
Abstract
In this paper we describe the first procedure for Agrobacterium tumefaciens-mediated genetic transformation of the desiccation tolerant plant Ramonda myconi (L.) Rchb. Previously, we reported the establishment of a reliable and effective tissue culture system based on the integrated optimisation of antioxidant and growth regulator composition and the stabilisation of the pH of the culture media by means of a potassium phosphate buffer. This efficient plant regeneration via callus phase provided a basis for the optimisation of the genetic transformation in R. myconi. For gene delivery, both a standard (method A) and a modified protocol (method B) have been applied. Since the latter has previously resulted in successful transformation of another resurrection plant, Craterostigma plantagineum, an identical protocol was utilized in transformation of R. myconi, as this method may prove general for dicotyledonous resurrection plants. On this basis, physical and biochemical key variables in transformation were evaluated such as mechanical microwounding of plant explants and in vitro preinduction of vir genes. While the physical enhancement of bacterial penetration was proved to be essential for successful genetic transformation of R. myconi, an additional two-fold increase in the transformation frequency was obtained when the above physical and biochemical treatments were applied in combination. All R 0 and R 1 transgenic plants were fertile, and no morphological abnormalities were observed on the whole-plant level.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Environmental factors that impose water-deficit stress, such as drought, salinity and temperature extremes, place major limits on plant productivity (Boyer 1982). To overcome these limitations and improve production efficiency in the face of a burgeoning world population, more stress tolerant crops must be developed (Khush 1999). Traditional breeding strategies that have attempted to utilize genetic variation arising from varietal germplasm, interspecific or intergeneric hybridisation, induced mutations and somaclonal variation of cell and tissue cultures have met with only limited success owing to the genetic complexity of stress responses (Flowers and Yeo 1995). In contrast with traditional breeding and marker assisted selection programs, the direct introduction of a small number of genes by genetic engineering seems to be a more attractive and rapid approach to improve stress tolerance. Products of these genes protect, either directly or indirectly, against osmotic stresses. Engineered over-expression of biosynthetic enzymes for osmoprotectants, scavengers of reactive oxygen species, stress-induced or late embryogenesis abundant proteins are among the approaches reported (for review see Cushman and Bohnert 2000). Although the above resistance genes can be identified and isolated from any living organisms in theory, there are special desiccation-tolerant systems like seeds and resurrection plants, in which the targeted gene isolations and differential screening of dehydration-associated genes have resulted in numerous hits (for review see Ingram and Bartels 1996). Since the physiological role of proteins encoded by these genes are largely unknown, their functional analysis must be completed to allow their utilization in molecular breeding programs. The most frequently applied approach to improve our knowledge on gene functions in plant systems through genetic transformation is the use of sense/antisense gene constructs (Birch 1997). For this purpose, in vitro plant regeneration protocols had been established in resurrection plants such as Craterostigma plantagineum (Furini et al. 1994; Toldi et al. 2002), Ramonda myconi (Tóth et al. 2004) and Haberlea rhodopensis (Djilianov et al. 2005) of which only C. plantagineum was transformed successfully until now (Furini et al. 1994; Toldi et al. 2002). Here we describe the first genetic transformation procedure for R. myconi as the continuation of our previous work on its tissue culture system (Tóth et al. 2004). Additionally, the similarity in genetic transformation between the two resurrection plants C. plantagineum and R. myconi as a consequence of their common physiological specialisation was investigated and discussed.
Materials and methods
Chemicals
Restriction endonucleases, Taq polymerase and dNTPs were bought from Promega (Madison, WI), while α[32P]-dCTP was obtained from Izinta Kft. (Budapest, Hungary). Plant Mini Kit for total DNA extraction was purchased from Qiagen Gmbh (Hilden, Germany). Antibiotics, plant growth regulators and X-GlcA were obtained from Duchefa Biochemie (Haarlem, The Netherlands). All other chemicals were from Reanal Kft. (Budapest, Hungary).
