
Abstract
Ribosome biogenesis is a crucial process for growth and constitutes the major consumer of cellular resources. This pathway is subjected to very stringent regulation to ensure correct ribosome manufacture with a wide variety of environmental and metabolic changes, and intracellular insults. Here we summarise our current knowledge on the regulation of ribosome biogenesis in Saccharomyces cerevisiae by particularly focusing on the feedback mechanisms that maintain ribosome homeostasis. Ribosome biogenesis in yeast is controlled mainly at the level of the production of both pre-rRNAs and ribosomal proteins through the transcriptional and post-transcriptional control of the TORC1 and protein kinase A signalling pathways. Pre-rRNA processing can occur before or after the 35S pre-rRNA transcript is completed; the switch between these two alternatives is regulated by growth conditions. The expression of both ribosomal proteins and the large family of transacting factors involved in ribosome biogenesis is co-regulated. Recently, it has been shown that the synthesis of rRNA and ribosomal proteins, but not of trans-factors, is coupled. Thus the so-called CURI complex sequesters specific transcription factor Ifh1 to repress ribosomal protein genes when rRNA transcription is impaired. We recently found that an analogue system should operate to control the expression of transacting factor genes in response to actual ribosome assembly performance. Regulation of ribosome biogenesis manages situations of imbalanced ribosome production or misassembled ribosomal precursors and subunits, which have been closely linked to distinct human diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Regulation of ribosome biogenesis
Ribosomes are highly complex ubiquitous ribonucleoprotein particles that, in eukaryotes, are composed of four ribosomal RNAs (rRNAs) and 79–80 ribosomal proteins (r-proteins). Production of ribosomes is a fundamental multi-step process and an extraordinarily complicated cellular challenge. This process has been extensively studied in the yeast Saccharomyces cerevisiae in which, in addition to rRNAs and r-proteins, more than 300 non-ribosomal trans-acting proteins, also known as ribosome assembly factors, have been described (Woolford and Baserga 2013).
Ribosome biogenesis occurs primarily in the nucleolus, although late events occur in the nucleoplasm, where the pre-ribosomal particles gain export competence, and also in the cytoplasm where the last steps in the maturation of the ribosomal subunits (r-subunits) take place. In the yeast nucleolus, mature 18S, 5.8S and 25S rRNAs are co-transcribed by RNA polymerase I (RNAP I) as a single precursor of rRNA (pre-rRNA) that undergo either co- or post-transcriptional processing. Pre-5S is independently transcribed by RNAP III. Pre-rRNA processing occurs concomitantly to most rRNA modification reactions, the folding of pre-rRNAs and the formation of pre-ribosomal particles, which contain different pre-rRNAs, r-proteins and ribosome assembly factors depending on their maturation step. These factors provide the overall process with the appropriate speed, accuracy, timing and directionality, and participate in its regulation (for reviews on yeast ribosome biogenesis, see de la Cruz et al. 2015; Fernandez-Pevida et al. 2015; Henras et al. 2008; Kressler et al. 2010, 2017; Nerurkar et al. 2015; Sharma and Lafontaine 2015; Watkins and Bohnsack 2012). It is important to stress that the processes of rRNA transcription, pre-rRNA processing and r-subunit assembly not only coincide temporally and spatially, but are also highly interdependent events. Thus, for instance, the mutations that result in elongation-defective RNAP I transcription significantly impair pre-rRNA processing and ribosome assembly. Likewise, either derepression or attenuation of RNAP I activity leads to an equivalent response on the transcription regulation of r-protein genes by RNAP II and 5S rRNA by RNAP III (Laferte et al. 2006; Schneider et al. 2007). Different subsets of essential ribosome assembly factors are also required for optimal rRNA transcription and/or pre-rRNA stability (Gallagher et al. 2004). The stability of nascent pre-rRNAs is also strongly affected upon the depletion of most r-proteins (i.e. Deshmukh et al. 1993; Rosado et al. 2007).
Ribosome biogenesis is a crucial process for growth and constitutes the major consumer of cellular resources. S. cerevisiae contains about 200,000 ribosomes per cell (von der Haar 2008). Ribosome biogenesis involves nearly 65–70% of global cellular transcriptional activity since 60% of all transcription is devoted to rRNA, 10% of all RNAP II transcription is dedicated to r-protein and ribosome assembly factor genes, and it can be assumed that about 30% of all RNAP III transcription is dedicated to 5S rRNA and MRP RNA genes. About 90% of all mRNA splicing, and approximately 30% of global translation, are employed for ribosome biogenesis in rapidly growing yeast cells (Metzl-Raz et al. 2017; Pelechano and Perez-Ortin 2010; Warner 1999). Thus it is not surprising to realise that the synthesis of ribosomes is subjected to such stringent regulation to ensure correct ribosome manufacture with a wide variety of environmental and metabolic changes, and intracellular insults. Regulation occurs at all different gene expression flux levels, allows the proper stoichiometry of all ribosomal components and coordinates optimal production and the activity of the many factors involved in the assembly of r-subunits (Fig. 1). The regulatory pathways that manage imbalanced ribosome production or misassembled ribosomal precursors and subunits are particularly interesting as they are normally linked to human disease, including cancer (Teng et al. 2013).

