
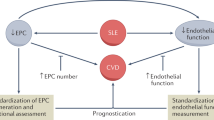
Abstract
Circulating endothelial progenitor cells (EPCs) were first described in 1997 by Asahara et al. as “putative endothelial cells” from human peripheral blood. The study of endothelial progenitors is also intensifying in several pathologies associated with endothelial damage, including diabetes, myocardial infarction, sepsis, pulmonary arterial hypertension, obstructive bronchopneumopathy and transplantation. EPCs have been studied in several autoimmune diseases with endothelial involvement such as systemic lupus erythematosus, thrombotic thrombocytopenic purpura, antineutrophil cytoplasmic antibodies, vasculitis, rheumatoid arthritis, Goujerot-Sjögren and antiphospholipid syndrome. Factors involved in endothelial damage are due to overexpression of pro-inflammatory cytokines and/or autoantibodies. Management of these pathologies, particularly the long-term use of glucocorticoids and methotrexate, promote atherosclerosis. A lack of standardized assessment of the number and function of EPCs represents a serious challenge for the use of EPCs as prognostic markers of cardiovascular diseases (CVD). The objective of this review was to describe EPCs, their properties and their involvement in several autoimmune diseases.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Blood vessels consist of three layers which are the tunica intima, the tunica media, and the tunica adventitia. The tunica intima forms a barrier between the vessel and blood flow. It is composed of a layer of endothelial cells and an internal elastic lamina. The vascular compartment is subject to various lesional processes of physiological and/or pathological origin, requiring reconstitution in order to maintain its integrity. Data have surfaced to show that reconstitution of endothelial cells (ECs) may prominently develop from locally residing progenitor cells [1].
The term endothelial progenitor cells (EPCs) refers to populations of cells that are capable of differentiating into mature endothelial cells and vasculogenesis. In physiological condition, a small fraction, of the order of 0.01% of medullary endothelial progenitors, enters the general circulation to allow endothelial regeneration [2]. In 1997, Asahara et al. reported the isolation of putative EPCs from human peripheral blood, based on the cell-surface expression of CD34 and other endothelial markers and introduced the novel concept of circulating EPCs [3]. In addition, the presence of CD133, a hematopoietic marker, appears to be a sign of immaturity [4]. Progressively, CD133 expresion is lost and EPCs express phenotypic endothelial markers like VE cadherin or von Willebrand factor (vWF). Several works confirm the presence of EPCs in the general circulation as well as in cord blood and fetal liver [5].
EPCs and Mobilization
Physiologically, ischemic models with tissue hypoxia induce mobilization of endothelial progenitors, initiating neovascularization. This tissue hypoxia induces an increase in the expression of the transcription factor hypoxia inducible factor 1α (HIF-1α), which is a proangiogenic factor. HIF-1α promotes the secretion of the vascular endothelial growth factor (VEGF) [6] as well as the stromal derived factor 1 (SDF-1) [7]. Administration of VEGF in a mouse model resulted in an increase in CD34+, VEGFR2+, VE cadherin + cells [8]. Several pro-inflammatory and immunosupressor cytokines like IL-6, IFNα, TNF-α, IL-1β, IL-8, TGF-β but equally hematopoietic growth factors (GM-CSF, EPO) or gonadotropins (FSH, LH) are involved in EPCs recruitment, mobilization and survival [9, 10] .
EPCs and Nomenclature
EPCs have been studied in various pathologies over the last two decades. However, the definition of EPCs has long lacked consensus, making comparison of results difficult [11]. This can be explained by the heterogeneity of the cell populations that can be isolated within EPCs. Two distinct methods are used to study EPCs (Fig. 1). First, a quantitative measurement of the number of EPCs is done by flow cytometry. As there is no specific marker for EPCs, it is essential to associate several expressed targets in order to selectively target the population of interest. Indeed, CD34+/VEGFR2 + can also identify mature endothelial cells detached from the vascular wall. A marker like CD133 + can be used, with a progressive appearance showing ECPs precocity and a progressive disappearance showing cell maturity. Second, functional aspects of EPCs (migration, adhesion and differentiation), are studied in cell culture models, after isolation by gradient density of mononuclear cells.

Characteristics, lineage and physiological roles of endothelial progenitor cells. MACs: myeloid angiogenic cells; ECFCs: endothelial colony forming cells
This distinction of endothelial progenitors is made according to the time of culture with early endothelial progenitors (Colony Forming Unit-Hill (CFU-Hill) and late endothelial progenitors (endothelial colony forming cells (ECFCs)) [12]. A distinction is made between “myeloid angiogenic cells” (MACs) and ECFCs. The phenotypic expression shows a lineage that is hematopoietic for MACs and endothelial for ECFCs [12]. The role of these two cell contingents in angiogenesis is different.
MACs derive from cultivated peripheral blood mononuclear cells. They specifically express surface cell markers CD45+, CD14 + CD31+, and CD146−, CD133−, and Tie2−. MACs are not able to differentiate into ECs. They promote angiogenesis by paracrine secretion of proangiogenic factors leading to proliferation, migration and tube formation. ECFCs derive from cultivated umbilical cord blood and peripheral blood mononuclear cells. They express CD31+, CD146+, CD105+, and CD45- and CD14- immunophenotype [12] (Fig. 1). ECFCs differentiate into ECs and are involved in vasculogenesis, angiogenesis and maintenance of vascular integrity [13]. The combination of these two cellular contingents amplifies mature EC formation. This distinction of cell line, names, phenotypes and functions is depicted in more detail by Medina et al. [12].
Methods
We performed a systematic review literature in PubMed using terms “endothelial progenitor cells” and ‘thrombotic thrombocytopenic purpura, Sjögren’s syndrome, antiphospholipid syndrome, systemic lupus erythematosus, ANCA vasculitis, rheumatoid arthritis or systemic sclerosis” covering the period of January 1st 1997 to December 31th 2022. We included studies with EPCs comparison in several autoimmune disorders. We only included articles written in English. Two investigators assessed the articles for eligibility and data extraction (GF and PB). We only evaluated EPC. We excluded articles based on titles and abstract in order to select eligible articles for full text review. Literature review, animal studies were excluded (Fig. 2).

Flowchart of articles selection
EPCs and Endothelial Dysfunction
During vascular endothelium damage, EPCs are released from the bone marrow to blood and promote vascular reendothelialization. Several studies reported correlation between EPC activity with endothelial damage and cardiovascular risk factors [14,15,16,17,18]. They observed a significant decrease in the number of EPC forming colonies in patients with elevated plasma cholesterol levels, hypertension and diabetes. There is an inverse correlation between the number of EPCs and the Framingham score used to determine cardiovascular risk. Their results suggest a greater cellular senescence of EPCs in patients with a high cardiovascular risk [19].
Several autoimmune diseases are associated with endothelial damage, including thrombotic thrombocytopenic purpura (TTP), rheumatoid arthritis, systemic lupus erythematosus, antiphospholipid syndrome, systemic scleroderma and anti-neutrophil cytoplasmic autoantibody (ANCA) vasculitis. Endothelial damage is multifactorial and is associated with premature atherosclerosis [20]. Factors involved in endothelial damage are due to overexpression of pro-inflammatory cytokines and/or autoantibodies. The management of these pathologies, particularly the long-term use of glucocorticoids and methotrexate, promote atherosclerosis. A part of endothelial dysfunction is a defect in vascular regeneration linked to an alteration in EPCs [11, 15]. This EPCs alteration, whether quantitative or functional, induces a decrease in vascular endothelium restoration and promotes a greater endothelial dysfunction.
EPCs in Thrombotic Thrombocytopenic Purpura
Thrombotic thrombocytopenic purpura is a specific form of thrombotic microangiopathies. The pathophysiology of TTP is a severe deficiency of the von Willebrand factor (vWF)–cleaving protease ADAMTS13 (a disintegrin and metallo-proteinase with a thrombospondin), that cleave ultralarge multimers of vWF. The outcome is the adhesion and aggregation of platelets to ultralarge von Willebrand multimers, capillaries, and arterioles, resulting in thrombosis and associated microvascular symptoms.
The very low concentrations of ADAMTS13 are not sufficient to cause TTP crisis. During acute idiopathic TTP, several triggers (pregnancy, infections, and drugs) induce endothelial activation and crisis. During crisis, biomarkers of endothelial injury were released, with increased P-selectin, vWF and circulating endothelial cells (CECs) which are mature endothelial cells in circulation.
In 2014, Widemann et al. investigated CECs and circulating EPCs (CD34+/KDR+) in 22 patients with autoimmune TTP (Tables 1 and 2) [21]. Seventeen patients of 22 received repeated rituximab infusions. Blood samples were collected before any treatment, at day 0 (D0) and at day 7 (D7) of hospitalization and at 3–6 months. Patients were considered in two groups: severe group (n = 5) death /neurological sequelae or less severe group (n = 17). Endothelial dysfunction markers correlated with TTP severity with increased CEC counts at D0 which significantly decreased at D7. In the severe group, CEC counts at D0 were five-fold higher than in the less severe group supporting a correlation between endothelial injury and disease severity. Associated with CECs, EPCs counts significantly increased at D0 and significantly decreased at three and six months. Unlike endothelial lesion markers, endothelial regeneration was not correlated with illness severity. Authors noted an inverse correlation between EPCs counts at the onset of the crisis and the number of plasma exchanges necessary to obtain remission. Indeed, patients with elevated EPCs required fewer plasma exchanges.
