
Abstract
An Agrobacterium tumefaciens-based transformation procedure was developed for the desiccation tolerant species Lindernia brevidens. Leaf explants were infected with A. tumefaciens strain GV3101 harbouring a binary vector that carried the hygromycin resistance gene and an eGFP reporter gene under the control of a native dehydration responsive LEA promoter (Lb2745pro). PCR analysis of the selected hygromycin-resistant plants revealed that the transformation rates were high (14/14) and seeds were obtained from 13/14 of the transgenic lines. A combination of RNA gel blot and microscopic analyses demonstrated that eGFP expression was induced upon dehydration and ABA treatment. Comparison with existing procedures used to transform the well studied resurrection plant and close relative, Craterostigma plantagineum, revealed that the transformation process is both rapid and leads to the production of viable seed thus making L. brevidens a candidate species for functional genomics approaches to determine the genetic basis of desiccation tolerance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although often observed in seeds, spores and pollen, desiccation tolerance is rare in vegetative tissues of vascular plants. Of the quarter of a million species of vascular plants, approximately 330 species have been documented as having the potential to survive desiccation in the vegetative growth phase (Porembski and Barthlott 2000). The acquisition of desiccation tolerance requires the induction of a co-ordinated programme of genetic and biochemical processes. Among the metabolic changes that take place during drying is the synthesis of proteins and sugars, which are postulated to form the basis of protective mechanisms that limit damage to cellular constituents. A group of proteins that are predicted to play a major role in desiccation tolerance are the Late Embryogenesis Abundant (LEA) proteins (Cuming 1999).
Plant systems have to be developed to elucidate desiccation tolerance associated gene functions through stable genetic transformation. So far only two species have proven amenable to gene transfer namely, Craterostigma plantagineum (Furini et al. 1994; Toldi et al. 2002) and Ramonda myconi (Tóth et al. 2006). T-DNA tagging strategies have subsequently been employed to identify C. plantagineum genes that play a role in abscisic acid (ABA) signalling, although a major drawback to this approach is that it is both time consuming and difficult to produce seeds (Furini et al. 1997; Smith-Espinoza et al. 2005). Thus there is a need to develop improved genetic transformation technology in order to over-express and silence genes in a desiccation tolerant species.
Previously, phylogenetic analysis has been used to demonstrate monophyly of the lineage that includes the genera Craterostigma and Lindernia (Rahmanzdeh et al. 2005). Recent data demonstrates that Lindernia brevidens is desiccation tolerant (Phillips, Fischer, Baron, van den Dries, Kutzer, Rahmanzdeh, Remus and Bartels unpublished). Mechanisms that transcriptionally regulate and confer cellular protection during extreme water loss appear well conserved in L. brevidens, and genes that play a putative role in desiccation tolerance have a high degree of sequence homology to the corresponding genes from C. plantagineum. In this context a homologue of the LEA gene CDeT27-45 from C. plantagineum (Bartels et al. 1990) was isolated from L. brevidens. Using an Agrobacterium tumefaciens-mediated procedure, a plasmid construct that contained the Lb27-45 gene promoter (Lb2745pro) driving eGFP expression was introduced. The regulatory parameters were investigated and Lb2745pro was found to be (1) abscisic acid responsive and (2) active in guard cells of desiccated leaves and in embryos of mature seeds.
Materials and methods
In vitro culture
Lindernia brevidens plants were collected by Professor Eberhard Fischer (University of Koblenz, Germany) from the Usambara Mountains, Tanzania. Seeds were surface sterilised in 70% (v/w) ethanol for 2 min and in 1.6% (w/v) sodium hypochlorite, 0.1% Tween 20 for 10 min. The seeds were rinsed five times in sterile deionised water and then germinated on MS medium pH 5.8 containing half-strength MS elements (Murashige and Skoog 1962), sucrose (20 g/l) and agar (8 g/l). Germination took place after stratification at 4°C for 3 days and maintained in a climate chamber at day/night temperatures of 22 and 18°C, respectively. Plants were grown under a 16 h day per 8 h night regime, with a light intensity of 80 μE/m2 s. Leaves (1–2 cm in length) were aseptically transferred to Leaflet Proliferation medium containing half-strength MS elements (Murashige and Skoog 1962), sucrose (20 g/l), naphthalene acetic acid (NAA; 0.1 mg/l), benzyl amino purine (BAP; 0.5 mg/l), myo-inositol (100 mg/l), nicotinic acid (1 mg/l), pyridoxine HCl (5 mg/l) and Gelrite (Duchefa Biochemie, Haarlem, Netherlands) (6 g/l) to generate a proliferation of leaflets (5–10 mm in length).
