
Abstract
Circulating endothelial progenitor cells (CEPCs) play an important role in the process of atherosclerosis. Most previous studies on CEPC in systemic lupus erythematosus (SLE) patients were on their number and some functions and the results were not consistent. No studies on their anti-inflammatory function and integrated status were reported. The purpose of this study was to determine the number, function (including anti-inflammatory function), and the integrated status of CEPCs in active SLE patients. The study was performed in 35 active SLE patients (28 females, 7 males) and 35 age-and gender-matched healthy controls. CEPC number was determined by Fluorescence-Activated Cell Sorting. Proliferation capacity of CEPC was assessed by PCNA staining. Adhesion capacity of CEPC to fibronectin and adhesion capacity of THP1 cell to CEPC were determined by cell adhesion assay. Migratory capacity of CEPC was measured by transwell chamber assay and the potential to form tubes on Matrigel of CEPC was determined by in vivo tube formation on Matrigel test. The expression of inducible nitric oxide synthase (iNOS) and interleukin-6 (IL-6) assessed by quantitative PCR as well as the expression of intercellular adhesion molecule-1 (ICAM-1) and phosphorylated-Akt (p-Akt) assessed by western-blotting were used to evaluate the anti-inflammatory function and cell status of CEPCs. The number of CEPC in SLE patients was not different from that in control (p > 0.05). Proliferation capacity of CEPC was decreased in active SLE patients (p = 0.027). Adhesion capacity of CEPC to fibronectin was decreased (p = 0.04) in SLE patients and adhesion capacity of THP1 cell to CEPC was increased in SLE patients (p < 0.001). Migratory activity was reduced in patient CEPCs (p < 0.001). Capacity of CEPCs to form tube on Matrigel was decreased in SLE patients (p < 0.001). Expression of iNOS and IL-6 (p < 0.001, p = 0.006, respectively) and ICAM-1 were increased in CEPC of SLE patients and expression of p-Akt was decreased in CEPC of SLE patients. Our data show that CEPC number in active SLE patients was not significantly different from healthy controls, but their functions were partly impaired, including proliferation, adhesion, migration, and tube formation. Bad cell status and increased susceptibility to inflammatory process of CEPCs in active SLE were also observed in our study.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Systemic lupus erythematosus (SLE) is associated with increased risk of cardiovascular events leading to enhanced morbidity and mortality [1]. Although there was increased incidence of traditional cardiovascular risk factors, such as hypertension and hypercholesterolaemia in SLE patients, recent observations indicated that traditional risk factors failed to account for the full cardiovascular risk [2].
Endothelial progenitor cell (EPC) is originated from hematopoietic stem cells (HSCs) and it has many functions such as vasculogenesis (including adhesion and migration through endothelium, differentiation to vascular endothelial cell and tube formation) and anti-inflammatory effects. The abnormality of the number and function of EPC plays an important role in the process of atherosclerosis [3–5]. Previous studies [6–9] on the circulating EPC (CEPC) from SLE patients indicated that some functions such as adhesion and migration were partly impaired, yet there was no consistent result on the quantification of CEPC. No report was found about the anti-inflammatory function and the integrated status of CEPC from SLE patients, which are also very important to CEPC and atherosclerosis. The purpose of our study is to comparatively study the number and function as well as the anti-inflammatory capacity and integrated status of CEPC from active SLE patients for the first time.
Patients and method
Characteristics of patients and controls
CEPCs were quantified in 28 female and 7 male active SLE patients, fulfilling the ACR criteria [10] and in 35 healthy age and gender-matched healthy controls (HCs) (Table 1). None of the patients had a history of coronary artery disease, diabetes, or cardiac insufficiency, which could have influenced the EPC quantities [11, 12].
CEPC isolation, culture, and identification
A 20-mL sample of venous blood was collected with the use of heparin (20 U/mL) from donors with their written agreement. The protocol was approved by Peking University Institutional Human Sample Use Committee. Mononuclear cells were isolated by density-gradient centrifugation with Ficoll (1.077 g/mL) and plated on plates coated with fibronectin (50 μg/mL; Sigma). The culture media M199 (Gibico) was supplemented with 20% fetal bovine serum (FBS; Hyclone), human vascular endothelial growth factor (VEGF, 10 ng/mL; Sigma), human basic fibroblast growth factor (bFGF, 1 ng/mL; Sigma), human epidermal growth factor (EGF, 10 ng/mL; Sigma), insulin-like growth factor-II (IGF, 2 ng/mL; PetroTech), and leukemia inhibitory factor (LIF, 10 ng/mL; Chemicon).