Circulating EPCs and markers of endothelial activation could be used to evaluate the response to treatment. Even if this study was based on a small cohort, several results were of interest, including different parameters as endothelial damage and endothelial repair. Therefore, this study highlights the importance of initial EPCs counts in the prognosis for plasma therapy.
EPCs and Sjögren’s Syndrome
Sjögren’s syndrome (SS) is a systemic autoimmune disease, characterized by lymphoid infiltration and affecting certain glands, particularly lacrimal and salivary saliva glands. Manifestations are dry syndrome and significant functional impairment. Visceral manifestations (lungs, kidneys, nerves), are less frequent but are more associated with anti-SSA/SSB autoantibodies [61]. There is no parallel between the severity of dry syndrome and the existence or severity of systemic manifestations. Sjögren’s syndrome may be primary or associated with another systemic disease (rheumatoid arthritis, systemic lupus erythematosus, inflammatory myopathies or scleroderma) [62]. During SS, neovessels contribute to inflammation. Patients with primary SS and without cardiovascular events described endothelial dysfunction [63,64,65,66]. Several actors are involved in endothelial damage like anti-SSA/SSB which promote atherosclerosis, endothelial cell detachment leading to anti-endothelial cell antibodies (AECAs) able to mediate cytoxicity and apoptosis [67].
In 2015, Bartoloni et al. investigated endothelial microvesicles (EMs) and EPCs to evaluate the balance between endothelial damage and endothelial repair in 34 patients with primary SS [22] (Table 1). Thristy-2% of patients were treated with hydroxychloroquine and/or low doses of corticosteroids (< 10 mg/day). They made a distinction between EPCs (CD34 + KDR+/CD133+) and late EPCs (CD34 + KDR + CD133-) in flow cytometry phenotype. An increased number of circulating EMs in SS patients was described. EPCs and late EPCs were significantly higher in primary SS compared to healthy controls. Moreover, there was an inverse correlation between circulating EPCs and disease duration from symptoms and diagnosis. EPCs and EPCs/late EPCs ratio were not correlated with clinical scores of disease activity (Eular SS Disease Activity Index (ESSDAI), SS Disease Damage Index SSDDI).
Alunno et al. investigated the involvement and cooperation of EPCs (CD34 + CD133 + VEGFR2+) with angiogenic T (Tang) cells (CD3 + CD31 + CXCR4+) in 36 patients with primary SS [23]. No correlation was observed between circulating EPCs and patient age, disease duration, autoantibody titers and serological status. EPCs were directly correlated with ESSDAI and Tang cells (Table 1).
In these two studies, the mean age was similar. The mean disease duration was higher in the Alunno study (11 years) compared to the Bartoloni study (5 years), with 53% and 32% of patients treated by hydroxychloroquine (HCQ), respectively.
Thus, contradictory results were obtained in these two studies regarding a correlation between EPCs, disease duration, and ESSDAI. Antibody profiles and titers did not be correlated with the decrease in EPCs. In primary SS, leading to endothelial damage, EPCs mobilization may preserve vascular integrity. Disease duration progressively decreased the reparative capacities of EPCs [22]. We were not able to determine if there was an impact of HCQ and/or corticosteroids on EPCs. HCQ appeared to improve endothelial dysfunction [68,69,70] limiting EPCs recruitment and angiogenesis.
EPCs and Antiphospholipid Syndrome
Antiphospholipid syndrome (APS) is a systemic autoimmune pathology defined by arterial and/or venous thrombotic manifestations, obstetrical complications, associated with the persistent presence of anti-phospholipid antibodies (aPLs) which are circulating lupus anticoagulants (LA), anticardiolipin antibodies (aCL), and anti-β2Glycoprotein I antibodies (aβ2GPI) [71]. These aPLs cause endothelial damage with cellular activation of endothelial cells, monocytes, neutrophils and platelets [72, 73]. Endothelial cells switch from an anticoagulant phenotype to a procoagulant phenotype through the expression of adhesion molecules (E-selectin, VCAM-1, ICAM-1), vWF secretion, tissue factor and pro-inflammatory cytokines (IL-1, IL-6, TNFα) [73, 74]. This cellular activation also generates pro-inflammatory and pro-coagulant microparticles [73]. This mechanism also activates the vascular endothelium and decreases fibrinolysis [73]. Preliminary studies have demonstrated endothelial dysfunction associated with arterial remodelling in patients with primary APS [75]. This alteration is secondary to an increase in production of TLR2 and TLR4. The consequence of this permanent endothelial dysfunction is hypercoagulability [76, 77], pro-inflammatory [78] and pro-aggressive/adhesive state [74, 79]. The pro-thrombotic state is described as a first trigger for vascular damage. The second trigger occurs when an inflammatory event (sepsis or trauma) occurs, inducing vascular thrombosis. In addition to hypercoagulability, APS is associated with increased markers of vascular remodeling leading to accelerated atherosclerosis [75].
In 2009, Gresele et al. studied endothelial function in 20 cases of primary antiphospholipid syndrome compared to 39 sex- and age-related healthy controls [24] (Table 1). They described no significant difference in endothelial dysfunction between primary APS and healthy controls evaluated by flow mediated dilation (FMD) of the brachial artery diameter, hyperemic blood flow and plasmatic markers with vWF and soluble P-selectin. In a subgroup, they compared endothelial damage and regeneration with CECs and circulating EPCs (CD34 + CD133 + VEGFR2+), respectively. No differences were observed for these two parameters. Authors concluded that APS was not a risk factor for endothelial damage. However, due to the small size of the sample, and the absence of endothelial damage recovered, these results need to be extended to a larger cohort. Green et al. found similar results with only a trend to decreased circulating EPCs (CD34 + CD133+) counts in 43 patients with primary APS compared to healthy controls, but not statistically different [25] (Table 1). They noted a significant reduction of EPCs to differentiate into endothelial cells in patients with primary APS. It appears that this alteration of the differentiation potential of EPCs was not induced by aPL. Type I IFN was overexpressed like in SLE. In vitro, IFN-I depletion restored EPCs differentiation into ECs. Treatment reducing specifically IFN-I like hydroxychloroquine, already used in SLE, is a promising track for the management of APS [70].
Thus, APS seem do not decreased EPCs number but reduced differentiation into ECs. Results required to be confirm on larger cohort.
EPCs and Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a systemic antuoimmune disorder with heterogeneous clinical manifestations. SLE increases cardiovascular risk which contributes to cardiovascular morbidity and mortality. The relative risk of coronary heart disease in SLE is increased 8–10 fold [80]. The main hypothesis is that systemic chronic inflammation, associated or not with immunosuppressive therapies, contributes to endothelial dysfunction with apoptosis of ECs and a higher remodelling of vascular wall. Lee et al., in 70 patients with SLE compared to 31 healthy controls, observed a significant decrease in the number of circulating EPCs (CD34 + VEGFR2+) as well as early EPCs [26] (Table 1). No correlation between early EPCs and autoantibody profile was described. MX1 gene expression coding for type I interferon (IFN-I), known to be involved in SLE pathophysiology, was associated with EPCs depletion. Thus IFN-I contributes to endothelial dysfunction by altering EPCs, leading to premature atherosclerosis by antiproliferative effect on EPCs. Similar results confirmed the depletion of circulating EPCs (CD34 + VEGFR2+) in 15 women with SLE in clinical remission for at least 1 year [27] (Table 1). However, EPCs autoantibody anti-DNA, antiphospholipid antibodies and complement factors were not associated with a decrease in EPCs. Moonen et al. supported these findings, showing a decreased number of circulating EPCs CD34+/CD133 + in 44 patients with SLE associated with impaired functionality and reduced migratory capacity [28] (Table 1). Ebner et al. summarized the results of several studies and reported an excessive apoptosis of EPCs associated with a decreased number and an impaired functionality of circulating EPCs with reduced differentiation into ECs, migratory capacity and proliferative rate [81].
Contradictory results were reported by Grisar et al. and Deng et al. in 31 and 35 patients with SLE, respectively. There were no differences in EPC numbers compared to healthy controls [29, 30] (Table 1). Grisar et al. described EPCs by flow cytometry (CD34 + CD133 + VEGFR2+) and early EPCs. EPCs from SLE presented impaired functionality with a lack of adhesion to HUVEC and migratory ability [29]. Deng et al. reported a significant reduction in proliferative capacity, adhesion to fibronectin, Matrigel tube formation assay and migratory capacity to VEGF associated with overexpression of ICAM, IL-6 and iNOS [30] (Table 1). Ablin et al. confirmed no difference in early EPCs and no correlation between EPCs depletion and SLE disease activity index (SLEDAI) score [31] (Table 1). Interestingly, EPCs increased adherence to fibronectin. Authors hypothesized that adhesion molecules on vascular endothelium were up-regulated and contributed to homing cell in vascular injury. Faced with these apparently contradictory results, authors suggested the involvement of the methodology used and the difference in treatment able to mobilize EPCs from bone marrow [30].