Cloning, bacterial strain and culture conditions
The 5’-flanking region of the Lb27-45 gene (GenBank Accession number EF364563) was cloned by genome walking, using the Universal Genome Walker kit (Clontech Laboratories, Palo Alto, CA, USA) and following the instructions of the manufacturer. Genomic DNA was prepared according to the cetyl trimethyl ammonium bromide (CTAB) method described by Ausubel et al. (2006) and digested with EcoRV. The blunt-end fragments generated were then ligated to a Genome Walker adaptor to construct a Genome Walker library. A 1,049 bp fragment putatively containing the 975 bp Lb27-45 promoter was isolated by two successive PCR-based DNA amplifications of the Genome Walker library. The primary PCR was performed with a gene-specific primer (5’-AGCATCGTCTTATCGTTTCCCTTCAAC-3’) and the outer adaptor primer (AP1). The primary product served as template for the secondary PCR with a nested gene-specific primer (5’-ACGCAGATCTTGGCGAGGTAAGTGAT-3’) and the nested adaptor primer (AP2). The 1,049 bp secondary PCR product was cloned into the pCR 2.1 TOPO vector (Invitrogen, Carlsbad, CA, USA) and the DNA sequence was determined. The search for putative cis elements in the promoter region was carried out with PLACE (http://www.dna.affrc.go.jp/PLACE/signalscan.html).
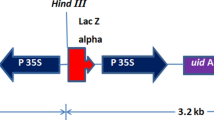
The Lb2745proGFP construct was used for transformation (see Fig. 1). The Lb27-45 promoter including the 5′ untranslated region (1,049 bp in total) was amplified using the following oligonucleotide primers: Lb27-45 proF 5′CTGCAGAGATCTTGGCGAGGTAAG 3′ and Lb27-45 proR 5′GAATTCCCTTGGAAGTTCTTCTCCT 3 and cloned into the EcoRI – PstI sites. The binary vector backbone was pH7GWIWG2(I) (Karimi et al. 2002) and the Gateway expression cassette was replaced by eGFP (Clontech Living Colors ®, Mountain View, CA; Cormack et al. 1996) and a pea RbcS terminator sequence (Coruzzi et al. 1984). The construct was transferred into A. tumefaciens via electroporation (Wen-jun and Forde 1989). The A. tumefaciens strain GV3101 carrying the helper plasmid pMP90RK was used as the host strain (Koncz and Schell 1986). The plasmid bearing A. tumefaciens were grown overnight at 28°C at 200 rpm in YEB medium containing rifampicin (100 μg/ml), gentamycin (25 μg/ml) and spectinomycin (150 μg/ml). The cells were pelleted and resuspended for plant infection in MS medium pH 5.8 containing half-strength MS elements (Murashige and Skoog 1962), sucrose (20 g/l) and an antioxidant mixture (0.15 g/l ascorbic acid; 0.1 g/l citric acid) to a final density of 0.5 at OD550.