Establishment and maintenance of axenic culture
Seeds of R. myconi (L.) Rchb. were obtained from Chiltern Seeds, UK. For the establishment of in vitro tissue culture, seeds were surface sterilized in 70% (v/w) ethanol for 1 min and in 30–50% (v/w) Domestos solution (Unilever Hungary Kft.) for 20 min. To remove the remaining disinfectant, the seeds were rinsed five times in sterile deionised water and then germinated on sugar-free GM medium (Toldi et al. 1994) containing half-strength MS elements (Murashige and Skoog 1962), 10 mg/l thiamine, filter sterilized GA3 (0.1 mg/l) and 8 g/l agar (Plant agar, Duchefa Biochemie bv.) in plastic Petri dishes. The pH of germination medium was adjusted to 5.8 with 1 M KOH. Germination occurred at 24±2°C under a 16 h light (approximately 4500 lx)/8 h dark regime. Epicotyls of the 2–3 week-old seedlings were excised and transferred into basal RA medium (Tóth et al. 2004) for micropropagation. Plantlets were grown in Vitro Vent containers (Duchefa Biochemie) at 24±2°C under a 16 h light (approximately 4500 lx)/8 h dark regime and were subcultured in every 3 weeks.
Bacterial strains
The A. tumefaciens strain GV2260 (Deblaere et al. 1985) with the binary vector p35SGUSINT (Vancanneyt et al. 1990) was used for genetic transformation (kindly provided by Dr. Rainer Höfgen MPI of Molecular Plant Physiology, Golm, Germany). The T-DNA region of p35SGUSINT vector contained a p35S-gus-intron-t35S transformation cassette in which the β-glucuronidase (gus)-coding region is interrupted by the second intron of the potato st-ls1 gene.
Genetic transformation
Vigorously growing plantlets were selected systematically for at least a year for the purpose of genetic transformation.
Two protocols were followed during the Agrobacterium-mediated transformation. Half of the plant material was transformed in a conventional way by using a general protocol developed for A. tumefaciens susceptible dicot plants (method A). The other half of the plant material was transformed according to the protocol by which monocot cereals, previously thought to be resistant to Agrobacterial infection, could be transformed effectively (method B).
Method A was applied according to Horsch et al. (1988) with slight modifications: About 50 ml of liquid ABM medium (Chilton et al. 1974) supplemented with the proper antibiotics (25 mg/l kanamycin, 100 mg/l rifampicin) was poured into 250 ml Erlenmeyer flasks to propagate transforming Agrobacteria. Bacterial cultures were kept in darkness at 28°C overnight on a shaker adjusted to 200 rpm. Healthy-white and dense bacterial cultures were used for transformation. Plant explants were prepared in two ways. Half of the leaves were sectioned by excising their edges that resulted in 5–7 mm × 5–7 mm leaf segments. These leaf segment explants (sgm explants) were placed into Petri dishes (10 cm in diameter) containing 10 ml liquid MS medium without further wounding. Subsequently, 1 ml of Agrobacterium suspension was added into each Petri dish. The other halves of the leaves were microwounded by sharp scalpel tips (4–5 wounds per leaf) but were not sectioned as the first half. They were applied for transformation as intact, but wounded leaves (int explants). The infection was carried out during a 20 min gentle shaking in dim light at 22°C.
Method B was applied according to Hiei et al. (1994) with slight modifications: To propagate transforming Agrobacteria, 50 ml of liquid CPY-Infection medium (CPY-I medium, pH: 5.6) containing 1 g/l yeast extract, 5 g/l triptone, 5 g/l sucrose, 500 mg/l MgSO4·7H2O, 2.0 g/l MES, kanamycin (25 mg/l) and rifampicin (100 mg/l) was poured into 250 ml Erlenmeyer flasks. Bacterial cultures were kept in darkness at 28°C overnight on a shaker adjusted to 200 rpm. Healthy-white and dense bacterial cultures were pelleted by centrifugation (10 min, 8000 rpm). These cultures were then resuspended in 40 ml liquid MS-Infection medium (Borsics et al. 2002) supplemented with one-third strength MS macro- and microelements, 10 mg/l thiamine-HCl, 1.0 g/l casein hydrolysate, 10 g/l glucose and 50 μM acetosyringone (MSI medium, pH 5.0). About 40 ml of MSI medium containing the resuspended bacterial cells and the plant explants were added into sterile Petri dishes to carry out bacterial infection.
