Chory 1997; Yan et al. 2015). We first cloned the BRI1 sgRNA expression cassette into the binary vector p35S: Cas9-P2A-GFP (Gent), harboring Cas9-P2A-GFP driven by a double CaMV 35S promoter, which was delivered into wild-type by Agrobacterium transformation. Among 60 T1 transgenic plants, five seedlings with strong GFP fluorescence all displayed severe defects in growth and infertile phenotypes, three of which are shown in Fig. 3A. In contrast, among the T1 plants without obvious GFP fluorescence, only three showed semidwarf phenotypes (Fig. 3E). Through Sanger sequencing, we further confirmed that all five lines with high GFP expression showed editing of the target locus leading to mosaic mutations (Fig. 3B, E).

Cas9-P2A-GFP edited the genome efficiently. A, C The fluorescence intensity of transgenic plants correlated with the phenotype resulting from BRI1 editing. 4-day-old transgenic T1 plants were photographed using a Leica fluorescence stereoscope (A, left and middle; C, upper). The seedlings were then grown in the soil, and their phenotypes were recorded after several weeks. White bar = 1 mm. Red bar = 1 cm. Cas9-P2A-GFP was driven by the double CaMV 35S promoter (A) or the UBQ10 promoter (C). B, D Sanger sequencing results of representative T1 transgenic lines in panels A and C. BRI1 sgRNA sequences are underlined, and protospacer-adjacent motif (PAM) sequences are labeled in red. E Comparison of the editing efficiency of the 35S and UBQ10 promoters driving Cas9-P2A-GFP. The homozygous genotypes were based on Sanger sequencing in the T1 generation and were not confirmed in the T2 generation, since the T1 dwarf plants were sterile
Then, the BRI1 sgRNA expression cassette was inserted into the binary vector pUBQ10:Cas9-P2A-GFP (Gent) harboring Cas9-P2A-GFP driven by the Arabidopsis ubiquitin 10 (UBQ10) gene promoter. Among 47 T1 transgenic seedlings, 10 plants exhibited strong GFP fluorescence, and they all exhibited a severe dwarf phenotype resembling that of bri1 mutants when grown in soil (Fig. 3C, E). Sequencing data showed that 3 out of the 10 seedlings were homozygous mutants (Fig. 3D, E). Unexpectedly, 12 T1 seedlings without obvious GFP fluorescence also showed severe dwarf phenotypes, and two of them were homozygous mutants. A possible explanation for this result is that the Cas9-P2A-GFP protein was expressed in the early embryonic development stage, but its expression was reduced when GFP fluorescence was examined. The summarized results clearly showed that UBQ10 is better than the 35S promoter for driving Cas9-P2A-GFP to edit the genome (Fig. 3E). Under both approaches, T1 transgenic plants with strong GFP fluorescence exhibited a much higher possibility of showing effective genome editing than those without obvious fluorescence (Fig. 3E).
Discussion
Coexpression of multiple proteins using 2A peptides
Generally, 2A sequences encode short peptides of 18–22 amino acids that mediate translational skipping from the glycyl-prolyl peptide bond near their 3′ ends (Donnelly et al. 2001; de Felipe et al. 2010). 2A peptides have been widely used in animal cells because of their high self-cleavage efficiency and the stoichiometric expression of proteins flanking them. Although 2A peptides have been successfully applied in several studies in plants (Cermak et al. 2017; Wang et al. 2017), the cleavage efficiency of various 2A peptides in plant cells has been unclear. In this study, we constructed constitutive expression vectors harboring Cas9-2A-GFP and analyzed the cleavage efficiency of five 2A peptides in Arabidopsis protoplasts (Fig. 1A–C). The P2A peptide exhibited an approximately 95% self-cleavage efficiency in protoplasts (Fig. 1A–C), which was similar to the results in animal cells (Kim et al. 2011). During the preparation of the manuscript, the 2A peptide was reported to be used for fusing Cas9 and GFP in the protoplasts of Nicotiana benthamiana. Fluorescence-activated cell sorting (FACS)-mediated selection of GFP-expressed protoplasts has been shown to facilitate Cas9-mediated mutation enrichment (Petersen et al. 2019), which is consistent with our study showing that Cas9-P2A-GFP improves the selection of transgenic plants with a high editing efficiency. In addition to the Cas9-P2A-GFP construct used in this study, the P2A peptide may be widely used to coexpress multiple proteins in plants for various purposes.