Schematic diagram of the Lb2745proGFP plasmid. A 1,049 bp 5′ upstream region that included the 975 bp Lb27-45 promoter was cloned via EcoRI—PstI sites as a translational fusion with the eGFP gene. Dehydration/ABA related cis-elements are annotated. Blue boxes indicate ABREs (ABA Responsive Elements; ACGTG) (Simpson et al. 2003). Yellow boxes indicate DRE (dehydration-responsive elements) (Dubouzet et al. 2003). The purple box indicates a R18-SAP domain transcription factor binding site (Nelson et al. 1994; Hilbricht et al. 2002 )
Genetic transformation
Leaflets (5–10 mm in length; Fig. 2a) were excised and lightly pressed on a sterile fine sand paper to increase the wounded area. The leaflets were then incubated with the A. tumefaciens suspension for 20 min in darkness and co-cultivated for 2 days on Leaflet Proliferation medium that also contained the antioxidant mixture (0.15 g/l ascorbic acid; 0.1 g/l citric acid). After co-cultivation, the infected leaflets were subcultured for 2 weeks on Leaflet Proliferation medium with the addition of the antioxidant mixture, cefotaxime (500 mg/l) and hygromycin (15 mg/l). Five further rounds of subculturing on identical medium were performed to obtain shoot clusters, each time with a 100 mg/l reduction in the level of cefotaxime. Shoots were rooted on Root Formation medium that contained half-strength MS elements (Murashige and Skoog 1962), sucrose (20 g/l), antioxidant mixture, hygromycin (15 mg/l), myo-inositol (100 mg/l), nicotinic acid (1 mg/l), pyridoxine HCl (5 mg/l) and Gelrite (Duchefa Biochemie, Haarlem, Netherlands) (6 g/l) for 3–6 weeks. Rooted plants were hardened in water for 2 days, transplanted to potting mix (FloraGard, Oldenburg, Germany), and grown to maturity in the regime described for in vitro-cultured plants. Vigorous growth and floral initiation was promoted by pruning. After 2–3 months, the first flowers appeared, however these were sterile–a phenomenon also observed in wild-type plants. The second flush of flowers followed 1 month later and gave rise to viable seed. 5–10 siliques were obtained per transgenic line, and approximately 100 seeds were contained in each silique

Generation of transgenic Lindernia brevidens plants obtained by Agrobacterium tumefaciens-mediated infection of in vitro cultured leaflet tissue. a In vitro leaflet formation. b Co-cultivation of leaflets with A. tumefaciens. c Hygromycin-based selection of transgenic shoot tissue. d Early root formation; arrow shows the formation of a root structure. e Example of a transgenic plant transferred to soil. f A L. brevidens flower. Bar = 1 cm
PCR analysis to verify the transgene insertion
A 0.7 kb fragment that includes the enhanced green fluorescent protein (eGFP) coding region (Clontech Living Colors ®, Mountain View, CA; Cormack et al. 1996) was amplified using the following oligonucleotide primers designed by Willige and Bartels (unpublished): eGFP_5′ CGAACGATAGCCATGGGTAAAG and eGFP_3′CTCTAGAGGATCCTTACAGCTCATCCTTACCGGTACCATTTGTATAGTTCATCCCATGC. Note that the eGFP_3′ primer contains additional 5′ sequence that encodes for a C-terminal endoplasmic reticulum retention signal (KDEL) which is incidental to this study. The 1,049 bp Lb27-45 promoter/5′ untranslated region (GenBank Accession number EF364562) was amplified using the oligonucleotide primers Lb27-45 proF and Lb27-45 proR. Genomic DNA was isolated from 1 g of leaf material harvested from mature plants according to the CTAB method described by Ausubel et al. (2006) and 100 ng was used in each PCR amplification.
Dehydration treatment
Young expanding leaves were detached from the hydrated transformed plant lines and dried on filter paper for 48 h under approximately 50% relative humidity in a climate chamber at 16 h day per 8 h night temperatures of 22 and 18°C, respectively, with a light intensity of 80 μE/m2 s. Following drying, the relative water content of the leaves was measured according to the method described by Bernacchia et al. (1996) and found to be approximately 5%.
RNA gel blot analysis
RNA extraction and RNA blot hybridisations were performed according to Bartels et al. (1990). About 10 μg total RNA was loaded in each lane from hydrated (relative water content ∼80%) and desiccated (relative water content ∼5%) leaf material. The filter was sequentially probed with the eGFP coding region (Clontech Living Colors ®, Mountain View, CA; Cormack et al. 1996), the Lb27-45 cDNA (Accession number EF364563) and rDNA (pTA71; Gerlach and Bedbrook 1979).
Confocal microscopy
Fluorescence imaging was performed using a confocal laser scanning microscope (e-C1 confocal microscope system, Nikon GmbH, Düsseldorf, Germany). Hydrated and dried samples were mounted in water and visualised within 5 min of preparation. Tissues were treated for 16 h with 100 μM ABA or water as described by (Bartels et al. 1990). With each figure, images were obtained at constant settings (i.e. laser power and pinhole size) to facilitate comparisons in fluorescence levels. Images were prepared using EZ-C1 3.20 software (Nikon). eGFP fluorescence was analysed using a blue argon ion laser (488 nm) and detection at 515/530 nm. Background fluorescence from chlorophyll and the testa was visualised using a green helium/neon laser (543 nm) and detection at 570 nm.