Sgm and int explants, transformed according to method A or method B, were dried shortly on sterile filter paper discs after the infection and were placed onto RA callus induction media (Tóth et al. 2004) for an additional 2 days of co-culture. Subsequently, explants were transferred onto selective callus induction medium (basal RA medium containing 0.2 mg/l BAP and 2.0 mg/l picloram) supplemented with kanamycin (1.0 mg/l) and cefotaxime (400 mg/l). Subculturing took place in every 2 weeks until fast growing, fresh green cell colonies appeared on the browning surface of dying leaf segments. After 4–6 weeks selection, leaf segment explants with surviving calli were transferred onto selective RA regeneration medium supplemented with 0.2 mg/l BAP, 1.0–2.0 mg/l NAA, as well as filter sterilized kanamycin (1.0 mg/l) and cefotaxime (350 mg/l). Putative transgenic plants were then transferred onto selective basal RA medium (contained 1.0 mg/l kanamycin) for micropropagation after the 4-week long regeneration process. Replicates of each independent primary transformant plus appropriate controls were grown in a containment glasshouse under conditions of 20°C day and 16°C night temperature, with supplementary light to maintain 16-h daylight period. Microplants were planted out in 9.0 cm pots containing a 1:1 mix of sand and perlite and were watered every second day with liquid medium that contained half-strength RA macro- and microelements (pH 6.0). For testing the fertility and for the analysis of progenies, stigmas were pollinated manually by using fine brushes.
PCR analysis
For PCR reactions, total DNA was extracted from the greenhouse-grown wild-type and putative transgenic plants using Qiagen Plant Mini Kit (Qiagen Gmbh, Hilden, Germany), plasmid DNA isolation from E. coli was performed using Qiagen Plasmid Mega Kit following the manufacturer's instructions. The PCR analysis was carried out in a Perkin Elmer Cetus thermocycler in 50 μl reaction mixtures containing commercial buffer, 2,5 mM MgCl2, 200 μM dATP, dTTP, dCTP, dGTP, 3 U Taq polymerase, DNA (100 ng total, or 10 pg plasmid), and oligonucleotide primers in 1 μM final concentration. To amplify an internal nptII fragment (781 bp) the following primer pair was used according to Beck et al. (1982):
Reactions were started with a denaturation at 94°C for 4 min, followed by 35 cycles with the following parameters: 94°C for 1 min, 55°C for 1 min and 72°C for 1.5 min. The program was terminated by an extension at 72°C for 7 min. Amplification products were analysed by electrophoresis in 1% agarose-ethidium bromide gels.
Southern hybridisations
Purified DNA (10 μg) was digested overnight with BamHI and HindIII, as these enzymes cut once within the T-DNA insert, and separated by electrophoresis on 0.8% agarose gels. DNA was blotted onto nitrocellulose (Hybond-C extra, Amersham) membranes. A 1.9 kb long gel-purified PstI fragment from the plasmid pFF19G (Timmermans et al. 1990), representing the gus coding sequence, was labelled by the random priming approach (Feinberg and Vogelstein 1983) using α[32P]-dCTP. Hybridization and autoradiography were carried out according to standard protocols (Sambrook et al. 1989) and instructions of the manufacturers.
Histochemical GUS analysis
Gus expression was detected by immersing intact leaves of putative transgenic and wild-type plants into the substrate (1 mM X-Glca: 5-bromo-4-chloro-3-indolyl-β-d-glucuronic acid cyclohexyl-ammonium) that was vacuum infiltrated into the samples. The incubation for enhancement of the specific enzyme reaction was performed as reported by Jefferson (1987) and Jefferson et al. (1987).
Statistical analysis
Students' t-tests were performed using MS Excel 2000 (Microsoft Corporation, Seattle, USA). Differences between results are described as being significant where P≤0.05, and not significant where P>0.05.