UBQ10 is the best promoter for driving Cas9-P2A-GFP
Promoters are one of the key elements in the CRISPR/Cas9 system that can affect editing efficiency. Several previous studies showed that the genome editing efficiency of the 35S-controlled Cas9 system is low and that many of the induced somatic mutations are non-inheritable (Feng et al. 2014; Yan et al. 2015). To overcome the shortcomings of the 35S promoter, several egg cell-specific and germline-specific promoters were used to drive Cas9 in an improved CRISPR/Cas9 system (Wang et al. 2015b; Mao et al. 2016). Here, for the development of a system that can select transgenic lines with high Cas9 expression based on fluorescence, egg cell- or germline-specific promoters could not be used, since their fluorescence in gametes is difficult to detect. Thus, the CaMV 35S and UBQ10 promoters were tested, and our study clearly showed that the efficiency of the UBQ10 promoter was much higher than that of 35S in driving Cas9-P2A-GFP to edit the genome (Fig. 3). In addition, UBQ10 promoter-driven Cas9-P2A-GFP was capable of generating homozygous lines in the T1 generation (Fig. 3C–E), which implies that the UBQ10 promoter may function in the early stages of embryo development. Overall, UBQ10 is more effective than the 35S promoter for driving Cas9-P2A-GFP to edit the genome. For the targeting of BZR1 and BES1, Cas9-P2A-GFP driven by the 35S promoter was successfully used to generate T2 Cas9-free mutants with homozygous mutations in all four sgRNA loci (Fig. 2). A higher efficiency could be expected if the UBQ10 promoter were to be used. Thus, two binary vectors [pUBQ10:Cas9-P2A-GFP (Gent) and pUBQ10:Cas9-P2A-GFP (Hyg)] and the sgRNA assembly vector pAtU6-26-M, can be used to efficiently generate Cas9-free multiplex mutants in Arabidopsis and possibly in other plant species.
Main advantage of our novel CRISPR/Cas9 system
Generally, our study indicates that high expression of Cas9 is important for efficient editing, especially for multiple sgRNAs that may compete for the Cas9 protein, and that our novel CRISPR/Cas9 system using Cas9-P2A-GFP provides the advantage of the convenient selection of plants with high Cas9 expression. Thus, when using our novel CRISPR/Cas9 system, the selection and retention of several T1 lines with strong fluorescence is sufficient to obtain Cas9-free T2 plants with a high editing efficiency. In contrast, under previous methods, multiple T1 transgenic plants need to be genotyped for the selection of lines with a high editing efficiency (Feng et al. 2013; Yan et al. 2015). Furthermore, the ratio of edited Cas9-free plants in the T2 generation could be very low if the T1 plants are not selected (Gao et al. 2016). Thus, our novel CRISPR/Cas9 system dramatically reduces labor required for generating Cas9-free multiplex mutants.
Materials and methods
Plant materials and growth conditions
In this study, wild-type Arabidopsis thaliana Columbia-0 was used. Seeds were sterilized with 15% NaClO for 10 min and then washed 5 times with sterilized ddH2O. Sterilized T1 seeds were grown in MS plates (4.4 g L−1 Murashige Skoog powder, 1% sucrose, 8 g L−1 Agar, pH 5.8) containing 50 mg L−1 hygromycin or 200 mg L−1 gentamicin sulfate. T2 seeds were sown on MS plates without antibiotics. After stratification for 4–5 days at 4 °C, the seeds were germinated and grown under continuous white light (80 μmol m−2 s−1) at 22 °C. 7-day-old seedlings were transferred to the soil and grown under long-day conditions (60–80 μmol m−2 s−1 white light, 16 h light 22 °C/8 h dark 18 °C cycles).