After culture for 2–3 weeks, clusters of EPC colonies grew at a high proliferative rate into a monolayer with a “cobblestone-like” morphology. This cell population appeared to be homogenous and maintained a similar morphology for several passages when cultured with growth factors and LIF. The population exhibited high levels of CD34 and VEGF receptor-2 (VEGFR-2; KDR) and had high capacity for taking up Dil-labeled acetylated LDL and binding to Ulex europaeus agglutinin-1 (UEA-1, Sigma).
Cells were incubated with 1, 1-dioctadecyl-3, 3, 3, 3-tetramethylindo- carbocyanine perchlorate-labeled acetylated LDL (DiI-acLDL; 10 μg/mL; Sigma) at 37°C for 4 h. After being fixed with 2% paraformaldehyde, the cells were incubated with FITC-labeled Ulex europeus agglutinin (UEA, 10 μg/mL; Sigma) for 1 h. The cells were also incubated with FITC conjugated primary antibodies against VEGFR-2 (KDR, Sigma). The results were obtained by Leica confocal fluorescence microscope.
Quantification of CEPC
Fluorescence-Activated Cell Sorting (FACS) analysis was used for quantification of CEPC. The percentage of cells expressing EPC markers was quantified by FACS assay. Briefly, the cells were trypsinized and resuspended in PBS staining buffer to obtain a concentration of 1 × 105 cells/mL, and then fixed with 2% paraformaldehyde. The markers of EPC detected were phycoerythrin (PE)-conjugated mouse anti-human CD34 (BD Pharmingen), and Dil-acLDL, FITC-conjugated UEA. Isotype-identical antibodies served as negative controls. 10,000 events per sample were analyzed with a Beckman Coulter EPICS XL flow cytometer with EXPO32 ADC software. The data were reported as the percentage of EPCs that expressed these markers.
Assessment of proliferation capacity of CEPC
Cells were washed in PBS and fixed with 4% Paraformaldehyde for 10 min at room temperature, then washed three times in PBS for 10 min and incubated directly with fluorescent conjugated-PCNA (BD Pharmingen, Buccinasco, Milano, Italy) antibody for 60 min at 4°C, washed, and stained with Hoechst 33342 (5μg/mL, Sigma). 3 random fields of each group were captured under a fluorescence microscope (Nikon Instruments) and proliferation capacity of CEPC was expressed as the percentage of PCNA-positive cells. The PCNA-positive cells were counted by three investigators blinded to the study.
CEPC adhesion assay
To assess cell adhesion to immobilized fibronectin, CEPCs were plated onto fibronectin-coated 24-well plates and cultured until confluent. The cells were trypsinizated and reseeded in fibronectin-coated wells at 5 × 105 cells per well. After incubation for 1 h at 37°C, the non-adherent cells were removed by washing three times with PBS. Then the rest of cells were labeled with Hoechst 33342. To quantify the number of adhesion cells, five random fields of each group were captured under a fluorescence microscope and the numbers of stained cells were counted, respectively.
To assess THP-1 cell adhesion to EPCs, the cell adhesion assay was performed as previously reported [13] with modification. CEPCs were grown to confluence on slides in a chamber (Lab-Tek) and incubated for 4 days. THP-1 cells were coincubated with the CEPCS for 30 min. The nonadhering cells were washed off, and the adhering THP-1 cells were observed and counted by use of a fluorescence microscope.
CEPC migration assay
The migration of CEPCs was performed in a Transwell Chamber (Corning Costar). Briefly, EPCs were gently detached, harvested, and resuspended in chemotaxis buffer (M199 supplemented with 20% FBS and 100 ng/mL VEGF). One hundred microliters of chemotaxis buffer containing 1 × 105 cells was added to the upper compartment, and 600 μL of chemotaxis buffer with 100 ng/mL VEGF was added to the lower compartment. After incubation at 37°C for 4 h, the filters were removed and the cells in the lower compartment were labeled with Hoechst 33258 and then counted under a fluorescence microscope. The migratory rate was expressed as the percentage of input cells migrating into the lower chamber. All groups of experiments were performed in triplicate.