Results
Since R. myconi showed an extreme sensitivity to tissue culture manipulations, first the effects of antibiotics used either as suppressers of the Agrobacterium overgrowth or as selective agents were tested on its in vitro morphogenesis. As a preliminary experiment, CPY media containing a series of timentin (50, 100, 150, 200, 250 mg/l) and cefotaxime (300, 350, 400, 450, 500 mg/l) were inoculated with the Agrobacterium strain to select those concentrations that inhibit bacterial growth. It was found that selective media should contain at least 150 mg/l timentin or 350 mg/l cefotaxime for that purpose (data not shown). At the same time, besides effective elimination of Agrobacterium after transformation, timentin and cefotaxime had to fulfil another important criteria in terms of special requirements of R. myconi. Due to a duality of its in vitro responses, a rapid and dramatic reaction to negative effects and, on the contrary, an extended reaction to positive effects, those concentrations of timentin and cefotaxime were screened that can give rise to maximum rate of survival during the whole selection procedure. Since higher than 300 mg/l in timentin and 500 mg/l in cefotaxime result in severe tissue necrosis in R. myconi, a concentration range of 100–300 mg/l was applied in the case of the former, while another concentration range of 300–500 mg/l was applied in the case of the latter. Figure 1 shows the rate of survival of the non-transformed plant explants in the presence of timentin or cefotaxime calculated both after the first 2 weeks of the incubation (white columns) and after the second 2 weeks of incubation (grey columns). Explants were transferred once into fresh medium that contained the same components as the first media between the two 2-week-long incubation periods. As it can be seen in Fig. 1A, there were no such concentrations of timentin that would satisfy the dual criteria of the evaluation simultaneously. While in the presence of 100 mg/l timentin the survival rates of the explants were nearly 95% both after the second and fourth weeks of incubation. With concentrations over 150 mg/l, which are sufficient enough to protect plant explants against Agrobacterium, the survival rates dropped dramatically by 30–40%. At the same time, there was a narrow range of concentration in cefotaxime (350–400 mg/l) that provided proper protection against Agrobacterium during the 4-week-long incubation (Fig. 1B) and where the survival rates of plant explants were still high enough (90%). Therefore, 350–400 mg/l cefotaxime was applied to suppress the overgrowth of transforming Agrobacterium during the selection procedure.

The influence of inhibitors (A, timentin; B, cefotaxime) of the Agrobacterium overgrowth on the survival rates (%) of non-transformed R. myconi explants. Experiments were repeated three times by using 50 explants/treatment in each repetition
Besides suppressing the overgrowth of Agrobacterium, antibiotics that are often utilized for positive selection of transgenic cell lines and plants have determining importance during a genetic transformation procedure. In our experiments the nptII gene, encoding neomycin phosphotransferase, was chosen as selection marker that can inactivate aminoglycoside-type antibiotics by phosphorylation. The application of nptII had two reasons. First, it shows moderate tissue toxicity in sub-lethal concentrations, second, there are numerous aminoglycoside-type antibiotics are available with slightly different chemical structures that render different site effects on the morphogenesis of R. myconi. Thus, we had a possibility to choose from selective agents (kanamycin, G418, neomycin, paramomycin) that meet the following dual criteria. First, the favoured concentration range of the given antibiotics should allow as high rate of explant survival as it is possible during the whole transformation procedure. However, it should suppress, but not totally inhibit callus development or plant regeneration simultaneously in non-transformed plant explants. The first information that Table 1 reveals is that the effective concentrations of all the antibiotics examined were unusually low. In the case of kanamycin and G418, the callus development and plant regeneration were suppressed at 1.0 mg/l concentration that was still accompanied with satisfactory survival rates of plant explants (kanamycin: 75%, G418: 60%). In contrast, survival rates dropped below 50% when neomycin (40%) or paramomycin (49%) were applied in concentrations suppressing morphogenesis (1.0 mg/l). On this basis, 1.0 mg/l kanamycin was used for the selection of transgenic cell lines and plantlets in genetic transformation experiments.
All putative transgenic plants were subjected to PCR tests and Southern hybridisations in order to confirm the presence of transgenes in their nuclear genomes. Primers designed into the nptII gene gave bands, besides the positive control (C+), only in those transformed R. myconi lines (Fig. 2E lanes T1, T3, T6) that displayed morphologically normal phenotype on selective media (Fig. 2D). However, no homologous fragments from non-transformed plants (Wt) and from transformed regenerants possessing abnormal phenotype (Fig. 2D) could be amplified (Fig. 2E lanes T2, T4, T5).