Plasmid construction
To generate the constructs containing 2A peptides fusing Cas9 and GFP for protoplast transformation, the intermediate cloning vectors pJim19 (Gent) and pBS were used. First, GFP was amplified from pJim19 (Bar)-GFP (Sun et al. 2016) and cloned into pJim19 (Gent) (modified from pZP222) with Spe I and Sac I to generate the pJim19 (Gent)-GFP. rbcS-E9 terminator was cloned from pHEE401E (Wang et al. 2015a) and inserted between the Sac I and EcoR I sites of pJim19 (Gent)-GFP to replace the Nos terminator, resulting in pJim19 (Gent)-GFP-rbcS-E9t. Five 2A peptides and a control peptide (CP) with Kpn I and Spe I restriction enzyme sites were synthesized, annealed and cloned into Kpn I and Spe I-digested pJim19 (Gent)-GFP-rbcS-E9t to produce pJim19 (Gent)-Linker-GFP-rbcS-E9t. The linker was CP, F2A-1, E2A, F2A-2, P2A, or T2A. Then, Cas9 without a stop codon was amplified from p35S-Cas9-SK (Feng et al. 2013) and inserted into pBS to produce pBS-Cas9 (without a stop codon). Fragments of the Linker-GFP-rbcS-E9t construct were amplified from the pJim19(Gent)-Linker-GFP-rbcS-E9t vector and cloned into pBS-Cas9 (without a stop codon) to generate pBS-Cas9-Linker-GFP-rbcS-E9t. Finally, p35S-Cas9-SK was modified by deleting one Xho I site upstream of the double CaMV 35S promoter and adding an Nhe I site between Hind III and Sal I. Both the modified p35S-Cas9-SK and pBS-Cas9-Linke-GFP-rbcS-E9t constructs were digested using Xho I and EcoR I to generate the final plasmid p35S-Cas9-Linker-GFP-rbcS-E9t. The linker sequences (CP/F2A-1/E2A/F2A-2/P2A/T2A) are shown in Supplemental Table 1.
The binary vector pJim19 (Gent) was modified by replacing the CaMV 35S promoter and Nos terminator with the sequences of Hind III, Stu I, Spe I, Kpn I, and EcoR I to generate the pJim19 (Gent)-M vector. The UBQ10 NheI F and UBQ10 XhoI R primers were used to clone the UBQ10 promoter from Arabidopsis genomic DNA. The UBQ10 promoter and p35S:Cas9-P2A-GFP-rbcS-E9t vector were digested with Nhe I and Xho I and assembled into the pUBQ10-Cas9-P2A-GFP-rbcS-E9t vector. UBQ10-Cas9-P2A-GFP-rbcS-E9t was cloned into EcoR I- and Kpn I-digested pJim19 (Gent)-M to generate the binary vector pJim19-UBQ10:Cas9-P2A-GFP-rbcS-E9t, abbreviated as pUBQ10:Cas9-P2A-GFP (Gent). Similarly, the 2 × 35S-Cas9-P2A-GFP-rbcS-E9t module was digested from p35S-Cas9-P2A-GFP-rbcS-E9t and cloned into pJim19 (Gent)-M to obtain the binary vector pJim19-2 × 35S-Cas9-P2A-GFP-rbcS-E9t, abbreviated as p35S:Cas9-P2A-GFP (Gent). In addition, the Cas9-P2A-GFP cassette was inserted into pCAMBIA1300 with hygromycin resistance to generate two other binary vectors, pUBQ10:Cas9-P2A-GFP (Hyg) and p35S:Cas9-P2A-GFP (Hyg).
To generate multiple sgRNA expression modules, the pAtU6-26-SK vector (Feng et al. 2013) was modified by deleting a Spe I site downstream of the sgRNA scaffold and adding a Nhe I site between Xho I and Sal I, resulting in pAtU6-26-M (Supplemental Fig. 2A). sgRNAs were inserted into the Bbs I site of pAtU6-26-M. Multiple sgRNA expression cassettes were assembled using Sal I and isocaudomer pair Spe I/Nhe I, as described in Fig. 1D.
To generate the final genome-editing plasmids, multiple sgRNA expression modules assembled using isocaudomer restriction enzymes were digested using Kpn I and Sal I and then inserted into binary vectors containing Cas9-P2A-GFP as described above.
All the primers are listed in Supplemental Table S3.