Scanning electron microscopy
Young expanding leaves were fixed in 2% (v/v) glutaraldehyde in phosphate buffered saline (PBS pH 7.2) and dehydrated through a graded ethanol series (10–100%) diluted in PBS prior to critical point drying according to the method of Svitkina et al. (1984). Leaf specimens were viewed after coating with gold using a Philips 501 scanning electron microscope.
Results
Isolation and analysis of the Lb27-45 promoter
Genomic DNA flanking the Lb27-45 gene was cloned and shown to contain the 5′ promoter region (GenBank Accession number EF364562). Cis-elements were identified that have been shown to mediate dehydration and ABA transcriptional responses (Fig. 1). These are: ACGTG, an ABRE sequence required for etiolation-induced expression of erd1 in Arabidopsis thaliana (Simpson et al. 2003) (blue boxes); RCCGAC, a core motif of DRE (dehydration-responsive element) (Dubouzet et al. 2003) (yellow boxes); and AGCCC, R18-SAP domain transcription factor binding sites in the desiccation responsive CDeT27-45 promoter (Nelson et al. 1994; Hilbricht et al. 2002) (purple box). To assay the function, we constructed an Lb27-45 promoter eGFP fusion (Lb27-45proGFP; Fig. 1) and analyzed the expression of the reporter gene. This allowed promoter activity to be observed in L. brevidens cells in a non-destructive manner by eGFP fluorescence microscopy.
Genetic transformation of Lindernia brevidens and selection of transgenic tissue and plants
Excised leaflets were wounded and infected with the Agrobacterium strain harbouring the Lb27-45proGFP plasmid construct (Fig. 1). The wounded explants and bacteria were co-cultivated (Fig. 2b) and then transferred to medium that inhibits bacterial growth and promotes the growth of transformed shoot tissue. Because of the high amount of phenolic compounds produced from the wounded tissue, it was necessary to add an antioxidant mixture to the culture media. In the absence of the antioxidant, the explants released phenolic substances whose oxidation products darkened both the tissue and the medium and subsequently the explants died within a few days.
Individual embryoids developed from the explants growing on medium containing NAA and BAP that gave rise to green shoots (Fig. 2c). During further subculturing, the shoots continuously proliferated and did not display any differences to that observed for wild-type shoots regenerated on the same medium in the absence of hygromycin. The established shoots were transferred to medium without hormones and supplemented with hygromycin for root formation and further development (Fig. 2d). More than 80% of the subcultured shoots gave rise to plantlets. The continuous regeneration of plants on selective medium was a positive indication that the putative transformants were stable. Rooted plants, with 6–8 leaves and with roots 2–3 cm long, were transferred to soil and cultivated in a growth chamber where they grew to maturity.
Presence and expression of the transgene
PCR analysis of transgenic tissue
PCR analysis was performed on putatively transformed, hygromycin resistant lines. The presence of the eGFP gene was confirmed in all of the 14 hygromycin-resistant lines and was not detected in the non-transformed control (Fig. 3). As a positive control, PCR analysis was also used to demonstrate the presence of the Lb27-45 promoter in all lines including the wild-type genomic DNA sample.

Analysis of L. brevidens transgenic lines by genomic DNA PCR. Amplification of the eGFP cDNA and Lb27-45 promoter region
RNA gel blot analysis
RNA gel blot analysis was used to compare the levels of the eGFP and endogenous Lb27-45 steady state RNA levels in hydrated and dehydrated leaf samples (Fig. 4). Dehydration responsive eGFP expression was detected in all lines except for the non-transformed control. A similar transcript profile was observed when the Lb27-45 cDNA was used as a probe with the exception that a signal was also present in the wild-type samples. Subtle differences between the expression levels of eGFP and Lb27-45 were observed specifically in lines 4, 8 and 11. In these three lines, the level of eGFP was noticeably higher than that of Lb27-45 mRNA in the hydrated sample. Variability in the levels of Lb27-45 mRNA was also seen between plant lines that also correlated with reporter gene levels. For example, lines 2, 3, 6 and 10 displayed low levels of both Lb27-45 and eGFP expression, whereas lines 4, 5, 8, 12, 13 and 14 exhibited high levels of expression. The level of total RNA that was present in each lane was controlled using a probe that consisted of the intergenic spacer and fragments of the 18S and 26S rRNA sequence (pTA71; Gerlach and Bedbrook 1979).