In vitro tube formation assay
Matrigel (Becton–Dickinson) at 300 μL/well was added to a 24-well plate, which was overlaid with 2 × 105 cells and then incubated in M199 medium supplemented with 20% FBS. The formation of capillary tubes in Matrigel was examined by use of an inverted microscope equipped with a digital camera. The tube-like structures (those exceeding 6 cells in length) in five randomly selected fields of each well were counted by three investigators blinded to the study.
Quantitative reverse transcriptase polymerase chain reaction
Total RNA was isolated from CEPCs using TRIzol reagent (Invitrogen). The resulting cDNAs were used as templates for quantitative RT-PCR with the EVA Green fluorescent DNA stain (Biotium). 18S was used as an internal control. The nucleotide sequences of the primers were as follows: inducible nitric oxide synthase (iNOS): forward 5′- acaagcctacccctccagat-3′, reverse 5′-tcccgtcagttggtaggttc-3′; interleukin-6 (IL-6): forward 5′-tacccccaggagaagattcc-3′, reverse 5′-ttttctgccagtgcctcttt-3′.
Western blot analysis
Western blot analysis followed standard protocols. The primary antibodies were the polyclonal antibodies anti-intercellular adhesion molecule-1 (ICAM-1) and phosphorylated-Akt (p-Akt) (all 1:1,000; all Santa Cruz Biotechnology) and β-actin (Bioss, Beijing; 1:3,000).
Statistical analysis
The significance of variability was determined by Student t test for continuous variables and by χ 2 or Fisher’s exact test for nominal variables, as appropriate. Pearson’s correlation coefficient was used to test for relations between complement levels and levels of antibodies against double-stranded (ds) DNA and the experimental outcomes. All results are presented as mean ± SD from at least three independent experiments. A p < 0.05 was considered statistically significant.
Results
Results of CEPC identification
The results were illustrated in Fig. 1. After two passages, the EPCs were stained with Hochest and antibodies against VEGFR-2 and analyzed by confocal fluorescent microscopy. The presented data were representative of three separate experiments. EPCs within passages 2–6 after withdrawal of LIF were used in all studies.

Isolation and identification of CEPC. a Mononuclear cells adherence. b and c EPCs were stained with Hochest (blue) and antibodies against VEGFR-2 (KDR) (red) and analyzed by confocal fluorescent microscopy
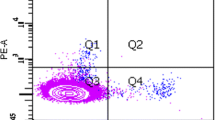
Circulating EPC levels are normal in SLE
EPC levels in SLE patients as analyzed by FACS were not significantly different in SLE when compared with that in HCs. Although the number of PBMNCs that stained positive for CD34, Dil-acLDL, and UEA was consistently lower in the SLE group, the difference did not reach statistical significance (p = 0.094, Fig. 2).

EPC levels in SLE patients as analyzed by FACS were not significantly different in SLE when compared with that in HCs
Impaired proliferation capacity of CEPC in SLE patients
The percentage of PCNA-positive cells in SLE group was significantly lower than that in control group (33.4 vs. 59.5%, p = 0.027, Fig. 3). The result indicated that proliferation capacity of CEPC was decreased in SLE patients.

Representative microscopic view of PCNA-positive cells (left) and Hoechst -positive cells (right) in control group (a) and SLE group (b). The percentage of PCNA-positive cells in SLE group was lower than that in control group (33.4 vs. 59.5%, p = 0.027)
Impaired adhesion of CEPC from SLE patients
Adhesion capacity of CEPC to fibronectin was significantly decreased (p = 0.04, Fig. 4) in SLE patients and adhesion capacity of THP-1 cell to CEPC increased significantly in SLE patients (p < 0.001, Fig. 5).

a Representative microscopic view of EPCs adhesion to fibronectin. b Adhesion capacity of CEPC to fibronectin was significantly decreased in SLE patients. (*p = 0.04)

Adhesion capacity of THP-1 cell to CEPC was increased significantly in SLE patients (***p < 0.001)
Migration capacity of CEPCs in SLE and HCs
Migratory activity was reduced in patient CEPCs (p < 0.001). The result was presented in Fig. 6.