Mesophyll protoplast isolation and transfection
3- to 4-week-old Arabidopsis were used for protoplast isolation by following previously described procedures (Yoo et al. 2007). Leaves were cut into 0.5–1 mm leaf strips and immediately digested in 15 mL of enzyme solution (1.87% cellulase R10, 0.32% macerozyme R10, 0.4 M mannitol, 20 mM MES, pH 5.7, 20 mM KCl, 10 mM CaCl2, 5 mM β-mercaptoethanol and 0.1% BSA) for 3 h. The digested products were diluted with 15 mL precooled W5 solution (154 mM NaCl, 125 mM CaCl2, 5 mM KCl and 2 mM MES, pH 5.7) and filtered through a 70 μm nylon mesh. The protoplasts were pelleted by centrifugation at 1400 rpm for 2 min and washed with 10 mL of W5 solution, then centrifuged again. Protoplasts were resuspended using 10 mL of precooling W5 buffer and incubated on ice for 30 min, after which the supernatant was discarded. MMG buffer (0.4 M mannitol, 15 mM MgCl2 and 4 mM MES, pH 5.7) was added to the protoplasts to obtain a protoplast concentration of 105–106 cells mL−1. A 2 mL round-bottom microcentrifuge tube was used to perform DNA transfection. A 200 μL aliquot of protoplasts was well mixed with 20 μg of plasmids and 220 μL of PEG solution (40% PEG 4000, 0.2 M mannitol and 0.1 M CaCl2). After 5 min of incubation at room temperature, the transformation reaction was quenched by adding 1 mL of precooling W5 buffer, and the transfected protoplasts were collected by centrifugation at 1200 rpm for 2 min. Next, the transfected protoplasts were resuspended using 100 μL of W5 solution and transferred to six-well culture plates that had been precoated with 1 mL of WI solution (0.5 M mannitol, 20 mM KCl, 5% fetal calf serum and 4 mM MES, pH 5.7). The plate was covered with toilet paper and placed in the incubator for 16 h before observation under a microscope or the extraction of total proteins.
Protein extraction and western blotting
For the protoplast experiments, the cultured protoplasts were collected by centrifugation at 100×g for 1 min. Total proteins were extracted by adding 20 μL of denaturing buffer (8 M urea, 100 mM NaH2PO4, 100 mM Tris–HCl, pH 8.0, 1 mM PMSF and 1× protease inhibitor) and 5 μL of 5× SDS loading buffer (50% glycerol, 10% SDS, 0.1% Bromo Phenol Blue, 10 mM DTT, 5% β-mercaptoethanol and 250 mM Tris–HCl, pH 6.8). Well-mixed protoplast and buffer mixtures were boiled at 100 °C for 10 min. For the plant analyses, 7-day-old light-grown seedlings were harvested and ground into powder in liquid nitrogen. Denaturing buffer was used to extract total proteins. After centrifugation at 13,000 rpm for 10 min, the supernatant containing the total proteins was mixed with 5× SDS loading buffer and then boiled at 100 °C for 10 min.
The protein samples were separated on 8% SDS-PAGE gels and transferred to a PVDF membrane. The membranes were incubated with anti-GFP, anti-RPN6 or anti-RPT5 antibodies after being blocked with 5% milk. Then, the membranes were washed three times and incubated with the corresponding secondary antibodies, followed by another three washes before detection by ECL Primer (GE Healthcare).
References
Buren S, Ortega-Villasante C, Otvos K, Samuelsson G, Bako L, Villarejo A (2012) Use of the foot-and-mouth disease virus 2A peptide co-expression system to study intracellular protein trafficking in Arabidopsis. PLoS One 7:e51973
Cermak T, Curtin SJ, Gil-Humanes J, Cegan R, Kono TJY, Konecna E, Belanto JJ, Starker CG, Mathre JW, Greenstein RL, Voytas DF (2017) A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 29:1196–1217
de Felipe P, Luke GA, Brown JD, Ryan MD (2010) Inhibition of 2A-mediated ‘cleavage’ of certain artificial polyproteins bearing N-terminal signal sequences. Biotechnol J 5:213–223
Donnelly MLL, Luke G, Mehrotra A, Li XJ, Hughes LE, Gani D, Ryan MD (2001) Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal ‘skip’. J Gen Virol 82:1013–1025
Feng ZY, Zhang BT, Ding WN, Liu XD, Yang DL, Wei PL, Cao FQ, Zhu SH, Zhang F, Mao YF, Zhu JK (2013) Efficient genome editing in plants using a CRISPR/Cas system. Cell Res 23:1229–1232