RNA gel blot analysis of L. brevidens transgenic lines. RNA was isolated from hydrated and desiccated leaf tissue and sequentially probed with the eGFP cDNA, Lb27-45 cDNA and rDNA fragment (pTA71; Gerlach and Bedbrook 1979 )
eGFP expression analysis
To observe the spatial distribution of eGFP expression in leaves and seeds, confocal laser scanning microscopy was used. Typical fluorescent patterns observed in the transgenic L. brevidens lines are shown in Figs. 5–7. Strong eGFP fluorescence was detected specifically in stomatal guard cells of dehydrated leaves (Fig. 5b) and the embryos of mature seeds (Fig. 6a) in all 14 transformed lines in comparison to hydrated leaves of transgenic plants (Fig. 5a) and wild type controls (Figs. 5c,d, 6b). Weak eGFP fluorescence was observed in the endosperm (Fig. 6a). eGFP labelling of the guard cells of desiccated leaves revealed that the stomata were fully open, a feature that has also been observed in other resurrection plants such as C. plantagineum (Schwab et al. 1989). The red background fluorescence in desiccated leaves is due to chlorophyll, which is indicative of homiochlorophyllous (chlorophyll-retaining) desiccation tolerant plant species, whereas in the case of seeds the red autofluorescence is due to the testa.

Localisation of eGFP expression in desiccated tissues of the Lb2745proGFP transgenic L. brevidens lines. a eGFP expression was barely detectable in hydrated leaves. b Following desiccation, eGFP expression was observed specifically in the guard cells of desiccated leaves. c and d No eGFP expression was observed in wild-type hydrated and desiccated leaves, respectively. Bar = 50 μm

Localisation of eGFP expression in mature seeds of the Lb2745proGFP transgenic L. brevidens lines. a eGFP expression was observed in the embryo and to a lesser extent in the endosperm of mature seeds. b No eGFP signal was detected in wild-type seeds. Bar = 200 μm

Induction of Lb27-45 promoter activity in epidermal cells of transgenic L. brevidens young expanding leaves in response to exogenously applied ABA after 16 h. a eGFP expression was barely detectable in water treated control leaves. b Strong eGFP expression was observed in ABA(100 μM) treated leaves. c A surface view of the leaf epidermis. Bar = 50 μm
Upon exogenous application of ABA, an increase in eGFP expression was observed in all young, expanding leaf epidermal cell types relative to a water control (Fig. 7a,b). A scanning electron microscope image of the epidermal cells from an equivalent region of a L. brevidens leaf is shown in Fig. 7c for comparison. Expression of eGFP in L. brevidens tended to result in the formation of eGFP aggregates, this phenomenon may be due to factors such as protein solubility or the activity of the Lb2745pro promoter.
Discussion
Transgenic plants allow the targeted over-expression and silencing of dehydration-related genes in vivo and are therefore excellent systems to assess the function and tolerance conferred by the encoded proteins. Collections of dehydration responsive genes have been assembled from Lindernia brevidens and Craterostigma plantagineum (Bockel et al. 1998; Bartels unpublished). Functional information regarding many of these gene products is required to determine their role in desiccation tolerance. It has been observed that a large proportion of the desiccation-tolerance-associated genes are conserved between both species, therefore we propose that knowledge derived from one organism may be applied to the other via a comparative genomics approach. The objective of this study was to develop a system for the stable genetic transformation of L. brevidens and to test the functional properties of the Lb27-45 promoter in a homologous background. The transformation procedure is based largely on the protocol described by Furini et al. (1994) that was used to genetically transform C. plantagineum with several key differences. Unlike C. plantagineum, no extensive callus phase was required for the regeneration of plants. After co-cultivation with A. tumefaciens, shoots were immediately regenerated from the infected explants. Although not observed, the presence of a brief callus phase cannot be ruled out. The initiation and maintenance of C. plantagineum callus in the presence of selective agents prior to shoot differentiation is a time consuming stage of the procedure, often requiring 4–6 months. Furthermore, the ability to generate large quantities of viable seed enable traits to be potentially monitored in transgenic plants over multiple generations.