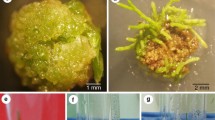
A Leaf segment explants (sgm explants) showed low rates of survival during the genetic transformation experiments. B Intact leaf explants (int explants), in which the starting phase of the callus development can be seen around the areas wounded by scalpel tip under selection pressure (yellow arrows), while conventional cutting resulted in severe tissue necrosis along the edges (black arrow). C Intact leaf explants possessed a satisfactory rate of survival in the presence of 1.0 mg/l kanamycin and 400 mg/l cefotaxime during the selection procedure. D Morphologically normal and abnormal plantlets were differentiated on int explants under selection pressure. E Amplification of a 781 bp long nptII-specific DNA fragment in morphologically normal (T1, T3, T6) and abnormal transformants (T2, T4, T5), and in wild-type Ramonda myconi plants (Wt). Purified binary vector p35SGUSINT served as positive control (C+), while the size marker (sm) was PstI digested λ phage DNA. F Identification of the integrated T-DNA and estimation of its copy number in the Ramonda genome by Southern blot analysis using the gus probe. The enclosed gel photograph demonstrates an experiment in which genomic DNA, extracted from putative transgenic (T1–T6) and wild-type plants (Wt), was digested with BamHI that made the estimation of the copy number of the integrated gus probe possible (see “Materials and methods” section). Abbreviations are same as in Figure 2E. For positive control, BamHI-digested p35SGUSINT vector was used. G Histochemical detection of the expression of the gusint reporter gene in a PCR and Southern positive transformant (T3). H Fertile and morphologically normal flowers of transgenic R. myconi plants grown in the glasshouse. Bars in each panel represent 1 cm
Southern hybridisations (Fig. 2F) confirmed the results of PCR analysis showing that the transformation cassette was integrated into the genome of morphologically normal transformants (lane T1, T3, T6) with copy number varying between one and three copies in the lines tested. At the same time, wild-type controls (Wt) and abnormal transformants did not posses the specific hybridisation signal (data not shown). Homologous fragments were also present in the positive controls, which were purified DNA from plasmids applied for transformation (C+).
The frequency of gene delivery and the proper translation of the bacterial T-DNA in the regenerated transgenic plants was estimated by histochemical localisation of the plant-specific GUSINT activity (Fig. 2G). The blue coloration of transgenic R. myconi plantlets could be visualized especially in leaf veins marking the actively growing areas (T3).
After providing evidence of successful genetic transformation, the frequency of the recovery in transgenic plants was evaluated in terms of different explant types and transformation protocols used. The impacts of physical (microwounding) and biochemical treatments (see the recipe of MSI medium: application of low pH, liquid medium supplemented with acetosyringone, aldose-type sugar source and organic nitrogen during the bacterial co-culture) that are thought to enhance the bacterial penetration into target tissues were monitored during the transformation procedure (Table 2). Two types of plant explants were applied as targets of transformation. Sgm explants were prepared in a conventional way excising 5–7 mm × 5–7 mm leaf segments by cutting off the edges of leaf blades. At the same time, int explants were detached intact leaves where the freshly wounded infection channels were provided by gentle punching of leaf blades with a new, sharp scalpel tip (four to five holes per leaf). Surprisingly, transgenic plants could be recovered on int explants only, proving that the physical enhancement (microwounding) of bacterial penetration was essential for the successful genetic transformation of R. myconi (Fig. 2B). An additional two-fold increase in the transformation frequency was obtained when the above physical and biochemical treatments were applied in combination. However, it appears that the survival rates of sgm explants were too low to produce transgenic plants independently of the transformation protocol applied (Fig. 2A).
Besides microwounding, the non-lethal selection strategy also had a pivotal role in producing transgenic R. myconi. Application of kanamycin at sub-lethal concentration not only made high survival rate of int explants possible (Fig. 2C) but it also allowed plant regeneration to occur even in non-transgenic explants. In this latter case, only the differentiation of morphologically abnormal plantlets could be obtained, possessing small, bleached leaves and poor root development (Fig. 2D). While these plantlets were non-transgenic according to PCR analyses, Southern hybridisations and GUS staining, the morphologically normal plantlets possessing healthy green, fully expanded leaves all carried the transgenes (Fig. 2 EFG, Table 2). After growing the R 0 generation of transgenic plants, self-pollinated R 1 and different R 0 × wild type crosses were analysed by germinating seeds on growth regulator-free RA medium containing 1.0 mg/l kanamycin. Progenies from selfed primary transformants (R 0) in all cases showed a typical 3:1 (KmR:KmS) Mendelian phenotypic segregation when seeds were germinated in the presence of kanamycin (data not shown). In a set of experiments, 21 plantlets from a transgenic × wild type cross were also analysed. The progeny showed a 11:10 KmR:KmS segregation, also indicating a monogenic dominant fashion for the transmission of the nptII gene. All R 0 and R 1 transgenic plants developed fertile flowers (Fig. 2H), and no morphological abnormalities were observed on the whole plant level.