Baker et al. studied the variation in EPCs (CD34 + CD133 + VEGFR2+) in 70 patients with SLE who previously developed atherosclerosis and coronary artery calcification evaluated by carotid intima-media thickness and tomography [32] (Table 1). EPCs decreased even if there was no coronary artery calcification. Thus, EPCs depletion preceded the development of atherosclerosis and was correlated with SLEDAI. In 46 female SLE patients, pathological values of pulse wave velocity (PWV) led to decrease of circulating EPCs compared to normal PWV [82]. Decreased EPC counts were also associated with fibrinogen, high-sensitivity CRP, hypertension, tobacco use, impaired glucose metabolism, and metabolic syndrome. To complete these results, metabolic syndrome, which is highly prevalent in patients with systemic lupus erythematosus SLE, is closely linked with a decreased percentage of circulating EPCs (CD34 + KDR+/CD133+) and late EPCs (CD34 + KDR + CD133-) [83].
A decrease in the number of circulating EPCs (CD34 + CD133+) and in their differentiation into ECs was observed in 19 childhood-onset SLE [33]. This deleterious effect was blocked by an inhibitor of type I IFN. Type I IFN was significantly elevated in SLE but not correlated with vascular dysfunction [33] (Table 1). In contrast to adults, the decrease in circulating EPCs (CD34 + CD133+) was correlated with SLEDAI. Type I IFN and specifically IFNα genes were overexpressed. TLR 7 and TLR 9 were induced by IFNα synthesis [84]. IFNα promoted defective vasculogenesis and was responsible for pleiotropic effects with premature atherosclerosis [84]. A correlation of EPCs increase was observed in 12 patients with intravenous immunoglobulin treatment [34] (Tables 1 and 2). The main hypothesis was the clearance of AECA which was able to induce EPCs apoptosis [34]. Finally, frequently used therapies for SLE have demonstrated beneficial effects on EPCs suggesting pleiotropic effects [35] (Tables 1 and 2). A regimen of ramipril 10 mg/day for 12 weeks increased the number of early EPCs in 37 female patients with SLE. These results suggest that angiotensin-converting enzyme inhibitor allows an extra benefit beyond the hypotensive action [35]. Recently, vitamin D substitution demonstrated an improvement on vascular endothelial function. Huang et al. reported an increase in the number, migration and proliferation of EPCs (CD34 + KDR+) after culturing in different concentrations of 1,25-(OH)2 D3 [36] (Tables 1 and 2).
Thus, in SLE, a great heterogeneity of results were reported, mainly linked to the diversity of the identification protocol [85]. These results do not allow us to conclude that EPCs are reduced in SLE or a negative correlation between EPCs and the SLEDAI score. A positive effect of pharmacological treatments used in this disease seem to improve the number and proliferation of EPCs.
EPCs and ANCA Vasculitis
Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is a small vessel vasculitis of autoimmune origin with autoantibodies directed against proteinase 3 (PR3) or myeloperoxidase (MPO) contained in neutrophils [86]. Three entities are currently described: microscopic polyangiitis, granulomatosis with polyangiitis and eosinophilic granulomatosis with polyangiitis.
Physiologically, endothelium-neutrophil interactions are involved, with migration to inflammatory site. This relationship regulates neutrophil recruitment [87]. ANCA disrupts mechanisms of regulatory diapedesis. An overexpression of adhesion molecules ICAM-1, and VCAM-1 by endothelial cell associated with β1/β2 integrins by neutrophil was observed in ANCA patients [87]. After adhesion, neutrophil activation leads to the release of inflammatory cytokines, oxidants and enzymes that are toxic for the endothelium. The consequences are endothelial damage with apoptosis of ECs, and vessel wall detachment with CECs [87]. All these changes in the vascular endothelium induce a hypercoagulability state and patients with AAV have an increased risk to develop venous thromboembolism [88].
Endothelial injury is not the only process involved in the balance between endothelial injury/endothelial repair, with decreased endothelial regeneration. In 2005, Holmén et al. investigated EPCs (CD34 + CD133 + VEGFR2+) in 36 patients with granulomatosis polyangiitis. They described a significantly lower number of early EPCs in patients with active granulomatosis polyangiitis as compared with those in remission and healthy controls [37] (Table 1). Závada et al. found a signifiantly decreased number of early EPCs in 41 patients with active AAV compared to healthy controls [38] (Table 1). In contrast, no difference was observed in the number of circulating EPCs in untreated patients with active disease, in treated patients with active disease and in patients in remission. There was no correlation between early EPCs and Birmingham Vasculitis Activity Score (BVAS), ANCA titers or CRP. They observed a trend toward a higher number of early EPCs in patients with anti-MPO antibodies compared to anti-PR3. A complementary study revealed that a decrease in EPCs count was predictive of early relapse in 41 AAV patients but not for disease progression and organs involved [39] (Table 1). De Groot et al. revealed contradictory outcomes in 31 patients [40] (Tables 1 and 2). They measured BVAS, CECs and EPCs before and 1, 3 and 6 months after the introduction of immunosuppressants. BVAS significantly decreased after 1, 3 and 6 months of treatment and the number of CECs significantly decreased at 3 and 6 months of treatment compared to baseline, suggesting a reduction in endothelial damage with the therapy. The median number of circulating EPCs was similar in patients before treatment and healthy controls. Therefore, one month after immunosuppressive therapy, the number of circulating EPCs increased significantly and with disease remission suggesting that the change in EPCs count was secondary to therapy. Závada et al. showed a trend toward higher early EPCs if patients were not treated by immunosuppresive therapies [38]. Wilde et al. confirmed Závada’s results with a decrease in the number of circulating EPCs (CD34 + CD133 + KDR+) in 53 patients with AAV compared to healthy controls with no difference between active disease or disease in remission [41] (Table 1). The number of ECFCs (after 10 days of culture) was decreased in active disease or disease in remission unlike early EPCs. ECFC impairment was associated with disease relapse. A lack of EPCs functionality (potential to form colonies, clusters) was reported [89] supporting the impairment of the regenerative capacities of EPCs in AAV. Impairment of migration and angiogenic capacity of ECFCs (after 40 days of culture) was confirmed in 13 patients with PR3-positive compared to controls [90].
All these results show that the number of bone marrow-derived circulating EPCs was significantly lower in patients with AAV than in healthy subjects. This decrease in EPCs number, in particular ECFCs, and function illustrate the lack of endothelial repair probably responsible for relapse, morbidity and mortality. Moreover, EPCs decreased is able to predict relapse. As previously reported, pharmacological treatments reported positive effect to restore EPCs count. The apparent contradiction between studies was more attributable to inclusion criteria, the therapeutics used, disease duration and the cardiovascular risks associated.
EPCs and Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a chronic inflammatory disease with a prevalence of about 1% in most parts of the world. The pathophysiology is based on chronic inflammation of synovial joints associated with synovial cell proliferation and infiltration by blood-derived cells like T cells and macrophages. The disease is associated with endothelial dysfunction which contributes to atherosclerosis. Patients with RA presented a higher incidence of cardiovascular disease [91]. RA is an independent factor of traditional cardiovascular risk like hypertension, smoking, and dyslipidemia. Clinically, RA is evaluated by Disease Activity Score 28 (DAS28).
Grisar et al. studied circulating EPCs (CD34+/CD133+/VEGFR2+) in 52 patients with RA compared to 16 healthy controls [42] (Table 1). A subgroup analysis was performed in RA patient, between active disease (DAS ≥ 3.2) and inactive disease. Active RA was associated with EPCs depletion while inactive RA presented similar results to healthy controls. The EPCs level was inversely correlated with disease activity assessed by DAS28. EPCs depletion is due to the overexpression of TNF-α. Results were confirmed in another study, including 13 patients, in which EPCs number and function were reduced in RA [43] (Table 1). EPCs in RA patients presented defective migratory activity to VEGF, attenuated ability to adhere to mature ECs and components of extracellular matrix. A closed correlation between IL-6 and EPCs count was reported. Therefore, in patients with RA and severe endothelial dysfunction, the ability to form early EPCs was significantly impaired and the disease duration was long, 9.2 years [92, 93]. Yiu et al., demonstrated that RA patients with coronary atherosclerosis have significantly lower levels of CD133/KDR + EPCs than those without [94].
Anti-TNFα treatment was evaluated in circulating EPCs. A single dose of infliximab led to an increase of 33.4% in EPCs (CD34 + KDR+) levels 14 days later in 14 patients with RA [44] (Tables 1 and 2). Moreover, authors observed an increase of 37.6% in the mean number of adhering EPCs and an increase of 60% incellular differentiation into ECs, suggesting an improvement in EPCs functionality. Simultaneously, they noted a decrease of 17.5% in DAS28 and a significant correlation between DAS28 and early EPCs, evaluated by in vitro culture. These finding are based on a short exposure to infliximab. Spinelli et al. studied the effect of etanercept and adalimumab on circulating EPCs (CD34 + KDR+) at three months in 17 patients [45] (Tables 1 and 2). A negative correlation was observed between the mean increase in EPCs count and the mean decrease in DAS28 after three months. The suppression of the inflammatory process might positively affect the endothelial function. Glucocorticoids used to treat 29 patients with active RA led to a decrease in TNF-α and IL-6 levels associated with a normalization of EPCs count and a decrease in DAS28 [46] (Tables 1 and 2). Similar results were described with a trend toward an increase in circulating EPCs (CD34 + CD133 + VEGFR2+) under anti-TNF-α and glucocorticoids [95]. TNF-α was increased in patients with early RA which was associated with DAS28. Authors suggested that a higher IFNα level was associated with endothelial repair failure [95]. The IFNα level was associated with higher disease activity (DAS28), presence of autoantibodies, higher levels of IL-1β, IL-6, IL-10 and MIP-1α, lower levels of TGF-β, and increased late EPCs (CD34 + CD133-VEGFR2+) / EPCs (CD34 + CD133 + VEGFR2+) ratio suggesting endothelial damage and defective repair [96]. Similarly, a three-month course of methotrexate restored circulating EPCs (CD34 + VEGFR2+) levels similar to those of the controls [97]. Conversely, three months treatment with fenofibrate did not significantly improve circulating EPCs in 15 RA patients [98].