Regulation of ribosome biogenesis in Saccharomyces cerevisiae. The TORC1 and PKA pathways channel the regulatory stimuli that regulate ribosome biogenesis. They control the transcription of the RP and RiBi regulons and rDNA. Posttranscriptional regulation is also exerted at different levels, including pre-rRNA processing, ribosome assembly, translation and ribosomal subunits degradation. On top of this, the actual performance of ribosome biosynthesis provides feedback on RP and assembly genes (RBB regulon)
Co-transcriptional and post-transcriptional pre-rRNA processing
It is well known that ribosome content in yeast cells is proportional to the growth rate. Ribosome production is controlled at the level of synthesis of both pre-rRNAs and r-proteins (Kief and Warner 1981). The yeast genome contains 100–200 copies in tandem of ribosomal DNA (rDNA). The rDNA copy number can vary, and the number of active/repressed copies can change in response to variations in environmental conditions (Dammann et al. 1993). This regulation involves the chromatin alterations carried out by histone deacetylase Rpd3 and the FACT complex (Johnson et al. 2013). However, it has been proposed that regulation of rRNA synthesis is not achieved by varying the number of active rDNA gene copies, but mainly by the actual number of RNAP I molecules that initiate (French et al. 2003) and successfully elongate (Zhang et al. 2010).
In eukaryotes, ribosome assembly begins co-transcriptionally. This was first evidenced by visualising the so-called terminal knobs on Miller chromatin spreads by electron microscopy, and by the demonstration that those knobs correspond to the ribonucleoprotein complexes that carry out the compaction of the earliest 90S pre-ribosomal particles on nascent 35S pre-rRNA (Mougey et al. 1993). The formation of these particles occurs in a stepwise and hierarchical manner (Dragon et al. 2002; Gallagher et al. 2004; Perez-Fernandez et al. 2007; Wery et al. 2009). Early 90S pre-ribosomal particles also contain a substantial number of 40S r-proteins, which assemble in a defined order (de la Cruz et al. 2015, and references therein). Moreover, pre-rRNA modifications on the nascent pre-rRNA occur co-transcriptionally, as evidenced by fast kinetic labelling analyses (Kos and Tollervey 2010). In contrast in higher eukaryotes, pre-rRNA processing occurs almost exclusively post-transcriptionally (discussed in Osheim et al. 2004; Talkish et al. 2016; Turowski and Tollervey 2015). However in yeast, pre-rRNA processing can occur either before or after the transcription of the 35S pre-rRNA is completed (Osheim et al. 2004). Quantitative measurements of pre-rRNA processing indicate that about 70% of all nascent pre-rRNA transcripts are co-transcriptionally cleaved at site A2 in early exponentially yeast growing cells (Kos and Tollervey 2010; Osheim et al. 2004). Among pre-rRNA processing reactions, cleavage at site A2 is special because it separates the intermediates on the large and small r-subunit synthesis pathways, which apparently follow independent nuclear maturation (Fernandez-Pevida et al. 2015). This cleavage does not take place immediately after the transcription of site A2, but instead appears to occur when RNAP I has travelled into the 25S rDNA about 1.5 kb downstream of this site (Axt et al. 2014; Osheim et al. 2004). Cleavage of the nascent transcript at site A2 releases the earliest 90S pre-ribosomal particles, and an incipient pre-60S r-particle starts to form in the 5′ region of nascent 27SA2 pre-rRNA before transcription termination (Osheim et al. 2004).