Lb27-45 is homologous to the CDet27-45 gene reported from C. plantagineum (Bartels et al. 1990; Phillips, Fischer, Baron, van den Dries, Kutzer, Rahmanzdeh, Remus and Bartels unpublished). The Lb27-45 promoter drives eGFP expression in a manner that is broadly similar in leaves to that observed for the endogenous Lb27-45 gene. Expression of the transgene was broadly observed to be similar to that of the endogenous Lb27-45 gene at the steady state mRNA level, indicating that the gene is predominantly regulated at the transcriptional, rather than at the level of transcript stability. In three different events, an increase in expression under hydrated conditions was observed, suggesting that the integration of the T-DNA may play some role on the induction kinetics. Factors such as chromatin structure may influence the promoter activity. In addition, variability in the expression of the endogenous Lb27-45 gene, which was broadly correlated with the reporter gene transcript levels (Fig. 4). This phenomenon is likely to be due to variability in the age of each plant, since the time spent in tissue culture and after transfer to soil varied slightly between the different lines.
The response of the promoter to desiccation and exogenously applied ABA was investigated in leaves using confocal laser scanning microscopy. The use of a GFP-based prokaryotic expression system as a tool to measure water availability has been established at least to a mild drop in water potential (−2.5 MPa) (Axtell and Beattie 2002). Axtell and Beattie (2002) state that “GFP is very stable, a factor which provides experimental flexibility when evaluating reporter activity following an induction event”. Our data support this statement to some degree, however further experiments are required to prove that GFP fluorescence is unaffected in desiccated cells. One disadvantage of GFP as a reporter protein in the study of dehydration tolerance, however, is that dying and dead plant tissue autofluoresces at similar emission wavelengths. Here we show that since L. brevidens leaf tissues are desiccation tolerant, no background fluorescence interfered with observations. This characteristic therefore makes GFP-based biosensors useful in monitoring promoter and gene function using this resurrection plant.
Abbreviations
- LEA:
-
Late Embryogenesis Abundant protein
- eGFP:
-
Enhanced green fluorescent protein
- ABA:
-
Abscisic acid
References
Ausubel FA, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (2006) Current protocols in molecular biology. Greene Publishing and Wiley Interscience, NY, USA
Axtell CA, Beattie GA (2002) Construction and characterization of a proU-gfp transcriptional fusion that measures water availability in a microbial habitat. Appl Environ Microbiol 68:4604–4612
Bartels D, Schneider K, Terstappen G, Piatkowski D, Salamini F (1990) Molecular cloning of ABA-modulated genes from the resurrection plant Craterostigma plantagineum which are induced during desiccation. Planta 181:27–34
Bernacchia G, Salamini F, Bartels D (1996) Molecular characterization of the rehydration process in the resurrection plant Craterostigma plantagineum. Plant Physiol 111:1043–1050
Bockel C, Salamini F, Bartels D (1998) Isolation and characterization of genes expressed during early events of the dehydration process in the resurrection plant Craterostigma plantagineum. J Plant Physiol 152:158–166
Cormack BP, Valdiva RH, Falkow S (1996) FACS optimized mutants of the green fluorescent protein (GFP). Gene 173:33–38
Coruzzi G, Broglie R, Edwards C, Chua NH (1984) Tissue-specific and light-regulated expression of a pea nuclear gene encoding the small subunit of ribulose-1, 5-bisphosphate carboxylase. EMBO J 3:1671–1679
Cuming A (1999) LEA proteins. In: Shewry PR, Casey R (eds) Seed proteins, Kluwer Academic Publishers, The Netherlands, pp 753–780
Dubouzet JG, Sakuma Y, Ito Y, Kasuga M, Dubouzet EG, Miura S, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2003) OsDREB genes in rice, Oryza sativa L, encode transcription activators that function in drought-, high-salt- and cold-responsive gene expression. Plant J 33:751–763