Discussion
Ramonda species are shade-adapted resurrection plants and are considered homoiochlorophyllous as well, since they preserve more than 80% of the chlorophyll content during dehydration (Markovska et al. 1994). For this reason, these resurrection plants becoming more and more important as experimental models in studying photosynthetic and metabolic processes resulting in an elevated tolerance to desiccation (Augusti et al. 2001; Müller et al. 1997; Quartacci et al. 2002). Exploring the unique physiology of Ramonda species will enable these plants to turn into subjects of targeted gene isolations (Pico et al. 2002) in a similar manner to C. plantagineum that has become the most extensively studied resurrection plant to date (Ingram and Bartels 1996; Norwood et al. 2003; Scott 2000). It is not an exaggeration to predict that functional analysis of these genes will inevitably follow the isolation work in the near future (Cushman and Bohnert 2000). This requires the development of a method for genetic transformation that allows the application of sense/antisense to unravel the physiological role of proteins encoded by the genes of interest (Birch 1997). Previously, we described methods both for in vitro micropropagation and plant regeneration of R. myconi (Tóth et al. 2004) that meant an applicable basis for its Agrobacterium-mediated genetic transformation. In a similar manner to other resurrection plants, R. myconi also possessed an extreme sensitivity during the establishment of its plant regeneration and genetic transformation system. Craterostigma plantagineum (Furini et al. 1994; Toldi et al. 2002), R. myconi (Tóth et al. 2004) and H. rhodopensis (Djilianov et al. 2005) have all been successfully tissue cultured. These tissues all displayed frequent evidence of tissue necrosis, development of hyperhydrated leaves and secretion of polyphenols into culture media under suboptimal conditions. In addition, R. myconi and H. rhodopensis displayed a sort of duality in their in vitro responses probably as a result of slow metabolism and growth. Rapid and dramatic reactions to negative effects were paralleled with extended and moderate reaction to positive effects (Djilianov et al. 2005; Tóth et al. 2004). This made the genetic transformation of R. myconi difficult, because most steps in a transformation procedure generate various types of physiological stresses that decrease survival rates of both transgenic and non-transgenic tissues. Indeed, the low survival rates of those plant explants that were made ready for genetic transformation by cutting off all edges of leaf blades (sgm explants) impeded the recovery of transgenic plants in our experiments (Fig. 2A, Table 2). At the same time, detached leaves where the Agrobacterial penetration was promoted by microwounding (int explants) survived the transformation procedure at a satisfactory frequency (Fig. 2C), which, in turn, allowed an efficient production of trangenic R. myconi plants (Fig. 2E–G, Table 2). In this latter case, we speculated that the proportion of wounded and non-wounded areas within an sgm or int explant was the most important factor in determining survival rates. While large wounded surfaces rendered a strong exposure to stresses in the case of sgm explants, large non-wounded surfaces might enhance the recovery of minor wounds in int explants after the bacterial infection (Fig. 2B).
Other important component of our ‘low stress’ transformation was the development of a non-lethal selection strategy for R. myconi. Most of the toxic drugs that are used as inhibitors of bacterial growth or as selective agents have side effects that can decrease or prevent plant regeneration not only in non-transgenic explants but also in transgenic ones (for review see Oreifig et al. 2004). This fact meant an additional risk knowing the in vitro behaviour of R. myconi. Therefore, concentrations of both Agrobacterium eliminating and selective agents were optimised with the intention to accomplish the essential preconditions of a non-lethal selection strategy. That is, on the one hand, survival rates of plant explants have to be as high as possible in the presence of antibacterial drugs to make successful genetic transformation possible. On the other hand, the optimal concentration of the selective agent has to suppress, but not totally inhibit morphogenesis on non-transformed explants to allow a simple visual pre-selection of transgenic regenerants. In other words, we were in search of such sub-lethal concentrations of the applied selective agent, in which the main distinctive features of transgenic plants might be the green colour, morphologically normal phenotype and a more intense growth, while non-transgenic regenerants are likely to be bleached showing morphological abnormalities and retarded growth. As it can be seen in Fig. 2D the moderate tissue toxicity of kanamycin applied at sub-lethal concentration (1.0 mg/l) made the differentiation of two types of regenerants possible in its presence. Examining all the abnormal regenerants no transgenic one was found among them, while normal regenerants carried the transgenes with no exceptions (Fig. 2E–G, Table 2).