Interestingly, the efficiency of the co-transcriptional cleavage of pre-rRNA strongly diminishes when cell growth declines or upon a short temperature shift to 37 °C (Osheim et al. 2004). The co-transcriptional cleavage of nascent pre-rRNA is inhibited upon the depletion or inactivation of specific 60S r-subunit biogenesis factors, such as Drs1, Rrp5 or Rat1 (Axt et al. 2014; Lebaron et al. 2013; Talkish et al. 2016). These phenomena clearly suggest that a link between pre-rRNA processing and ribosome assembly may exist (Talkish et al. 2016). It seems likely that the formation of some high-order structures at the 5′ end region of upcoming 27SA2 pre-rRNA is required for co-transcriptional cleavage to occur. While many early 60S r-subunit assembly factors are indirectly required, specific factors may play a direct role in promoting the co-transcriptional cleavage at site A2. For example, recent data indicate that Rrp5 binds to an evolutionarily conserved stem-loop located 3′ adjacent to site A2 in ITS1, which suggests that the binding of Rrp5 to nascent pre-ribosomal particles is necessary to define the correct timing of co-transcriptional pre-rRNA processing (Lebaron et al. 2013). It is feasible to imagine that upon the depletion of an early 60S r-subunit biogenesis factor, the improper folding of pre-rRNA around site A2, and the incapacity of loading or activation of the required nuclease(s), would delay cleavage at site A2 and permit the formation of a full-length 35S pre-rRNA product. Interestingly, the trans-acting factors required for the late steps in the formation of 60S r-subunits, which normally associate only with late pre-60S r-particles, appear to be associated with 35S pre-rRNA-containing particles when pre-rRNA processing occurs post transcriptionally (Talkish et al. 2016). Under these conditions, it seems that cleavage at site A3 tends to occur prematurely, which leads to the formation of a substantial amount of 23S pre-rRNA-containing particles, whose further maturation to productive 40S r-subunits is controversial (Kos-Braun et al. 2017; Talkish et al. 2016).
The efficiency of the co-transcriptional cleavage of pre-rRNA diminishes not only as cell growth slows down in the stationary phase, but also following a long list of environmental stresses, such as heat or cold shock, low pH, osmotic or oxidative stress, or notably as a result of nutrient limitation or rapamycin treatment (Kos-Braun et al. 2017; Talkish et al. 2016). The mechanism that lies behind this observation has remained unknown until very recently (Kos-Braun et al. 2017). The Koš laboratory has recently shown that 35S pre-rRNA can follow a distinct processing pathway when cells pass the diauxic transition, are starved, or are exposed to different environmental stresses. Under these conditions, the synthesis of pre-rRNA strongly reduces, but is not completely inhibited, and it follows post-transcriptional processing. This pathway has, however, the singularity that cleavage of 35S pre-rRNA occurs practically exclusively at site A3 and leads to unproductive 23S and 27SA3 pre-rRNAs, which are not processed, and thereby, do not finally yield mature rRNAs within newly synthesised ribosomes (Kos-Braun et al. 2017). This process is fully reversible as soon as the growth conditions go back to being optimal. Interestingly, an elegant genetic approach has demonstrated that the switch to this peculiar processing pathway is regulated at a post-transcriptional level and is specifically dependent on the activities of kinases CK2 and TORC1, but is independent of TOR effectors, such as Sch9 or Tap42 (Kos-Braun et al. 2017). As the TOR pathway controls many aspects of ribosome biogenesis, Kos-Braun and Koš carefully rule out that the switch to this peculiar pathway is due to the down-regulation of either RNAP I transcription or the side effect of the repression of the transcription of most r-protein or RiBi genes as a result of TOR inactivation following low nutrients or stress. Instead, the switch is the result of the activation of a novel post-transcriptional regulatory mechanism that depends on TORC1 and CK2, but whose downstream effectors, responsible for executing the switch, remain unknown. Interestingly, the CK2 kinase that consists of four subunits, Cka1, Cka2, Ckb1 and Ckb2, is a component of the so-called CURI complex, which is also composed of ribosome assembly factors Utp22 and Rrp7, and the transcriptional co-activator Ifh1 (Rudra et al. 2007). Whether coordination exists between the role of CK2 kinase in the CURI complex and as a regulator of the switch to this non-productive pre-rRNA processing pathway is an intriguing question that still needs further clarification. Finally, Kos-Braun and Koš discuss the rationalisation of a regulatory mechanism based on an apparent waste of valuable cellular resources: maintenance of pre-rRNA transcription, but at a low rate, and the execution of pre-rRNA processing that leads to non-productive pre-rRNA molecules (Kos-Braun and Kos 2017). These authors conclude that this manner of down-regulating ribosome biogenesis preserves the yeast nucleolar structure, and also impedes both the diffusion of ribosome assembly factors out of the nucle(ol)us and the full repression of r-protein synthesis, and thus allows cells to rapidly revert to co-transcriptional pre-rRNA processing and the formation of functional r-subunits as soon as the environmental conditions are favourable again.
This novel process is not the sole example of post-transcriptional regulation by TORC1 in ribosome biogenesis. Inactivation of TORC1 has also been described to tether the nucleoplasmic/cytoplasmic GTPase Nog1 to the nucleolus, and to thus inhibit the last steps of 60S r-subunit maturation and transport (Honma et al. 2006). Similarly, TORC1 regulates the subcellular localisation of nucleoplasmic 40S r-subunit assembly factors Dim2 and Rrp12, which are also trapped in the nucleolus upon its inactivation (Vanrobays et al. 2008). Finally, inactivation of TORC1 has also been reported to induce both a rapid cytoplasmic turnover of mature ribosomes, which does not imply autophagy (Pestov and Shcherbik 2012), and ribophagy (Waliullah et al. 2017).