More recently, in addition to endothelial dysfunction and premature atherosclerosis, EPCs depletion was associated with bone erosion. CXCL12, a marker of bone erosion, was inversely correlated with EPCs (CD34 + VEGFR2+) count in 126 patients [47] (Table 1). CXCL12 is a chemokine cognate CXCR4 receptor which promotes the migration of progenitor and inflammatory cells. CXCL12 is produced in RA by the synovium which facilitates the migration and sequestration into inflamed joints leading to a reduced number of circulating EPCs. The recovery of EPCs after 24 weeks of anti-TNFα therapy improved endothelial function. CXCL12 did not change with treatment. TNF-α promotes CXCR4 receptor expression which facilitates CXCL12 chemoattractant activity. Thus, anti-TNF-α allows to decrease CXCR4 expression and to limit EPCs retention in joints.
Similarly, EPCs have been proposed as a biomarker of RA with interstitial lung disease. Authors reported a significant increase in EPCs (CD34 + CD133 + VEGFR2+) number in 20 patients with RA and interstitial lung disease compared to healthy controls and to patients with RA and no interstitial lung disease [48] (Table 1). These results suggest that EPCs are recruited at the sites of vascular damage to exert a compensatory mechanism due to endothelial damage. Moreover, authors reported an association between depletion of circulating EPCs and decrease of Tang cells [99]. This decrease in Tang cell-abrogated EPCs functionality was correlated with disease activity (DAS28) and inversely with autoantibody positivity [99]. Tang cells collaborated with EPCs for angiogenesis, representing one part of the pathophysiology of endothelial dysfunction in RA.
EPCs number is decreased during PR more particularly during active disease and is inversely correlated with DAS28. Defective migratory activity is associated with EPCs depletion. The mechanism is probably multifactorial, mediated on the one hand by pro-inflammatory cytokines like IFN-I and on the other by joint sequestration linked to bone erosion. Immunosuppressive treatments, such as anti-TNF or glucocorticoids, improve the number and function of EPCs.
EPCs and Systemic Sclerosis
Systemic sclerosis (SSc) is a chronic autoimmune rheumatic disorder characterized by fibrosis and vascular obliteration of the skin and other organs, particularly the lungs, heart and digestive tract. There are two main forms of SSc: limited cutaneous form (lSSc) and diffuse cutaneous form (dSSc). The etiology is unknown but probably multifactorial. Pathogenesis is based on immunologic abnormalities with the presence of typical anti-nuclear autoantibodies, chronic inflammation, microangiopathy, excessive deposition of collagen, ischemic tissue and endothelial damage.The extensive fibrotic changes are responsable for morbidity with digital ulcer, Raynaud’s phenomenon (RP), chronic pain syndromes, pulmonary arterial hypertension in 15% of patients, lung fibrosis in 80% of cases, gastrointestinal complications, renal failure due to thrombotic microangiopathy and cardiac disease, which is probably underestimated [100, 101]. Recent data suggest an impairment of vasculogenesis [49], neovascularization, angiogenesis and vascular wall remodeling [102].
Contradictory results were reported during the past two decades in EPCs studies. Kuwana et al. were the first to describe a decrease in the number of circulating EPCs (CD34+/CD133+/VEGFR2+) and a lower ability to differentiate into endothelial cells in 11 patients with SSc, associated with higher angiogenic factors [49] (Table 1).
Avouac et al. reported in 100 SSc patients that high placental growth factor and low EPCs (CD34+/CD133+/VEGFR2+) count predict new digital ulcers [103]. Similar results in 60 SSc patients reported that decreased EPCs (CD34+/CD133+/VEGFR2+) count and increased VEGF were associated with the late nailfold videocapillaroscopy [104].
In 2006, Del Papa et al. reported a significant increase of circulating EPCs (CD133+) in 62 patients at early stage disease compared to healthy controls but not in late stage disease [50] (Table 1). They reported a close negative correlation between EPC level and disease duration. Indeed, when SSc patients were stratified by disease duration (under 5 years and 3 years for lSSc and dSSc, respectively), they observed that patients with recent disease had a significantly higher level of circulating EPCs than patients with chronic disease. These authors found no difference in EPC rate between lSSc or dSSc and no correlation with clinical or disease activity score.
Avouac et al. also described a higher level of circulating EPCs in 50 patients with SSc, with a mean disease duration of 9 years compared to healthy controls, with no difference in the number of late EPCs [51] (Table 1). EPCs count was inversely correlated with digital ulcers. Yamaguchi et al. reported higher MACs (CD34 + VEGFR1 + CD14+) level in 23 SSc patients that exert enhanced angiogenesis but are impaired in vasculogenesis [105]. Benyamine et al. described in 45 SSc patients compared to 41 controls EPCs (CD34+/CD45−). The increase of EPCs was correlated with serum fractalkine level and associated with disease severity [106].
Allanore et al. described a decrease in EPCs count in 32 patients with chronicity of disease explaining the results of the Kuwana study in which patients had a mean disease duration of 10 years and the Del Papa study in early stage disease [52] (Table 1). Another study reported similar results to Kuwana et al. with a significantly lower number of EPCs (CD133+/VEGFR2+) in SSc patients associated with endothelial dysfunction evaluated with FMD [107]. Patients with SSc with no concomitant cardiovascular risk factors also had a lower number of EPCs. Andrigueti et al. supported these findings with a decrease in EPCs in early-stage disease associated with endothelial dysfunction. Moreover, EPCs level was positively correlated with the mean number of RP, but not with RP duration, disease duration, autoantibody profiles and modified Rodnan Skin Score (RSS) in 39 patients [53] (Table 1). The number of early EPCs was lower in patients with SSc than in healthy controls.
Zhu et al. described lower circulating early EPCs (CD34 + CD133+, CD34 + VEGFR2 + or CD34 + CD133 + VEGFR2+) counts and CFU-Hill in 54 SSc patients than healthy subjects with no difference between patients with lSSc and dSSc or between early and intermediate/late disease duration [54] (Table 1). An in vitro culture of EPCs, isolated from bone marrow or cord blood, with sera of SSc patients exhibited increased apoptosis. The depletion of the IgG fraction of SSc sera abolished this effect. EPCs can be altered after their mobilization from bone marrow by autoantibodies. Apoptosis was induced by the Akt-FOXO3a pathway [54]. In 2010, Del Papa et al. showed the presence of bone marrow EPCs dysfunction in 28 patients with SSc, without hematopoiesis dysfunction, correlated with significant titers of AECA [55] (Table 1). Thus, autoantibodies may be involved very early in the pathophysiology, functionally impairing EPCs and leading to the failure of endothelial restoration. The loss of functional ability was partially illustrated by the fact that early EPCs progressively acquired a mesenchymal phenotype in SSc, due to overexpression of TGF-β, which led to vascular dysfunction [108].
EPCs are involved in the pathophysiology of vasculopathy and lung fibrosis in SSc [56]. Mobilization of EPCs during idiopathic pulmonary fibrosis has been previously described with a hypercoagulable state [109]. Indeed, 21 patients with SSc and interstitial lung disease had a higher number of EPCs (CD34 + CD133 + VEGFR2+) than healthy controls or patients with SSc and no interstitial lung disease. Like Del Papa et al.,in 2006, a negative correlation between disease duration and EPCs number was found [56] (Table 1). Administration in intra-venous of cyclophosphamide at two weeks to treat intersitital lung disease in SSc increased EPCs [58]. In contrast, in 20 and 41 SSc patients, simvastatine 20 mg/days 12 weeks and sildenafil 100 mg/day 8 weeks respectively failed to improve EPCs number (Table 2) [59, 60].
Pharmacological treatment are not only process which can be increased EPCs in SSc. Tinazzi et al. reported in 30 SSc patients that EPCs increased at 60 and 90 days after extracorporeal shock waves et improved RSS and skin vascular score [110]. However in this article, the gating strategy to differentiate CECs and EPCs was not specified.
Recently, Manetti reported a decrease in the number of circulating lymphatic EPCs (CD34 + CD133 + VEGFR3+) in 40 patients with complicated digital ulcer in SSc [57] (Table 1). Tang cell expansion was described, suggesting an ineffective proliferation unable to compensate the decrease in EPCs [111]. The association of lymphatic EPCs and Tang cells with EPCs needs to be more investigated.