In contrast to C. plantagineum where the physical enhancements of bacterial penetration had negative rather than positive effects (Toldi et al. 2002), such treatment (microwounding) had primary importance in the success of genetic transformation of R. myconi (Fig. 2B). An additional two-fold increase in the transformation frequency was obtained when the above physical and biochemical treatments (in vitro preinduction of vir genes) were applied in combination (Table 2). These modifications together with the appropriate tissue culture system resulted in a reliable method for genetic transformation of the physiological model plant Ramonda myconi.
Abbreviations
- GM medium:
-
Germination medium (Toldi et al. 1994)
- RA medium:
-
Ramonda medium (Tóth et al. 2004)
- MSI medium:
-
Modified MS medium for bacterial infection (Borsics et al. 2002)
- CPY medium:
-
Culture medium for Agrobacterium
- BAP:
-
6-Benzyladenine
- NAA:
-
1-Naphtaleneacetic acid
- sgm explants:
-
Leaf segment explants
- int explants:
-
Intact, but microwounded explants
References
Augusti A, Scartazza A, Navari-Izzo F, Sgherri CLM, Stevanovic B, Brugnoli E (2001) Photosystem II photochemical efficiency, zeaxanthin and antioxidant contents in the poikilohydric Ramonda serbica during dehydration and rehydration. Photosynthesis Res 67:79–88
Beck E, Ludwig G, Auerswald EA, Reiss B, Schaller H (1982) Nucleotide sequence and exact localization of the neomycin phosphotransferase gene from transposon Tn5. Gene 19:327–336
Birch RG (1997) Plant transformation: problems and strategies for practical application. Ann Rev Plant Phys Plant Mol Biol 48:297–326
Borsics T, Mihálka V, Oreifig AS, Bárány I, Lados M, Nagy I, Jenes B, Toldi O (2002) Methods for genetic transformation of the parasitic weed dodder (Cuscuta trifolii Bab. et Gibs) and for PCR-based detection of early transformation events. Plant Sci 162:193–199
Boyer JS (1982) Plant productivity and environment. Science 218:443–448
Chilton MD, Currier TC, Farrand SK, Bendich AJ, Gordon MP, Nester EW (1974) Agrobacterium tumefaciens DNA and P58 bacteriophage DNA not detected in crown gall tumors. Proc Natl Acad Sci USA 71:3672–3676
Cushman JC, Bohnert HJ (2000) Genomic approaches to plant stress tolerance. Curr Opin Plant Biol 3:117–124
Deblaere R, Bytebier B, De Greve H, Deboeck F, Schell J, Van Montagu M, Leemans J (1985) Efficient octopine Ti plasmid-derived vectors for Agrobacterium-mediated gene transfer to plants. Nucleic Acids Res 13:4777–4788
Djilianov D, Genova G, Parvanova D, Zapryanova N, Konstantinova T, Atanasov A (2005) In vitro culture of the resurrection plant Haberlea rhodopensis. Plant Cell Tissue Organ Culture 80:115–118
Feinberg AP, Vogelstein B (1983) A technique for radiolabelling DNA restriction endonuclease fragments to high specific activity. Anal Biochem 132:6–13
Flowers TJ, Yeo AR (1995) Breeding for salinity resistance in crop plants: where next? Aust J Plant Physiol 22:875–884
Furini A, Koncz C, Salamini F, Bartels D (1994) Agrobacterium-mediated transformation of the desiccation-tolerant plant Craterostigma plantagineum. Plant Cell Rep 14:102–106
Hiei Y, Ohta S, Komari T, Kumashiro T (1994) Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J 6:271–282
Hiei Y, Komari T, Kubo T (1997) Transformation of rice mediated by Agrobacterium tumefaciens. Plant Mol Biol 35:205–218
Horsch R, Fry J, Hoffmann N, Neidermeyer J, Rogers S, Fraley R (1988) Plant Mol Biol Manual A 5:1–9
Ingram J, Bartels D (1996) The molecular basis of dehydration tolerance in plants. Ann Rev Plant Physiol Plant Mol Biol 47:377–403
Jefferson RA (1987) Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol Biol Rep 5:387–405