Feng ZY, Mao YF, Xu NF, Zhang BT, Wei PL, Yang DL, Wang Z, Zhang ZJ, Zheng R, Yang L, Zeng L, Liu XD, Zhu JK (2014) Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc Natl Acad Sci USA 111:4632–4637
Gao XH, Chen JL, Dai XH, Zhang D, Zhao YD (2016) An effective strategy for reliably isolating heritable and Cas9-free Arabidopsis mutants generated by CRISPR/Cas9-mediated genome editing. Plant Physiol 171:1794–1800
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170
Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, Kim MK, Shin BA, Choi SY (2011) High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One 6:e18556
Li J, Chory J (1997) A putative leucine-rich repeat receptor kinase involved in brassinosteroid signal transduction. Cell 90:929–938
Li JF, Norville JE, Aach J, McCormack M, Zhang DD, Bush J, Church GM, Sheen J (2013) Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31:688–691
Ma XL, Zhang QY, Zhu QL, Liu W, Chen Y, Qiu R, Wang B, Yang ZF, Li HY, Lin YR, Xie YY, Shen RX, Chen SF, Wang Z, Guo JX, Chen LT, Zhao XC, Dong Z, Liu YG (2015) A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8:1274–1284
Mao YF, Zhang H, Xu NF, Zhang BT, Gou F, Zhu JK (2013) Application of the CRISPRCas system for efficient genome engineering in plants. Mol Plant 6:2008–2011
Mao YF, Zhang ZJ, Feng ZY, Wei PL, Zhang H, Botella JR, Zhu JK (2016) Development of germ-line-specific CRISPR-Cas9 systems to improve the production of heritable gene modifications in Arabidopsis. Plant Biotechnol J 14:519–532
Mao YF, Botella JR, Liu YG, Zhu JK (2019) Gene editing in plants: progress and challenges. Natl Sci Rev 6:421–437
Petersen BL, Moller SR, Mravec J, Jorgensen B, Christensen M, Liu Y, Wandall HH, Bennett EP, Yang Z (2019) Improved CRISPR/Cas9 gene editing by fluorescence activated cell sorting of green fluorescence protein tagged protoplasts. BMC Biotechnol 19:36
Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng G, Sharp PA, Zhang F (2014) CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159:440–455
Sun N, Wang JJ, Gao ZX, Dong J, He H, Terzaghi W, Wei N, Deng XW, Chen HD (2016) Arabidopsis SAURs are critical for differential light regulation of the development of various organs. Proc Natl Acad Sci USA 113:6071–6076
Wang C, Shen L, Fu YP, Yan CJ, Wang KJ (2015a) A simple CRISPR/Cas9 system for multiplex genome editing in rice. J Genet Genom 42:703–706
Wang ZP, Xing HL, Dong L, Zhang HY, Han CY, Wang XC, Chen QJ (2015b) Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation. Genome Biol 16:144
Wang M, Lu Y, Botella JR, Mao Y, Hua K, Zhu JK (2017) Gene targeting by homology-directed repair in rice using a geminivirus-based CRISPR/Cas9 system. Mol Plant 10:1007–1010
Yan LH, Wei SW, Wu YR, Hu RL, Li HJ, Yang WC, Xie Q (2015) High-efficiency genome editing in Arabidopsis using YAO promoter-driven CRISPR/Cas9 system. Mol Plant 8:1820–1823
Yoo SD, Cho YH, Sheen J (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2:1565–1572
Yu H, Zhao YD (2019) Fluorescence marker-assisted isolation of Cas9-free and CRISPR-edited Arabidopsis plants. Methods Mol Biol 1917:147–154
Zhang ZJ, Mao YF, Ha S, Liu WS, Botella JR, Zhu JK (2016) A multiplex CRISPR/Cas9 platform for fast and efficient editing of multiple genes in Arabidopsis. Plant Cell Rep 35:1519–1533
Acknowledgements
We thank Prof. Xing Wang Deng for lab resources, Prof. Jian-Kang Zhu for providing the p35S-Cas9-SK and pAtU6-26-SK plasmids, and Prof. Qi-Jun Chen for providing the pHEE401E vector. This study was supported by the National Key R&D Program of China (2018YFE0204700), the National Natural Science Foundation of China (31621001), the Peking-Tsinghua Center for Life Sciences, and the State Key Laboratory of Protein and Plant Gene Research.
Author information
Authors and Affiliations
Contributions
JW and HC designed the study. JW performed the experiments. JW and HC analyzed the data. JW and HC wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, J., Chen, H. A novel CRISPR/Cas9 system for efficiently generating Cas9-free multiplex mutants in Arabidopsis. aBIOTECH 1, 6–14 (2020). https://doi.org/10.1007/s42994-019-00011-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42994-019-00011-z