Furini A, Koncz C, Salamini F, Bartels D (1994) Agrobacterium-mediated transformation of the desiccation–tolerant plant Craterostigma plantagineum. Plant Cell Rep 14:102–106
Furini A, Koncz C, Salamini F, Bartels D (1997) High level transcription of a member of a repeated gene family confers dehydration tolerance to callus tissue of Craterostigma plantagineum. EMBO J 16:3599–3608
Gerlach WL, Bedbrook JR (1979) Cloning and characterization of ribosomal RNA genes from wheat and barley. Nucleic Acids Res 7:1869–1885
Hilbricht T, Salamini F, Bartels D (2002) CpR18, a novel SAP-domain plant transcription factor, binds to a promoter region necessary for ABA mediated expression of the CDeT27-45 gene from the resurrection plant Craterostigma plantagineum Hochst. Plant J 31:293–303
Karimi M, Inze D, Depicker A (2002) Gateway vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci 7:193–195
Koncz C, Schell J (1986) The promoter of TL-DNA gene 5 controls the tissue-specific expression of chimeric genes carried by a novel type of Agrobacterium binary vector. Mol Gen Genet 204:383–396
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio-assays with tobacco tissue cultures. Physiol Plant 15:473–497
Nelson D, Salamini F, Bartels D (1994) Abscisic acid promotes novel DNA-binding activity to a desiccation—related promoter of Craterostigma plantagineum. Plant J 5:451–458
Porembski S, Barthlott W (2000) Granitic and gneissic outcrops (inselbergs) as centers of diversity for desiccation—tolerant vascular plants. Plant Ecol 151:19–28
Rahmanzdeh R, Müller K, Fischer E, Bartels D, Borsch T (2005) The Linderniaceae and Gratiolaceae are further lineages distinct from the Scrophulariaceae (Lamiales). Plant Biol 7:1–12
Schwab KB, Schreiber U, Heber U (1989) Response of photosynthesis and respiration of resurrection plants to desiccation and rehydration. Planta 177:217–227
Simpson SD, Nakashima K, Narusaka Y, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2003) Two different novel cis-acting elements of erd1, a clpA homologous Arabidopsis gene function in induction by dehydration stress and dark-induced senescence. Plant J 33:259–270
Smith-Espinoza CJ, Phillips JR, Salamini F, Bartels D (2005) Identification of further Craterostigma plantagineum cdt mutants affected in abscisic acid mediated desiccation tolerance. Mol Genet Genomics 274:364–372
Svitkina TM, Shevelev AA, Bershadsky AD, Gelfand VI (1984) Cytoskeleton of mouse embryo fibroblasts. Electron microscopy of platinum replicas. Eur J Cell Biol 34:64–74
Toldi O, Tóth S, Pónyi T, Scott P (2002) An effective and reproducible transformation protocol for the model resurrection plant Craterostigma plantagineum Hochst. Plant Cell Rep 21:63–69
Toth S, Kiss C, Scott P, Kovacs G, Sorvari S, Toldi O (2006) Agrobacterium-mediated genetic transformation of the desiccation tolerant resurrection plant Ramonda myconi (L.) Rchb. Plant Cell Rep 25:442–449
Wen-jun S, Forde BG (1989) Efficient transformation of Agrobacterium spp. by high voltage electroporation. Nucleic Acids Res 17:8385
Acknowledgments
We thank Prof. Dr. Eberhard Fischer (University of Koblenz, Germany) for providing plant material, Dr. B. Buchen and H.-J. Ensikat for help with microscopy, Marjan Siam for help with tissue culture, Guillaume Pilot for help with plasmid construction, Niels van den Dries for help with promoter isolation, Christiane Buchholz for plant care and Birgit Schmitz for technical assistance. C.S.-E. was supported by a Lise-Meitner-Stipendium.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Fehér.
Rights and permissions
About this article
Cite this article
Smith-Espinoza, C., Bartels, D. & Phillips, J. Analysis of a LEA gene promoter via Agrobacterium-mediated transformation of the desiccation tolerant plant Lindernia brevidens . Plant Cell Rep 26, 1681–1688 (2007). https://doi.org/10.1007/s00299-007-0370-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-007-0370-3