RP and RiBi regulons: common nutritional control with different transcription factors
About one-tenth of yeast protein-coding genes encode ribosome transacting factors and r-proteins. Considering their associated non-coding RNAs (Beilharz 2016), ribosome-related genes represent a very substantial proportion of RNA pol II-dependent transcription. Their expression is tightly co-regulated and responds collectively to a variety of nutritional and environmental stimuli. These genes are referred to as members of RiBi (Ribosome Biogenesis) (Jorgensen et al. 2004; Wade et al. 2006; Wu et al. 2002a) and RP (Ribosomal Protein) regulons (Warner 1989, 1999; Warner et al. 1985). Notably, the RiBi regulon not only encompasses rRNA processing and ribosome assembly factors, but also a number of other genes that are functionally connected with ribosome biogenesis and function, such as those which encode translation factors, aminoacyl-tRNA synthetases, tRNA-modifying factors, the enzymes involved in nucleotide metabolism, and the subunits of RNAP I and III (Jorgensen et al. 2002, 2004). Both RP and RiBi regulons display very analogous expression profiles and respond similarly to different environmental conditions (i.e. Gasch et al. 2000; Ho and Gasch 2015) or genetic perturbations (i.e. Miyoshi et al. 2003; Wan et al. 2015). However, RP genes are expressed at a much higher level than RiBi ones and exert some independent regulatory features (Jorgensen et al. 2004; Wade et al. 2006; Wu et al. 2002b).
The regulation of RP and RiBi regulons by nutritional signals or different stresses is channelled mainly through the TORC1 and Protein kinase A (PKA) pathways (Lempiainen and Shore 2009; Loewith and Hall 2011; Martin et al. 2004; Powers and Walter 1999). TORC1 is known to indirectly regulate 35S pre-rRNA production since r-proteins are required to avoid the degradation of pre-rRNAs (Reiter et al. 2011). Long-term inactivation of TORC1 also results in a proteasome-dependent degradation of Rrn3, an RNAP I initiation factor (Philippi et al. 2010), and in the dephosphorylation of Maf1, which interacts with RNAP III and represses the production of pre-rRNA 5S and other RNAP III-dependent transcripts (i.e. Wei and Zheng 2009 and references therein).
TORC1 regulates the transcription of RP genes by modulating the association between DNA-binding protein Fhl1 and either Ifh1 or Crf1. Fhl1 is constitutively bound to RP gene promoters and this binding is facilitated by Rap1 (Rudra et al. 2007). When TORC1 is active, Ifh1 is phosphorylated and binds Fhl1 at the promoters of RP genes to stimulate their transcription, whereas Crf1 remains cytoplasmic. Inhibition of TORC1 results in the phosphorylation of Crf1, which enters the nucleus and displaces Ifh1 from the RP gene promoters to repress transcription (Martin et al. 2004; Schawalder et al. 2004; Zhao et al. 2006). Strikingly, transcription repression by the inactivation of TORC1 still operates in the absence of Crf1 (Martin et al. 2004; Schawalder et al. 2004; Zhao et al. 2006), which thus suggests additional regulators, such as Sfp1 or Sch9. The zinc-finger Sfp1 protein, a functional analogue of c-Myc in yeast, is a general transcriptional activator of the RP regulon, whose function depends on TORC1 (Fingerman et al. 2003; Jorgensen et al. 2004; Lempiainen and Shore 2009; Marion et al. 2004).
Another positive effector of the RP regulon is kinase Sch9, the putative orthologue of mammalian S6K1 (Jorgensen et al. 2004), which also plays an important role in the positive regulation of RiBi genes by TORC1 (Loewith and Hall 2011). Indeed Sch9 phosphorylates transcription factors Stb3 and Tod6/Dot6. When TORC1 is inactivated, Stb3 and Dot6/Tod6 are dephosphorylated, which permits them to bind to the promoters of RiBi genes and to recruit histone acetyltransferase complex Rpd3L to thus repress their transcription (Huber et al. 2011b). Sfp1 also contributes to the positive regulation of RiBi genes, in parallel to Sch9 (Jorgensen et al. 2004). This parallel control by Sfp1 and Sch9 indicates the certain level of regulatory redundancy that characterises regulons RP and RiBi (Jorgensen et al. 2004).