Jefferson RA, Kavanagh TA, Bevan M (1987) GUS fusions: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Khush GS (1999) Green revolution: preparing for the 21st century. Genome 42:646–655
Markovska YK, Tsonev TD, Kimenov GP, Tutekova AA (1994) Physiological changes in higher poikilohydric plants — Haberlea rhodopensis Friv. and Ramonda serbica Panc. during drought and rewatering at different light regimes. J Plant Physiol 144:100–108
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Müller J, Sprenger N, Bortlik K, Boller T, Wiemken A (1997) Desiccation increases sucrose levels in Ramonda and Haberlea, two genera of resurrection plants in the Gesneriaceae. Physiol Plant 100:153–158
Norwood M, Toldi O, Richter A, Scott P (2003) Investigation into the ability of roots of the poikilohydric plant Craterostigma plantagineum to survive dehydration stress. J Exp Bot 54:2313–2321
Oreifig AS, Kovács G, Jenes B, Kiss E, Scott P, Toldi O (2004) Development of a non-lethal selection system by using aadA marker gene for efficient recovery of transgenic rice (Oryza sativa L.). Plant Cell Rep 22:490–496
Pico FX, Möller M, Ouborg NJ, Cronk QCB (2002) Single nucleotide polymorphisms in the coding region of the developmental gene Gcyc in natural populations of the relict Ramonda myconi (Gesneriaceae). Plant Biol 4:625–629
Quartacci MF, Glisic O, Stevanovic B, Navari-Izzo F (2002) Plasma membrane lipids in the resurrection plant Ramonda serbica following dehydration and rehydration. J Exp Bot 53:2159–2166
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press.
Scott P (2000) Resurrection plants and the secrets of eternal leaf. Ann Bot 85:159–166
Timmermans MCP, Maliga P, Viera J, Messing J (1990) The pFF plasmids: cassettes utilizing CaMV sequences for expression of foreign genes in plants. J Biotechnol 14:333–344
Toldi O, Gyulai G, Preininger É, Várallyay É, Fári M, Balázs E (1994) Minibeet initiation from derooted sugarbeet (Beta vulgaris L.) seedlings in vitro. Plant Sci 97:217–224
Toldi O, Tóth S, Ponyi T, Scott P (2002) An effective and reproducible transformation protocol for the model resurrection plant Craterostigma plantagineum Hochst. Plant Cell Rep 21:63–69
Tóth S, Scott P, Sorvari S, Toldi O (2004) Effective and reproducible protocols for in vitro culturing and plant regeneration of the physiological model plant Ramonda myconi (L.) Rchb. Plant Sci 166:1027–1034
Vancanneyt G, Schmidt R, O'Connor-Sanchez A, Willmitzer L, Rocha-Sosa M (1990) Construction of an intron-containing marker gene: splicing of the intron in transgenic plants and its use in monitoring early events in Agrobacterium-mediated plant transformation. Mol Gen Genet 220:245–250
Acknowledgements
This project was supported by an OECD Co-operative Research Programme entitled as “Biological Resource Management for Sustainable Agriculture Systems.” The authors would like to thank Gábor Takács for the excellent photographic work and Dr. Rainer Höfgen (MPI of Molecular Plant Physiology, Golm, Germany) for kindly providing p35SGUSINT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D. Dudits
Collaborator via a fellowship under the OECD Co-operative Research Programme: Biological Resource Management for Sustainable Agriculture Systems
Rights and permissions
About this article
Cite this article
Tóth, S., Kiss, C., Scott, P. et al. Agrobacterium-mediated genetic transformation of the desiccation tolerant resurrection plant Ramonda myconi (L.) Rchb.. Plant Cell Rep 25, 442–449 (2006). https://doi.org/10.1007/s00299-005-0083-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-005-0083-4