Several studies presented contradictory results about EPCs number and correlation with disease duration or disease activity score. Due to clinical presentation, disease duration and medical care, further studies are requiring to determine EPCs evolution during SSc.
Conclusion
EPCs contribute to endothelial vascular repair and are involved in this homeostasis. Dysregulation, with decreased number or impaired functionality, leads to higher cardiovascular risk. Rheumatic diseases and autoimmune disorders are associated with a higher frequency of cardiovascular events and EPCs depletion is involved in this complex process. However, most studies have not evaluated the functional aspect of ECFCs. It would be necessary to assess the impact of therapies on early EPCs and ECFCs. Standardisation is needed for cardiovascular risk diseases, in particular APS, SLE and RA on the evaluation of EPCs. To conclude, EPCs could be used to evaluate early endothelial dysfunction and, thanks to their angiogenic properties, the therapeutic potential of EPCs is being increasingly investigated in several pathologies.
References
Alessandri, G., Girelli, M., Taccagni, G., Colombo, A., Nicosia, R., Caruso, A., Baronio, M., Pagano, S., Cova, L., & Parati, E. (2001). Human vasculogenesis ex vivo: Embryonal aorta as a tool for isolation of endothelial cell progenitors. Laboratory Investigation, 81, 875–885.
Rafii, S., Heissig, B., & Hattori, K. (2002). Efficient mobilization and recruitment of marrow-derived endothelial and hematopoietic stem cells by adenoviral vectors expressing angiogenic factors. Gene Therapy, 9, 631–641.
Asahara, T., Murohara, T., Sullivan, A., Silver, M., van der Zee, R., Li, T., Witzenbichler, B., Schatteman, G., & Isner, J. M. (1997). Isolation of putative progenitor endothelial cells for angiogenesis. Science, 275, 964–967.
Rossi, E., Poirault-Chassac, S., Bieche, I., Chocron, R., Schnitzler, A., Lokajczyk, A., Bourdoncle, P., Dizier, B., Bacha, N. C., Gendron, N., Blandinieres, A., Guerin, C. L., Gaussem, P., & Smadja, D. M. (2019). Human endothelial colony forming cells express intracellular CD133 that modulates their vasculogenic properties. Stem Cell Review and Reports, 15, 590–600.
Del Díaz, S., Barrena, S., Muñoz-Chápuli, R., & Carmona, R. (2020). Embryonic circulating endothelial progenitor cells. Angiogenesis, 23, 531–541.
Forsythe, J. A., Jiang, B. H., Iyer, N. V., Agani, F., Leung, S. W., Koos, R. D., & Semenza, G. L. (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Molecular and Cellular Biology, 16, 4604–4613.
Ceradini, D. J., Kulkarni, A. R., Callaghan, M. J., Tepper, O. M., Bastidas, N., Kleinman, M. E., Capla, J. M., Galiano, R. D., Levine, J. P., & Gurtner, G. C. (2004). Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nature Medicine, 10, 858–864.
Laufs, U., Werner, N., Link, A., Endres, M., Wassmann, S., Jürgens, K., Miche, E., Böhm, M., & Nickenig, G. (2004). Physical training increases endothelial progenitor cells, inhibits neointima formation, and enhances angiogenesis. Circulation, 109, 220–226.
Détriché, G., Gendron, N., Philippe, A., Gruest, M., Billoir, P., Rossi, E., Guerin, C. L., Lokajczyk, A., Brabant, S., Prié, D., Mirault, T., & Smadja, D. M. (2022). Gonadotropins as novel active partners in vascular diseases: Insight from angiogenic properties and thrombotic potential of endothelial colony-forming cells. Journal of Thrombosis and Haemostasis, 20, 230–237.
Urbich, C., & Dimmeler, S. (2004). Endothelial progenitor cells: Characterization and role in vascular biology. Circulation Research, 95, 343–353.
Mak, A., & Chan, J. K. Y. (2022). Endothelial function and endothelial progenitor cells in systemic lupus erythematosus. Nature Reviews Rheumatology, 18, 286–300.
Medina, R. J., Barber, C. L., Sabatier, F., Dignat-George, F., Melero-Martin, J. M., Khosrotehrani, K., Ohneda, O., Randi, A. M., Chan, J. K. Y., Yamaguchi, T., Van Hinsbergh, V. W. M., Yoder, M. C., & Stitt, A. W. (2017). Endothelial progenitors: A consensus statement on nomenclature. Stem Cells Translational Medicine, 6, 1316–1320.
Smadja, D. M., Melero-Martin, J. M., Eikenboom, J., Bowman, M., Sabatier, F., & Randi, A. M. (2019). Standardization of methods to quantify and culture endothelial colony-forming cells derived from peripheral blood: Position paper from the international society on thrombosis and haemostasis SSC. Journal of Thrombosis and Haemostasis, 17, 1190–1194.
Hill, J. M., Zalos, G., Halcox, J. P. J., Schenke, W. H., Waclawiw, M. A., Quyyumi, A. A., & Finkel, T. (2003). Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. New England Journal of Medicine, 348, 593–600.
Wils, J., Favre, J., & Bellien, J. (2017). Modulating putative endothelial progenitor cells for the treatment of endothelial dysfunction and cardiovascular complications in diabetes. Pharmacology & Therapeutics, 170, 98–115.
Nuzzolo, E. R., Iachininoto, M. G., & Teofili, L. (2012). Endothelial progenitor cells and thrombosis. Thrombosis Research, 129, 309–313.
Heinisch, P. P., Bello, C., Emmert, M. Y., Carrel, T., Dreßen, M., Hörer, J., Winkler, B., & Luedi, M. M. (2022). Endothelial progenitor cells as biomarkers of cardiovascular pathologies: A narrative review. Cells, 11, 1678.
Altabas, V., Altabas, K., & Kirigin, L. (2016). Endothelial progenitor cells (EPCs) in ageing and age-related diseases: How currently available treatment modalities affect EPC biology, atherosclerosis, and cardiovascular outcomes. Mechanisms of Ageing and Development, 159, 49–62.
Schmidt-Lucke, C., Rössig, L., Fichtlscherer, S., Vasa, M., Britten, M., Kämper, U., Dimmeler, S., & Zeiher, A. M. (2005). Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: Proof of concept for the clinical importance of endogenous vascular repair. Circulation, 111, 2981–2987.
Sherer, Y., & Shoenfeld, Y. (2006). Mechanisms of disease: Atherosclerosis in autoimmune diseases. Nature Clinical Practice Rheumatology, 2, 99–106.
Widemann, A., Pasero, C., Arnaud, L., Poullin, P., Loundou, A. D., Choukroun, G., Sanderson, F., Lacroix, R., Sabatier, F., Coppo, P., Dignat-George, F., Kaplanski, G., ENDO-13 study group. (2014). Circulating endothelial cells and progenitors as prognostic factors during autoimmune thrombotic thrombocytopenic purpura: Results of a prospective multicenter french study. Journal of Thrombosis and Haemostasis, 12, 1601–1609.
Bartoloni, E., Alunno, A., Bistoni, O., Caterbi, S., Luccioli, F., Santoboni, G., Mirabelli, G., Cannarile, F., & Gerli, R. (2015). Characterization of circulating endothelial microparticles and endothelial progenitor cells in primary Sjögren’s syndrome: New markers of chronic endothelial damage? Rheumatology (Oxford, England), 54, 536–544.
Alunno, A., Ibba-Manneschi, L., Bistoni, O., Cipriani, S., Topini, F., Gerli, R., & Manetti, M. (2019). Angiogenic T cells in primary Sjögren’s syndrome: A double-edged sword? Clinical and Experimental Rheumatology, 37(Suppl 118), 36–41.
Gresele, P., Migliacci, R., Vedovati, M. C., Ruffatti, A., Becattini, C., Facco, M., Guglielmini, G., Boscaro, E., Mezzasoma, A. M., Momi, S., & Pengo, V. (2009). Patients with primary antiphospholipid antibody syndrome and without associated vascular risk factors present a normal endothelial function. Thrombosis Research, 123, 444–451.
Grenn, R. C., Yalavarthi, S., Gandhi, A. A., Kazzaz, N. M., Núñez-Álvarez, C., Hernández-Ramírez, D., Cabral, A. R., McCune, W. J., Bockenstedt, P. L., & Knight, J. S. (2017). Endothelial progenitor dysfunction associates with a type I interferon signature in primary antiphospholipid syndrome. Annals of the Rheumatic Diseases, 76, 450–457.
Lee, P. Y., Li, Y., Richards, H. B., Chan, F. S., Zhuang, H., Narain, S., Butfiloski, E. J., Sobel, E. S., Reeves, W. H., & Segal, M. S. (2007). Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis and Rheumatism, 56, 3759–3769.
Westerweel, P. E., Luijten, R. K. M. A. C., Hoefer, I. E., Koomans, H. A., Derksen, R. H. W. M., & Verhaar, M. C. (2007). Haematopoietic and endothelial progenitor cells are deficient in quiescent systemic lupus erythematosus. Annals of the Rheumatic Diseases, 66, 865–870.
Moonen, J. R. A. J., de Leeuw, K., van Seijen, X. J. G. Y., Kallenberg, C. G. M., van Luyn, M. J. A., Bijl, M., & Harmsen, M. C. (2007). Reduced number and impaired function of circulating progenitor cells in patients with systemic lupus erythematosus. Arthritis Research & Therapy, 9, R84.