Despite the similar expression profiles of RP and RiBi genes in response to nutritional and environmental stimuli, the DNA elements and factors that control the transcription of these two regulons are clearly not the same (reviewed in Bosio et al. 2011). Generally whereas the RP genes are preferentially regulated in a positive manner, the RiBi ones are controlled mainly by repression. Transcription of RP genes is regulated by DNA binding activators Rap1 and Fhl1 (Berger et al. 2007; Hall et al. 2006; Li et al. 1999), although these genes are also subjected to repression, which is mediated by the binding of Stb3 to the T-rich elements of their promoters (Liko et al. 2007) and by the silencing domain of Rap1 (Buck and Shore 1995). This domain is also responsible for the repression of RP genes in response to a secretory defect, as previously shown since a functional secretory pathway is essential for continued yeast ribosome biogenesis (Mizuta and Warner 1994).
Transcription of RiBi genes is controlled mainly by the PAC (RNA Polymerase A and C promoters) and RRPE (ribosomal RNA-processing element) sequence elements found in their promoter regions (Bosio et al. 2011; Hughes et al. 2000; Jorgensen et al. 2004; Wade et al. 2001). The PAC element is recognised by transcription repressors Dot6 and Tod6 (Badis et al. 2008; Freckleton et al. 2009; Zhu et al. 2009), while the RRPE element is bound by the repressor Stb3 (Huber et al. 2011a; Liko et al. 2007; Lippman and Broach 2009; McKnight et al. 2015). It has been recently shown that the promoters of most RiBi genes also contain one binding site or more for Abf1, Reb1, and, less frequently, Rap1 and Tbf1, which are four transcription activators that belong to the so-called general regulatory factors (Bosio et al. 2017). In fact the association of these factors to the promoters of the RiBI genes is required for full expression in a rich medium, and for their modulation in response to glucose starvation, in a manner that counteracts the Rpd3L-dependent repression entailed by the above-mentioned negative factors (Bosio et al. 2017). Interestingly, it has been suggested that the frequent presence of Abf1 binding sites in the promoters of mitochondrial RP genes might facilitate the co-regulation of the whole network of growth-related genes in yeast cells (Fermi et al. 2017). In short, although repression dominates RiBi gene regulation and activation features RP gene control, both ribosome-related regulons combine positive and negative control systems that coordinately contribute to their tight nutritional regulation.
Apart from the modulation of RNAP II recruitment to promoters, the transcriptional regulation of RP and RiBi regulons also occurs at the elongation level, as formerly shown for yeast cells that grow in different carbon sources (Pelechano et al. 2009). Such transcription elongation regulation depends on PKA and is channelled, at least partially, through the above-mentioned silencing domain of Rap1 (Pelechano et al. 2009). The importance of transcription elongation in RP gene expression has also been evidenced by the high dependence that these genes have on Dst1, the yeast TFIIS transcription elongation factor, under transcriptional stress conditions (Gomez-Herreros et al. 2012).
Coupling rRNA and r-protein production
As previously mentioned, to ensure efficient ribosome biosynthesis, an equimolar production of the different ribosomal components is required. In fact, the imbalance between r-proteins and rRNA production is characteristic of the so-called nucleolar-stress response and leads to the impairment of cell growth and G1 cell cycle arrest in both yeast (Gomez-Herreros et al. 2013) and mammalian cells (see Boulon et al. 2010; Pelava et al. 2016 and references therein). This balance in yeast is achieved by the coordinated transcription of pre-rRNA and RP genes, which extends beyond the stimulation of the transcription of both rRNA and r-protein genes by the TOR signalling pathway. Several transcription factors have been proposed to play a role in coordinating the transcription of rRNA and RP genes. For instance, Hmo1, which is present in the rDNA locus and in the promoter of a subset of RP genes (Berger et al. 2007; Hall et al. 2006; Lempiainen and Shore 2009), is simultaneously released from both regions upon TORC1 inhibition (Berger et al. 2007).