Transwell chamber assay illustrated reduced capacity of migration in patient CEPCs (***p < 0.001)
Cluster formation on matrigel
Capacity of CEPCs to form tube on Matrigel was decreased in SLE patients (p < 0.001, Fig. 7).

a Representative microscopic view of tube formation (white arrow) in EPCs. b Invitro capillary tube formation assay indicated reduction of capacity to form tube in SLE CEPCs (***p < 0.001)
Expression of iNOS and IL-6
Expression of iNOS and IL-6 at mRNA level were significantly increased in CEPC of SLE patients (p < 0.001 and p = 0.006, respectively, Fig. 8).

Quantitative RT-PCR demonstrated increased expression of iNOS (a) (***p < 0.001) and IL-6 (b) (**p = 0.006) in CEPC from active SLE patients
Expression of ICAM-1 and phosphorylated-Akt
Expression of ICAM-1 at protein level was significantly increased in CEPC of SLE patients and phosphorylation of Akt was significantly decreased in CEPC of SLE patients (Fig. 9).

Phosphorylation of Akt detected by western blot was decreased in patients CEPC while expression of ICAM-1 determined by western blot was increased in SLE CEPC
Correlations between the degree of dysfunction in EPC with clinical SLE findings
To analyze the relation between clinical SLE findings and degree of dysfunction in CEPC (proliferation, adhesion capacity, migration and tube formation on matrigel, expression of iNOS, IL-6 and ICAM-1), we determined the correlation between complement C3 and C4 levels, levels of antibodies to dsDNA, organ involvement (renal involvement, Joint involvement and so on), and these experimental outcomes. No significant correlation was found between any of the clinical SLE findings and experimental outcomes (data not shown).
Discussion
Our study demonstrated the following: First, active SLE was associated with normal circulating EPC level. Second, the proliferation capacity, adhesion capacity, the migration behavior as well as the tube formation ability of EPC from SLE patients were significantly impaired. Third, expression of proinflammatory factors like IL-6 and ICAM-1 was increased significantly in CEPC from active SLE patients. Taking these results together with the decreased phosphorylation of Akt, increased expression of iNOS and increased adhesion of THP-1 cell to EPC, we could see, for the first time that CEPC from SLE patients is in a bad status with impaired function of anti-inflammation and predisposition to apoptosis.
The fact that our results showing normal circulating EPC levels were in accordance with those of J. Grisar et al. [6] yet contrasting some other observations [7, 8]. It requires further discussion.
First, the population studied here comprised totally Chinese, whereas the study of Lee et al. [8] involved African Americans, whites and others to nearly equal parts. In their study the ethnic distribution among controls was not matched to that among the patients. Yet there might be differences in EPC levels between various ethnic groups.
Second, the patients involved in our study were all at active stage while those involved in Peter E Westerweel’s study [7] were in clinical remission. As we all know, during active stage there are many immunocomplexes coming into being. The deposition of immunocomplex in the endothelium and the activation of complement will cause severe damage to the endothelial cell [14]. The increased impairment of endothelial cell needs more EPC to repair so the number of CEPC will be more than during quiescent stage.
Third, immunosuppressive therapy such as cyclophosphamide [15] and cyclosporin A [16] can induce the mobilization of endothelial progenitor cells. Intermediate doses of glucocorticoids for a short period of time also significantly increase CEPC [17]. All of our patients were treated with glucocorticoids and some of them were treated with cyclophosphamide at the same time. Maybe this is another factor contributing to the larger number of CEPC in our study than in those reported before [7, 8].
Last, chloroquin can induce apoptosis of human endothelial cells and depress cell proliferation, thus repressing angiogenesis [18]. The increased apoptosis of mature endothelial cell may require more CEPC to compensate. The majority of our patients were treated with hydroxychloroquin, so this may be another reason for the discrepancy with other studies.
Thus, we conclude that the levels of EPC, as characterized by CD34, Dil-acLDL, and UEA, are in the normal range in active SLE patients.
In our study, the proliferation capacity of CEPC, as demonstrated by PCNA staining, in SLE patients was decreased. This can partly explain the tendency of lower number of EPC in SLE patients than that in control, although the difference did not reach statistical significance (maybe because of the presence of the above four reasons).