Grisar, J., Steiner, C. W., Bonelli, M., Karonitsch, T., Schwarzinger, I., Weigel, G., Steiner, G., & Smolen, J. S. (2008). Systemic lupus erythematosus patients exhibit functional deficiencies of endothelial progenitor cells. Rheumatology (Oxford, England), 47, 1476–1483.
Deng, X. L., Li, X. X., Liu, X. Y., Sun, L., & Liu, R. (2010). Comparative study on circulating endothelial progenitor cells in systemic lupus erythematosus patients at active stage. Rheumatology International, 30, 1429–1436.
Ablin, J. N., Boguslavski, V., Aloush, V., Elkayam, O., Paran, D., Levartovski, D., Caspi, D., & George, J. (2011). Enhanced adhesive properties of endothelial progenitor cells (EPCs) in patients with SLE. Rheumatology International, 31, 773–778.
Baker, J. F., Zhang, L., Imadojemu, S., Sharpe, A., Patil, S., Moore, J. S., Mohler, E. R., & Von Feldt, J. (2012). Circulating endothelial progenitor cells are reduced in SLE in the absence of coronary artery calcification. Rheumatology International, 32, 997–1002.
Mohan, S., Barsalou, J., Bradley, T. J., Slorach, C., Reynolds, J. A., Hasni, S., Thompson, B., Ng, L., Levy, D., Silverman, E., & Kaplan, M. J. (2015). Endothelial progenitor cell phenotype and function are impaired in childhood-onset systemic lupus erythematosus. Arthritis & Rheumatology (Hoboken, NJ), 67, 2257–2262.
Coppolino, G., Campo, S., Bolignano, D., Sturiale, A., Giacobbe, M. S., Loddo, S., & Buemi, M. (2008). Effect of immunoglobulin treatment on endothelial progenitor cells in systemic lupus erythematosus. Annals of the Rheumatic Diseases, 67, 1047–1048.
Oliveira, A. C. D., Arismendi, M. I., Machado, L. S. G., & Sato, E. I. (2022). Ramipril improves endothelial function and increases the number of endothelial progenitor cells in patients with systemic Lupus Erythematosus. Journal of Clinical Rheumatology: Practical Reports on Rheumatic & Musculoskeletal Diseases, 28, 349–353.
Huang, Z., Liu, L., Huang, S., Li, J., Feng, S., Huang, N., Ai, Z., Long, W., & Jiang, L. (2020). Vitamin D (1,25 (OH)2D3) improves endothelial progenitor cells function via enhanced NO secretion in systemic lupus erythematosus. Cardiology Research and Practice, 16(2020), 6802562. https://doi.org/10.1155/2020/6802562
Holmén, C., Elsheikh, E., Stenvinkel, P., Qureshi, A. R., Pettersson, E., Jalkanen, S., & Sumitran-Holgersson, S. (2005). Circulating inflammatory endothelial cells contribute to endothelial progenitor cell dysfunction in patients with vasculitis and kidney involvement. Journal of the American Society of Nephrology, 16, 3110–3120.
Závada, J., Kideryová, L., Pytlík, R., Vanková, Z., & Tesar, V. (2008). Circulating endothelial progenitor cells in patients with ANCA-associated vasculitis. Kidney & Blood Pressure Research, 31, 247–254.
Závada, J., Kideryová, L., Pytlík, R., Hrusková, Z., & Tesar, V. (2009). Reduced number of endothelial progenitor cells is predictive of early relapse in anti-neutrophil cytoplasmic antibody-associated vasculitis. Rheumatology (Oxford, England), 48, 1197–1201.
de Groot, K., Goldberg, C., Bahlmann, F. H., Woywodt, A., Haller, H., Fliser, D., & Haubitz, M. (2007). Vascular endothelial damage and repair in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis and Rheumatism, 56, 3847–3853.
Wilde, B., Mertens, A., Arends, S. J., Rouhl, R. P., Bijleveld, R., Huitema, J., Timmermans, S. A., Damoiseaux, J., Witzke, O., Duijvestijn, A. M., van Paassen, P., van Oostenbrugge, R. J., & Cohen Tervaert, J. W. (2016). Endothelial progenitor cells are differentially impaired in ANCA-associated vasculitis compared to healthy controls. Arthritis Research & Therapy, 18, 147.
Grisar, J., Aletaha, D., Steiner, C. W., Kapral, T., Steiner, S., Seidinger, D., Weigel, G., Schwarzinger, I., Wolozcszuk, W., Steiner, G., & Smolen, J. S. (2005). Depletion of endothelial progenitor cells in the peripheral blood of patients with rheumatoid arthritis. Circulation, 111, 204–211.
Herbrig, K., Haensel, S., Oelschlaegel, U., Pistrosch, F., Foerster, S., & Passauer, J. (2006). Endothelial dysfunction in patients with rheumatoid arthritis is associated with a reduced number and impaired function of endothelial progenitor cells. Annals of the Rheumatic Diseases, 65, 157–163.
Ablin, J. N., Boguslavski, V., Aloush, V., Elkayam, O., Paran, D., Caspi, D., & George, J. (2006). Effect of anti-TNFalpha treatment on circulating endothelial progenitor cells (EPCs) in rheumatoid arthritis. Life Sciences, 79, 2364–2369.
Spinelli, F. R., Metere, A., Barbati, C., Pierdominici, M., Iannuccelli, C., Lucchino, B., Ciciarello, F., Agati, L., Valesini, G., & Di Franco, M. (2013). Effect of therapeutic inhibition of TNF on circulating endothelial progenitor cells in patients with rheumatoid arthritis. Mediators of Inflammation, 2013, 537539.
Grisar, J., Aletaha, D., Steiner, C. W., Kapral, T., Steiner, S., Säemann, M., Schwarzinger, I., Buranyi, B., Steiner, G., & Smolen, J. S. (2007). Endothelial progenitor cells in active rheumatoid arthritis: Effects of tumour necrosis factor and glucocorticoid therapy. Annals of the Rheumatic Diseases, 66, 1284–1288.
Park, Y. J., Kim, J. Y., Park, J., Choi, J. J., Kim, W. U., & Cho, C. S. (2014). Bone erosion is associated with reduction of circulating endothelial progenitor cells and endothelial dysfunction in rheumatoid arthritis. Arthritis and Rheumatology, 66, 1450–1460.
Pulito-Cueto, V., Remuzgo-Martínez, S., Genre, F., Mora-Cuesta, V. M., Iturbe-Fernández, D., Fernández-Rozas, S., Atienza-Mateo, B., Lera-Gómez, L., Alonso-Lecue, P., Rodríguez-Carrio, J., Prieto-Peña, D., Portilla, V., Blanco, R., Corrales, A., Gualillo, O., Cifrián, J. M., López-Mejías, R., & González-Gay, M. A. (2020). Endothelial progenitor cells as a potential biomarker in interstitial lung disease associated with rheumatoid arthritis. Journal of Clinical Medicine, 9, 4098.
Kuwana, M., Okazaki, Y., Yasuoka, H., Kawakami, Y., & Ikeda, Y. (2004). Defective vasculogenesis in systemic sclerosis. Lancet, 364, 603–610.
Del Papa, N., Quirici, N., Soligo, D., Scavullo, C., Cortiana, M., Borsotti, C., Maglione, W., Comina, D. P., Vitali, C., Fraticelli, P., Gabrielli, A., Cortelezzi, A., & Lambertenghi-Deliliers, G. (2006). Bone marrow endothelial progenitors are defective in systemic sclerosis. Arthritis and Rheumatism, 54, 2605–2615.
Avouac, J., Juin, F., Wipff, J., Couraud, P. O., Chiocchia, G., Kahan, A., Boileau, C., Uzan, G., & Allanore, Y. (2008). Circulating endothelial progenitor cells in systemic sclerosis: Association with disease severity. Annals of the Rheumatic Diseases, 67, 1455–1460.
Allanore, Y., Batteux, F., Avouac, J., Assous, N., Weill, B., & Kahan, A. (2007). Levels of circulating endothelial progenitor cells in systemic sclerosis. Clinical and Experimental Rheumatology, 25, 60–66.
Andrigueti, F. V., Arismendi, M. I., Ebbing, P. C. C., & Kayser, C. (2015). Decreased numbers of endothelial progenitor cells in patients in the early stages of systemic sclerosis. Microvascular Research, 98, 82–87.
Zhu, S., Evans, S., Yan, B., Povsic, T. J., Tapson, V., Goldschmidt-Clermont, P. J., & Dong, C. (2008). Transcriptional regulation of Bim by FOXO3a and akt mediates Scleroderma Serum-Induced apoptosis in endothelial progenitor cells. Circulation, 118, 2156–2165.
Del Papa, N., Quirici, N., Scavullo, C., Gianelli, U., Corti, L., Vitali, C., Ferri, C., Giuggioli, D., Manfredi, A., Maglione, W., Onida, F., Colaci, M., & Bosari, S. (2010). Lambertenghi Deliliers G. Antiendothelial cell antibodies induce apoptosis of bone marrow endothelial progenitors in systemic sclerosis. Journal of Rheumatology, 37, 2053–2063.