A key element for coupling rRNA and r-proteins synthesis, but not RiBi regulon proteins, is Ifh1, which is a co-activator of RP gene transcription (Martin et al. 2004; Rudra et al. 2007; Schawalder et al. 2004). Ifh1 is found to be associated with transcriptional factor Fhl1 at the promoters of RP genes when these are actively transcribed (Martin et al. 2004; Rudra et al. 2005; Schawalder et al. 2004), or it is alternatively found in the nucleolus in a complex so-called CURI, which comprises Ifh1 and the UTP-C subcomplex formed by the four subunits of CK2, and by ribosome assembly factors Utp22 and Rrp7 (Krogan et al. 2004; Rudra et al. 2007). A model for coupling rRNA and r-protein production was initially suggested by the Warner laboratory (Rudra et al. 2007) which has been recently validated by the Shore laboratory (Albert et al. 2016). In this model, Ifh1 is sequestered within the CURI complex to repress RP gene transcription when rRNA transcription is reduced. This, under optimal growth conditions, Ifh1 is bound to the promoter of RP genes to facilitate transcription, while the UTP-C subcomplex is co-transcriptionally engaged in early ribosome assembly steps. Under nutrient starvation (TORC1 inhibition), the transcription of rRNA and RP genes slow-down, Ifh1 is released from the RP gene promoters, and concomitantly the UTP-C subcomplex is also released from rDNA, which results in the formation of the CURI complex after the binding of Ifh1, presumably through Utp22 (Albert et al. 2016; Rudra et al. 2007). Although the CURI complex sequesters Ifh1, it is apparently not responsible of the direct release of Ifh1 from RP promoters, but is responsible of stabilizing it. Indeed TORC1 inhibition follows in the absence of Utp22 or upon conditions where RNAP I transcription is not fully down-regulated, which keeps Utp22 involved in ribosome biogenesis. Then Ifh1 can rapidly release from the promoter of RP genes upon TORC1 inactivation, but immediately returns to these promoters as it is unable to establish a long-term interaction within the CURI complex (Albert et al. 2016). The rapid release of Ifh1 depends on the inactivation of the Sch9 kinase, which simultaneously leads to the inhibition of RNAP I transcription. The reduction in RNAP I transcription, but also that of RP genes by RNAP II, makes enough free UTP-C subcomplex available to facilitate a long-term retention of Ifh1 (Albert et al. 2016). Strikingly, the UTP22 gene is one of the few non-r-protein genes whose transcription is dependent on Ifh1, and thus provides a feedback mechanism to tune this particular regulation (Albert et al. 2016).
In addition to this fine mechanism to coordinate r-protein gene transcription and early ribosome assembly steps, it is worth mentioning other post-transcriptional events that balance the accumulation of r-proteins versus that of rRNAs during ribosome biogenesis, including the autogenously regulation of mRNA splicing or translation, and an efficient turnover of those r-proteins that are unassembled into ribosomes. Regarding the first mechanism, the transcripts of several RP genes contain potential structures that mimic their respective binding sites in rRNA (i.e. Fewell and Woolford 1999; Vilardell et al. 2000; Vilardell and Warner 1997). Thus when an imbalance on ribosome biogenesis leads to excess r-proteins versus rRNAs, the former can bind with a low affinity to their own transcripts by inhibiting their splicing and further translation (Dabeva and Warner 1993; Vilardell and Warner 1994). Regarding the second mechanism, it is well known that overproduced r-proteins are efficiently eliminated by rapid degradation (elBaradi et al. 1986; Gorenstein and Warner 1977; Maicas et al. 1988; Tsay et al. 1988; Warner et al. 1985). By such a system, yeast cells avoid the negative effects of the accumulation of r-proteins, which are highly basic and especially prone to undergo non-specific interactions and aggregation when not bound to rRNA (Jakel et al. 2002). It has been recently demonstrated that degradation of excess r-proteins occurs in the nucleus and involves a ubiquitin–proteasome-depending system that comprises ubiquitin-conjugating enzymes Ubc4 and Ubc5 and ubiquitin-ligase Tom1 (Sung et al. 2016a, b). Interestingly, it appears that Tom1 selectively recognises and ubiquitinates residues of the r-proteins that are accessible only in the unassembled state, but are completely unreachable in the mature ribosome (Sung et al. 2016a). Finally, active surveillance systems also exist to degrade the fraction of pre-rRNAs that is not productively engaged in pre-ribosomal particles. These systems comprise the nuclear TRAMP, the RNA exosome and Nrd1–Nab3–Sen1 complexes (Allmang et al. 2000; Dez et al. 2006; Lepore and Lafontaine 2011; Wery et al. 2009).
Feedback regulation of ribosome trans-acting factors
RiBi factors also exhibit a feedback regulation that operates within a more restricted network of genes, which are directly related to ribosome assembly (Gomez-Herreros et al. 2017). We recently analysed the transcriptomic consequences of deleting 59 non-essential genes that encode distinct r-proteins, RNAP I subunits, or the protein factors involved in pre-rRNA processing and ribosome assembly. Unlike the mutations that affect general regulators like Sfp1, Sch9 and Hmo1, which globally impair the ribosome biogenesis process, many of the analysed null mutants only alter the expression of a limited number of genes within regulons RiBi and RP (Gomez-Herreros et al. 2017). For instance, we observed that the deletions of individual RP genes up-regulated a large subset of genes that encode ribosome assembly factors, and did not affect the RP genes themselves, nor the subset of the RiBi regulon genes that encode aminoacyl-tRNA synthetases or those involved in nucleotide metabolism (Gomez-Herreros et al. 2017). A similar specific up-regulation of assembly genes was observed upon the deletion of specific ribosome assembly factors such as Rsa1 and Utp30.