Consistent with previous studies [3], the migration as well as the adhesion capacity of CEPC from SLE patients turned out to be significantly impaired compared with those of healthy controls. Besides, there were two more findings in our study: first, the tube formation capacity, which represents the ability of in vitro angiogenesis of CEPC, was decreased in CEPC from SLE patients; second, the expression of ICAM-1 was increased in patients CEPCs, which indicated that the capability of EPC to differentiate into endothelial cell was damaged.
Adhesion to the vascular wall, migration through the endothelium, differention into mature endothelial cell, and neuvascularization contribute importantly to vasculogenesis, one of the major functions of EPC [19]. Thus, the impaired function of EPC rather than their number may be a more important factor contributing to the increased cardiovascular risk associated with SLE, suggesting a relative EPC deficiency in tissues in need for EPC. So taken together, the impaired function of EPC, including adhesion, migration, differentiation into endothelial cell, and tube formation, could partly explain why the vascular repair was inadequate in SLE. In fact, the normal circulating EPC levels in SLE patients may even be a result of the impairment of the above functions.
Anti-inflammatory effect of EPCs, next to vascular repair, was another important feature of EPCs, which also played a role in the prevention of development or progression of atherosclerosis. From our data we could see that the expression of proinlfammatory cytokines, such as IL-6 and ICAM-1 was increased in EPC from SLE patients. Besides, adhesion capacity of THP-1 cell to CEPC from SLE patients was increased. Both of these results indicated that EPC from SLE was prone to induce inflammatory response. The serial of inflammatory response might contribute to the dysfunction of endothelium. It has been reported that the dysfunction of endothelium is the key point in both the development of vascular inflammation [20] and atherosclerosis [21]. So the impaired anti-inflammatory effect of EPCs from lupus patients might be involved in the development of both vasculitis and atherosclerosis in SLE. As we all know, SLE has been associated with vascular inflammation and altered angiogenesis [8, 22]. So, maybe further study on the role of EPC in the development of vasculitis and the pathogenesis of SLE is warranted.
For the first time, our data illustrated that expression of phosphorylated Akt decreased in CEPC from SLE patients. Akt is a serine/threonine protein kinase that is activated by a number of growth factors and cytokines in a phosphatidylinositol-3 kinase (PI3K)-dependent manner [16]. Akt serves as a multifunctional regulator of cell biology. Phosphorylated Akt becomes available to phosphorylate its downstream substrates, such as endothelial nitric oxide synthase (eNOS), leading to eNOS activation and nitric oxide (NO) release [23]. PI3K-Akt signaling axis plays an important role in many aspects of EPC functions, such as survival, migration, tube formation, proliferation, and NO production, so the reduced expression of phosphorylated Akt in EPC implied a bad status of EPC, like more disposed to apoptosis and impaired capability of proliferation. In fact, maybe the impaired functions of proliferation, adhesion, migration, and tube formation, were part of the results of the decrease of Akt phosphorylation.
Expression of iNOS was increased in EPC from lupus patients according to our data. An increased expression of iNOS in EPCs is usually correlated to pathological conditions which are characterized by elevated and dangerous levels of NO [24].
Taken together, the increased adhesion of THP-1 cell to CEPC, increased expression of proinflammatory cytokines and iNOS and decreased phosphorylation of Akt all implied a bad cell status of CEPC from active SLE patients.
In summary, our data revealed that CEPC was affected in active SLE despite their apparently normal levels by exhibiting a bad status of function. Many capacities, such as proliferation, adhesion, migration, differentiation to endothelial cell, tube formation, and anti-inflammatory function, were impaired in CEPC from SLE patients. This abnormality might constitute one of the several reasons underlying the increased cardiovascular risk in SLE, which, if confirmed in larger cohorts of patients, could allow for therapeutic interventions aimed at further improving their function, for example, with statins.