Pulito-Cueto, V., Remuzgo-Martínez, S., Genre, F., Atienza-Mateo, B., Mora-Cuesta, V. M., Iturbe-Fernández, D., Lera-Gómez, L., Pérez-Fernández, R., Prieto-Peña, D., Portilla, V., Blanco, R., Corrales, A., Gualillo, O., Cifrián, J. M., López-Mejías, R., & González-Gay, M. A. (2021). Endothelial progenitor cells: Relevant players in the Vasculopathy and Lung Fibrosis Associated with the Presence of interstitial lung disease in systemic sclerosis patients. Biomedicines, 9, 847.
Manetti, M., Pratesi, S., Romano, E., Rosa, I., Bruni, C., Bellando-Randone, S., Guiducci, S., Maggi, E., Ibba-Manneschi, L., & Matucci-Cerinic, M. (2019). Decreased circulating lymphatic endothelial progenitor cells in digital ulcer-complicated systemic sclerosis. Annals of the Rheumatic Diseases, 78, 575–577.
Furuya, Y., Okazaki, Y., Kaji, K., Sato, S., Takehara, K., & Kuwana, M. (2010). Mobilization of endothelial progenitor cells by intravenous cyclophosphamide in patients with systemic sclerosis. Rheumatology (Oxford, England), 49, 2375–2380.
Del Papa, N., Cortiana, M., Vitali, C., Silvestris, I., Maglione, W., Comina, D. P., Lucchi, T., & Cortelezzi, A. (2008). Simvastatin reduces endothelial activation and damage but is partially ineffective in inducing endothelial repair in systemic sclerosis. The Journal of Rheumatology, 35, 1323–1328.
Andrigueti, F. V., Ebbing, P. C. C., Arismendi, M. I., & Kayser, C. (2017). Evaluation of the effect of sildenafil on the microvascular blood flow in patients with systemic sclerosis: A randomised, double-blind, placebo-controlled study. Clinical and Experimental Rheumatology, 35(Suppl 106), 151–158.
Mariette, X., & Criswell, L. A. (2018). Primary Sjögren’s syndrome. New England Journal of Medicine, 378, 931–939.
Mariette, X. (2010). Sjögren’s syndrome pathophysiology. Revue De Medecine Interne, 31(Suppl 1), S2-6.
Gerli, R., Vaudo, G., Bocci, E. B., Schillaci, G., Alunno, A., Luccioli, F., Hijazi, R., Mannarino, E., & Shoenfeld, Y. (2010). Functional impairment of the arterial wall in primary Sjögren’s syndrome: Combined action of immunologic and inflammatory factors. Arthritis Care Res (Hoboken), 62, 712–718.
Vaudo, G., Bocci, E. B., Shoenfeld, Y., Schillaci, G., Wu, R., Del Papa, N., Vitali, C., Delle Monache, F., Marchesi, S., Mannarino, E., & Gerli, R. (2005). Precocious intima-media thickening in patients with primary Sjögren’s syndrome. Arthritis and Rheumatism, 52, 3890–3897.
Pirildar, T., Tikiz, C., Ozkaya, S., Tarhan, S., Utük, O., Tikiz, H., & Tezcan, U. K. (2005). Endothelial dysfunction in patients with primary Sjögren’s syndrome. Rheumatology International, 25, 536–539.
Rachapalli, S. M., Kiely, P. D., & Bourke, B. E. (2009). Prevalence of abnormal ankle brachial index in patients with primary Sjogren’s syndrome. Clinical Rheumatology, 28, 587–590.
Bodolay, E., Koch, A. E., Kim, J., Szegedi, G., & Szekanecz, Z. (2002). Angiogenesis and chemokines in rheumatoid arthritis and other systemic inflammatory rheumatic diseases. Journal of Cellular and Molecular Medicine, 6, 357–376.
Potvin, F., Petitclerc, E., Marceau, F., & Poubelle, P. E. (1997). Mechanisms of action of antimalarials in inflammation: Induction of apoptosis in human endothelial cells. The Journal of Immunology, 158, 1872–1879.
Ghigo, D., Aldieri, E., Todde, R., Costamagna, C., Garbarino, G., Pescarmona, G., & Bosia, A. (1998). Chloroquine stimulates nitric oxide synthesis in murine, porcine, and human endothelial cells. The Journal of Clinical Investigation, 102, 595–605.
Miranda, S., Billoir, P., Damian, L., Thiebaut, P. A., Schapman, D., Le Besnerais, M., Jouen, F., Galas, L., Levesque, H., Le Cam-Duchez, V., Joannides, R., Richard, V., & Benhamou, Y. (2019). Hydroxychloroquine reverses the prothrombotic state in a mouse model of antiphospholipid syndrome: Role of reduced inflammation and endothelial dysfunction. PLoS One, 14, e0212614.
Miyakis, S., Lockshin, M. D., Atsumi, T., Branch, D. W., Brey, R. L., Cervera, R., Derksen, R. H. W. M., De Groot, P. G., Koike, T., Meroni, P. L., Reber, G., Shoenfeld, Y., Tincani, A., Vlachoyiannopoulos, P. G., & Krilis, S. A. (2006). International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). Journal of Thrombosis and Haemostasis, 4, 295–306.
Proulle, V., Furie, R. A., Merrill-Skoloff, G., Furie, B. C., & Furie, B. (2014). Platelets are required for enhanced activation of the endothelium and fibrinogen in a mouse thrombosis model of APS. Blood, 124, 611–622.
Chaturvedi, S., & McCrae, K. R. (2017). Diagnosis and management of the antiphospholipid syndrome. Blood Reviews, 31, 406–417.
Ahmed, K., Vianna, J. L., Khamashta, M. A., & Hughes, G. R. (1992). IL-2, IL-6 and TNF levels in primary antiphospholipid syndrome. Clinical and Experimental Rheumatology, 10, 503.
Benhamou, Y., Bellien, J., Armengol, G., Brakenhielm, E., Adriouch, S., Iacob, M., Remy-Jouet, I., Le Cam-Duchez, V., Monteil, C., Renet, S., Jouen, F., Drouot, L., Menard, J. F., Borg, J. Y., Thuillez, C., Boyer, O., Levesque, H., Richard, V., & Joannidès, R. (2014). Role of toll-like receptors 2 and 4 in mediating endothelial dysfunction and arterial remodeling in primary arterial antiphospholipid syndrome (Vol. 66, pp. 3210–3220). Arthritis and Rheumatology.
Schreiber, K., Sciascia, S., de Groot, P. G., Devreese, K., Jacobsen, S., Ruiz-Irastorza, G., Salmon, J. E., Shoenfeld, Y., Shovman, O., & Hunt, B. J. (2018). Antiphospholipid syndrome. Nature Reviews Disease Primers, 4, 17103.
López-Pedrera, C., Buendía, P., Cuadrado, M. J., Siendones, E., Aguirre, M. A., Barbarroja, N., Montiel-Duarte, C., Torres, A., Khamashta, M., & Velasco, F. (2006). Antiphospholipid antibodies from patients with the antiphospholipid syndrome induce monocyte tissue factor expression through the simultaneous activation of NF-kappaB/Rel proteins via the p38 mitogen-activated protein kinase pathway, and of the MEK-1/ERK pathway. Arthritis and Rheumatism, 54, 301–311.
Kinev, A. V., & Roubey, R. A. S. (2008). Tissue factor in the antiphospholipid syndrome. Lupus, 17, 952–958.
Pierangeli, S. S., Espinola, R. G., Liu, X., & Harris, E. N. (2001). Thrombogenic effects of antiphospholipid antibodies are mediated by intercellular cell adhesion molecule-1, vascular cell adhesion molecule-1, and P-selectin. Circulation Research, 88, 245–250.
Esdaile, J. M., Abrahamowicz, M., Grodzicky, T., Li, Y., Panaritis, C., du Berger, R., Côte, R., Grover, S. A., Fortin, P. R., Clarke, A. E., & Senécal, J. L. (2001). Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis and Rheumatism, 44, 2331–2337.
Ebner, P., Picard, F., Richter, J., Darrelmann, E., Schneider, M., Strauer, B. E., & Brehm, M. (2010). Accumulation of VEGFR-2+/CD133 + cells and decreased number and impaired functionality of CD34+/VEGFR-2 + cells in patients with SLE. Rheumatology (Oxford, England), 49, 63–72.
Castejon, R., Jimenez-Ortiz, C., Valero-Gonzalez, S., Rosado, S., Mellor, S., & Yebra-Bango, M. (2014). Decreased circulating endothelial progenitor cells as an early risk factor of subclinical atherosclerosis in systemic lupus erythematosus. Rheumatology (Oxford, England), 53, 631–638.
Castejon, R., Jimenez-Ortiz, C., Rosado, S., Tutor-Ureta, P., Mellor-Pita, S., & Yebra-Bango, M. (2016). Metabolic syndrome is associated with decreased circulating endothelial progenitor cells and increased arterial stiffness in systemic lupus erythematosus. Lupus, 25, 129–136.
Denny, M. F., Thacker, S., Mehta, H., Somers, E. C., Dodick, T., Barrat, F. J., McCune, W. J., & Kaplan, M. J. (2007). Interferon-alpha promotes abnormal vasculogenesis in lupus: A potential pathway for premature atherosclerosis. Blood, 110, 2907–2915.