We also found that some deletion mutants changed the expression of a very specific subset of assembly genes. Interestingly, the set of genes that specifically respond to ribosome assembly insults better fits the RRB (rRNA and Ribosome Biosynthesis) regulon than the wider RiBi regulon (Gomez-Herreros et al. 2017). The RRB regulon was originally defined as the set of co-regulated genes directly related to ribosome assembly (Wade et al. 2001, 2006). Its extension to the RiBi regulon was done by analysing the transcriptomic responses to nutritional and environmental changes, as well as to alterations produced by genetic perturbations of the TOR signalling pathway (Jorgensen et al. 2004). An example of these deletion mutants is dbp7∆, which lacks a putative RNA helicase, required for 60S r-subunit biogenesis (Daugeron and Linder 1998). dpb7∆ produced a strong up-regulation of a very limited set of assembly genes. These up-regulated genes encode factors that contribute specifically to 60S r-subunit biogenesis and some have a direct functional link with Dbp7 (Gomez-Herreros et al. 2017). These, and other complementary results, show that a feedback system specifically regulates RRB genes, which is highly mutant specific. Thus the regulation in response to assembly perturbation is expected to follow its own regulatory dynamics within the frame of genes that directly participate in this process.
The clustering analysis of transcriptomic data allowed us to detect a regulatory structure within the RRB regulon, which divides into two main groups of genes. The first group includes the factors present in 90S and pre-40S r-particles, which are required for 40S r-subunit biogenesis. The second is enriched in the ribosome biogenesis factors present in pre-60S r-particles, required for 60S r-subunit biogenesis (Fig. 2; Gomez-Herreros et al. 2017). While no obvious rules for regulation were detected, it is worth stressing that most of the mutations that specifically induce the genes which belong to the second group are themselves linked to the assembly of 60S r-subunits (Gomez-Herreros et al. 2017). Strikingly, the distribution of PAC and RRPE elements differs in the genes of each group; thus RPPE elements are less frequently present in the promoter regions of the genes that belong to the second group. In contrast, both clusters exhibit comparable frequencies of PAC elements (Gomez-Herreros et al. 2017). Since the Dieci laboratory has recently published an updated map of the regulatory elements present in RiBi genes, including the binding sites of four transcriptional activators (see above, Bosio et al. 2017), we investigated whether the asymmetry between the two main clusters of RBB genes that we identified could be extended to other regulators. As shown in Fig. 2, we could not find a significant bias for the presence of the binding sites for individual activators. Intriguingly however, we found that repressors significantly more predominated the activators in the first cluster than in the second one. These results indicate that the heterogeneity of the regulatory elements within the RBB regulon may well explain its internal regulatory structure and the plasticity of the feedback control that we have just revealed.

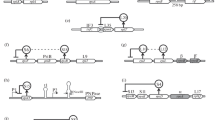
Internal regulatory structure of RBB genes. a The genes that encode pre-rRNA processing and ribosome assembly factors distribute into two main clusters according to the transcriptome of the set of ribosome-related mutants analysed in (Gomez-Herreros et al. 2017). Within these clusters, genes are ordered according to the regulatory elements present in their upstream regions (Bosio et al. 2017). Only the elements that fit the consensus or have a single mismatch were considered. The difference between the number of activating and repressive elements is also indicated (Activ–Repr). The red/purple code distinguishes the factors that operate in the 90S or pre-40S particles from those that act in pre-60S. b The genes dominated by repressive elements are significantly more abundant in Cluster I. c Presence of 90S/pre-40S versus pre-60S factors significantly differs in the two clusters. p values correspond to the Chi square test
Another aspect that seems to characterise this feedback regulatory network of the ribosome assembly process is that it involves the modulation of the elongation step of transcription. Our experiments, which adopted an in vivo depletion strategy, show that shortly after ribosome assembly impairment, a substantial set of RBB genes increases the density of RNAP II molecules that are able to produce a run-on signal, with no parallel change in total RNAP II occupancy (Gomez-Herreros et al. 2017). This pattern reflects the decrease presence of backtracked RNAP II molecules, which suffices to explain the moderate increase in mRNA levels (Gomez-Herreros et al. 2017). This change in transcription elongation involves the recruitment of chromatin factor Spt6 to regulated genes, which contributes to the optimal dynamics of histones during activated transcription (Gomez-Herreros et al. 2017). Two main clues can explain this control at the elongation level. Since it operates in the gene body, it does not require any change in the formation of the preinitiation complex on promoter regions. Therefore, it is completely compatible with any other regulation at this level. At the same time, transcription elongation alterations are related to changes in mRNA stability. The increased expression of RiBi and RP regulons with growth is dictated by the relative changes of transcription and mRNA degradation rates (Chavez et al. 2016; Garcia-Martinez et al. 2016). Within the different steps of transcription, elongation has been proposed to participate in the cross-talk with mRNA degradation (Gupta et al. 2016; Haimovich et al. 2013; Medina et al. 2014). In agreement with this proposal, we find that the transcription elongation changes in RBB genes are linked to parallel changes in mRNA stability (Gomez-Herreros et al. 2017).