References
Bessant R, Hingorani A, Patel L, MacGregor A, Isenberg DA, Rahman A (2004) Risk of coronary heart disease and stroke in a large British cohort of patients with systemic lupus erythematosus. Rheumatology 43:924–929
Esdaile JM, Abrahamowicz M, Grodzicky T et al (2001) Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 44:2331–2337
Hristov M, Weber C (2009) Progenitor cell trafficking in the vascular wall. J Thromb Haemost 7(Suppl 1):31–34
Hristov M, Zernecke A, Schober A et al (2008) Adult progenitor cells in vascular remodeling during atherosclerosis. Biol Chem 389(7):837–844
Moreno PR, Sanz J, Fuster V (2009) Promoting mechanisms of vascular health: circulating progenitor cells, angiogenesis, and reverse cholesterol transport. J Am Coll Cardiol 53(25):2315–2323
Grisar J, Steiner CW, Bonelli M et al (2008) Systemic lupus erythematosus patients exhibit functional deficiencies of endothelial progenitor cells. Rheumatology 47:1476–1483
Westerweel PE, Luijten RK, Hoefer et al (2007) Haematopoietic and endothelial progenitor cells are deficient in quiescent systemic lupus erythematosus. Ann Rheum Dis 66:865–870
Lee PY, Li Y, Richards HB et al (2007) Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis Rheum 56:3759–3769
Robak E, Kierstan M, Cebula B et al (2009) Circulating endothelial cells and angiogenic proteins in patients with systemic lupus erythematosus. Lupus 18:332–341
Hochberg MC (1997) Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 40:1725
Urbich C, Dimmeler S (2005) Risk factors for coronary artery disease, circulating endothelial progenitor cells, and the role of HMG-CoA reductase inhibitors. Kidney Int 67:1672–1676
Tepper OM, Galiano RD, Capla JM et al (2002) Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 26:2781–2786
Wang N, Verna L, Liao H, Ballard A, Zhu Y, Stemerman MB (2001) Adenovirus-mediated overexpression of dominant-negative mutant of c-Jun prevents intercellular adhesion molecule-1 induction by LDL: a critical role for activator protein-1 in endothelial activation. Arterioscler Thromb Vasc Biol 21:1414–1420
Bruce IN (2005) Atherogenesis and autoimmune disease: the model of lupus. Lupus 14:687–690
Murakami J, Li TS, Ueda K, Tanaka T (2009) Inhibition of accelerated tumor growth by blocking the recruitment of mobilized endothelial progenitor cells after chemotherapy. Int J Cancer 124(7):1685–1692
Wang CH, Cherng WJ, Yang NI et al (2008) Cyclosporine increases ischemia-induced endothelial progenitor cell mobilization through manipulation of the CD26 system. Am J Physiol Regul Integr Comp Physiol 294(3):R811–R818
Grisar J, Aletaha D, Steiner CW et al (2007) Endothelial progenitor cells in active rheumatoid arthritis: effects of tumour necrosis factor and glucocorticoid therapy. Ann Rheum Dis 66(10):1284–1288
Potvin F, Petitclerc E, Marcau F, Poubelle PE (1997) Mechanisms of action of antimalarials in inflammation: induction of apoptosis in human endothelial cells. J Immunol 158:1872–1879
Asahara T, Murohara T, Sullivan A et al (1997) Isolation of putative progenitor endothelial cells for angiogenesis. Science 275:964–967
Hingorani AD, Cross J, Kharbanda RK et al (2000) Acute systemic inflammation impairs endothelium-dependent dilatation in humans. Circulation 102:994–999
Ross R (1999) Atherosclerosis: an inflammatory disease. N Engl J Med 340:115–126
Szekanecz Z, Koch AE (2007) Mechanism of disease: angiogenesis in inflammatory diseases. Nat Clin Pract Rheumatol 3:635–643
Shiojima I, Walsh K (2002) Role of Akt Signaling in vascular homeostasis and angiogenesis. Circ Res 90:1243–1250
Kleinert H, Pautz A, Linker K, Schwarz PM (2004) Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol 500:255–266
Acknowledgments
This paper is supported by the following funds: (1) Seed Foundation of Peking University Third Hospital; (2) Key Program Foundation of Peking University Third Hospital; (3) National Natural Science Foundation of China (30600209). Department of Rheumatology and Immunology, Peking University Third Hospital is the department to which the work should be attributed.
Conflict of interest statement
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Both Xiao Li Deng and Xiao Xia Li are the first authors of this paper.
Rights and permissions
About this article
Cite this article
Deng, X.L., Li, X.X., Liu, X.Y. et al. Comparative study on circulating endothelial progenitor cells in systemic lupus erythematosus patients at active stage. Rheumatol Int 30, 1429–1436 (2010). https://doi.org/10.1007/s00296-009-1156-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-009-1156-4