Ferrante, A., Guggino, G., Di Liberto, D., Ciccia, F., Cipriani, P., Balistreri, C. R., Sireci, G., Giacomelli, R., & Triolo, G. (2016). Endothelial progenitor cells: Are they displaying a function in autoimmune disorders? Mechanisms of Ageing and Development, 159, 44–48.
Lapraik, C., Watts, R., Bacon, P., Carruthers, D., Chakravarty, K., D’Cruz, D., Guillevin, L., Harper, L., Jayne, D., Luqmani, R., Mooney, J., & Scott, D. (2007). BSR and BHPR Standards, Guidelines and Audit Working Group. BSR and BHPR guidelines for the management of adults with ANCA associated vasculitis. Rheumatology (Oxford, England), 46, 1615–1616.
Halbwachs, L., & Lesavre, P. (2012). Endothelium-neutrophil interactions in ANCA-associated diseases. Journal of the American Society of Nephrology, 23, 1449–1461.
Stassen, P. M., Derks, R. P. H., Kallenberg, C. G. M., & Stegeman, C. A. (2008). Venous thromboembolism in ANCA-associated vasculitis–incidence and risk factors. Rheumatology (Oxford, England), 47, 530–534.
Hong, Y., Eleftheriou, D., Klein, N. J., & Brogan, P. A. (2015). Impaired function of endothelial progenitor cells in children with primary systemic vasculitis. Arthritis Research & Therapy, 17, 292.
Del Rio, A. P. T., Frade-Guanaes, J. O., Ospina-Prieto, S., Duarte, B. K. L., Bertolo, M. B., Ozelo, M. C., & Sachetto, Z. (2022). Impaired repair properties of endothelial colony-forming cells in patients with granulomatosis with polyangiitis. Journal of Cellular and Molecular Medicine, 26, 5044–5053.
Westerweel, P. E., & Verhaar, M. C. (2009). Endothelial progenitor cell dysfunction in rheumatic disease. Nature Reviews Rheumatology, 5, 332–340.
Egan, C. G., Caporali, F., Garcia-Gonzalez, E., Galeazzi, M., & Sorrentino, V. (2008). Endothelial progenitor cells and colony-forming units in rheumatoid arthritis: Association with clinical characteristics. Rheumatology (Oxford, England), 47, 1484–1488.
Adawi, M., Pastukh, N., Saaida, G., Sirchan, R., Watad, A., & Blum, A. (2018). Inhibition of endothelial progenitor cells may explain the high cardiovascular event rate in patients with rheumatoid arthritis. QJM, 111, 525–529.
Yiu, K. H., Wang, S., Mok, M. Y., Ooi, G. C., Khong, P. L., Lau, C. P., Lai, W. H., Wong, L. Y., Lam, K. F., Lau, C. S., & Tse, H. F. (2010). Role of circulating endothelial progenitor cells in patients with rheumatoid arthritis with coronary calcification. Journal of Rheumatology, 37, 529–535.
Rodríguez-Carrio, J., Prado, C., de Paz, B., López, P., Gómez, J., Alperi-López, M., Ballina-García, F. J., & Suárez, A. (2012). Circulating endothelial cells and their progenitors in systemic lupus erythematosus and early rheumatoid arthritis patients. Rheumatology (Oxford, England), 51, 1775–1784.
Rodríguez-Carrio, J., de Paz, B., López, P., Prado, C., Alperi-López, M., Ballina-García, F. J., & Suárez, A. (2014). IFNα serum levels are associated with endothelial progenitor cells imbalance and disease features in rheumatoid arthritis patients. PLoS One, 9, e86069.
Cafaro, G., Petito, E., Bistoni, O., Falcinelli, E., Cipriani, S., Borghi, M. C., Bonifacio, A. F., Giglio, E., Alunno, A., Perricone, C., Gerli, R., Gresele, P., & Bartoloni, E. (2022). Methotrexate improves endothelial function in early rheumatoid arthritis patients after 3 months of treatment. Arthritis Research & Therapy, 24, 236.
Shirinsky, I., Polovnikova, O., Kalinovskaya, N., & Shirinsky, V. (2013). The effects of fenofibrate on inflammation and cardiovascular markers in patients with active rheumatoid arthritis: A pilot study. Rheumatology International, 33, 3045–3048.
Rodríguez-Carrio, J., Alperi-López, M., López, P., Alonso-Castro, S., Ballina-García, F. J., & Suárez, A. (2015). Angiogenic T cells are decreased in rheumatoid arthritis patients. Annals of the Rheumatic Diseases, 74, 921–927.
Denton, C. P., & Khanna, D. (2017). Systemic sclerosis. Lancet, 390, 1685–1699.
Ota, Y., & Kuwana, M. (2020). Endothelial cells and endothelial progenitor cells in the pathogenesis of systemic sclerosis. European Journal of Rheumatology, 7(Suppl 3), S139–S146. https://doi.org/10.5152/eurjrheum.2019.19158
Flavahan, N. A., Flavahan, S., Mitra, S., & Chotani, M. A. (2003). The vasculopathy of Raynaud’s phenomenon and scleroderma. Rheumatic Diseases Clinics of North America, 29, 275–291. vi.
Avouac, J., Meune, C., Ruiz, B., Couraud, P. O., Uzan, G., Boileau, C., Kahan, A., Chiocchia, G., & Allanore, Y. (2012). Angiogenic biomarkers predict the occurrence of digital ulcers in systemic sclerosis. Annals of the Rheumatic Diseases, 71, 394–399.
Avouac, J., Vallucci, M., Smith, V., Senet, P., Ruiz, B., Sulli, A., Pizzorni, C., Frances, C., Chiocchia, G., Cutolo, M., & Allanore, Y. (2013). Correlations between angiogenic factors and capillaroscopic patterns in systemic sclerosis. Arthritis Research & Therapy, 15, R55.
Yamaguchi, Y., Okazaki, Y., Seta, N., Satoh, T., Takahashi, K., Ikezawa, Z., & Kuwana, M. (2010). Enhanced angiogenic potency of monocytic endothelial progenitor cells in patients with systemic sclerosis. Arthritis Research & Therapy, 12, R205.
Benyamine, A., Magalon, J., Cointe, S., Lacroix, R., Arnaud, L., Bardin, N., Rossi, P., Francès, Y., Bernard-Guervilly, F., Kaplanski, G., Harlé, J. R., Weiller, P. J., Berbis, P., Braunstein, D., Jouve, E., Lesavre, N., Couranjou, F., Dignat-George, F., Sabatier, F., … Granel, B. (2017). Increased serum levels of fractalkine and mobilisation of CD34 + CD45- endothelial progenitor cells in systemic sclerosis. Arthritis Research & Therapy, 19, 60.
Mok, M. Y., Yiu, K. H., Wong, C. Y., Qiuwaxi, J., Lai, W. H., Wong, W. S., Tse, H. F., & Lau, C. S. (2010). Low circulating level of CD133 + KDR + cells in patients with systemic sclerosis. Clinical and Experimental Rheumatology, 28, S19-25.
Patschan, S., Tampe, D., Müller, C., Seitz, C., Herink, C., Müller, G. A., Zeisberg, E., Zeisberg, M., Henze, E., & Patschan, D. (2016). Early endothelial progenitor cells (eEPCs) in systemic sclerosis (SSc) - dynamics of cellular regeneration and mesenchymal transdifferentiation. Bmc Musculoskeletal Disorders, 17, 339.
Billoir, P., Blandinières, A., Gendron, N., Chocron, R., Gunther, S., Philippe, A., Guerin, C. L., Israël-Biet, D., & Smadja, D. M. (2021). Endothelial colony-forming cells from idiopathic pulmonary fibrosis patients have a high procoagulant potential. Stem Cell Rev Rep, 17, 694–699.
Tinazzi, E., Amelio, E., Marangoni, E., Guerra, C., Puccetti, A., Codella, O. M., Simeoni, S., Cavalieri, E., Montagnana, M., Adani, R., Corrocher, R., & Lunardi, C. (2011). Effects of shock wave therapy in the skin of patients with progressive systemic sclerosis: A pilot study. Rheumatology International, 31, 651–656.
Manetti, M., Pratesi, S., Romano, E., Bellando-Randone, S., Rosa, I., Guiducci, S., Fioretto, B. S., Ibba-Manneschi, L., Maggi, E., & Matucci-Cerinic, M. (2017). Angiogenic T cell expansion correlates with severity of peripheral vascular damage in systemic sclerosis. PLoS One, 12, e0183102.
Acknowledgements
Authors are grateful to Nikki Sabourin-Gibbs, CHU Rouen, for her help in editing the manuscript.
Author information
Authors and Affiliations
Contributions
G Feugray and P Billoir wrote the manuscript, analyzed and interpreted the data. S Miranda, V Lecam Duchez and J Bellien revised the manuscript and results. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Authors state no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Feugray, G., Miranda, S., Le Cam Duchez, V. et al. Endothelial Progenitor Cells in Autoimmune Disorders. Stem Cell Rev and Rep 19, 2597–2611 (2023). https://doi.org/10.1007/s12015-023-10617-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-023-10617-y