Conclusions
In this review, we focus on the main aspects of the different regulatory circuits that govern the synthesis of productive ribosomes in S. cerevisiae. Given that a vast majority of cellular resources are dedicated to the biogenesis of ribosomes in rapidly growing yeast cells, it is not surprising that evolution has led to the selection of different and redundant mechanisms for the regulation of ribosome production, which operates at all different gene expression levels in response to changing environmental conditions and intracellular insults. Although some overlapping/redundancy is clearly present between different regulatory pathways, most proceed in parallel to each other, but maintain a certain degree of coordination. Some of these systems operate globally and are driven by well-known master regulators, such as TORC1 and its downstream effectors Sch9, Sfp1 or Crf1. Others are more specific and serve as fine-tuning mechanisms of regulation, and often imply both feedback loops and feed-forward situations. We have attempted to summarise the important advances made in the field of ribosome biogenesis regulation that have been described in recent years. Many questions, however, remain unsolved; among others, the elements that work in the coordination between different processes in ribosome biogenesis are still largely unknown or described at a low resolution. Additionally, many regulators or their downstream targets that operate on these pathways have not yet been identified. Finally, it is important to examine the degree of conservation that these systems show in higher eukaryotes as a way to understand how dysregulation of human ribosome biogenesis could lead to pathogenesis.
References
Albert B, Knight B, Merwin J, Martin V, Ottoz D, Gloor Y, Bruzzone MJ, Rudner A, Shore D (2016) A molecular titration system coordinates ribosomal protein gene transcription with ribosomal RNA synthesis. Mol Cell 64:720–733. doi:10.1016/j.molcel.2016.10.003
Allmang C, Mitchell P, Petfalski E, Tollervey D (2000) Degradation of ribosomal RNA precursors by the exosome. Nucleic Acids Res 28:1684–1691
Axt K, French SL, Beyer AL, Tollervey D (2014) Kinetic analysis demonstrates a requirement for the Rat1 exonuclease in cotranscriptional pre-rRNA cleavage. PloS One 9:e85703. doi:10.1371/journal.pone.0085703
Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, Gebbia M, Talukder S, Yang A, Mnaimneh S, Terterov D, Coburn D, Li Yeo A, Yeo ZX, Clarke ND, Lieb JD, Ansari AZ, Nislow C, Hughes TR (2008) A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell 32:878–887. doi:10.1016/j.molcel.2008.11.020
Beilharz TH (2016) Understanding the regulation of coding and noncoding transcription in cell populations. Curr Genet 62:317–319. doi:10.1007/s00294-015-0547-1
Berger AB, Decourty L, Badis G, Nehrbass U, Jacquier A, Gadal O (2007) Hmo1 is required for TOR-dependent regulation of ribosomal protein gene transcription. Mol Cell Biol 27:8015–8026. doi:10.1128/MCB.01102-07
Bosio MC, Negri R, Dieci G (2011) Promoter architectures in the yeast ribosomal expression program. Transcription 2:71–77. doi:10.4161/trns.2.2.14486
Bosio MC, Fermi B, Spagnoli G, Levati E, Rubbi L, Ferrari R, Pellegrini M, Dieci G (2017) Abf1 and other general regulatory factors control ribosome biogenesis gene expression in budding yeast. Nucleic Acids Res 45:4493–4506. doi:10.1093/nar/gkx058
Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI (2010) The nucleolus under stress. Mol Cell 40:216–227. doi:10.1016/j.molcel.2010.09.024
Buck SW, Shore D (1995) Action of a RAP1 carboxy-terminal silencing domain reveals an underlying competition between HMR and telomeres in yeast. Genes Dev 9:370–384
Chavez S, Garcia-Martinez J, Delgado-Ramos L, Perez-Ortin JE (2016) The importance of controlling mRNA turnover during cell proliferation. Curr Genet 62:701–710. doi:10.1007/s00294-016-0594-2
Dabeva MD, Warner JR (1993) Ribosomal protein L32 of Saccharomyces cerevisiae regulates both splicing and translation of its own transcript. J Biol Chem 268:19669–19674
Dammann R, Lucchini R, Koller T, Sogo JM (1993) Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res 21:2331–2338
Daugeron MC, Linder P (1998) Dbp7p, a putative ATP-dependent RNA helicase from Saccharomyces cerevisiae, is required for 60S ribosomal subunit assembly. RNA 4:566–581
de la Cruz J, Karbstein K, Woolford JL Jr (2015) Functions of ribosomal proteins in assembly of eukaryotic ribosomes in vivo. Annu Rev Biochem 84:93–129. doi:10.1146/annurev-biochem-060614-033917
Deshmukh M, Tsay YF, Paulovich AG, Woolford JL Jr (1993) Yeast ribosomal protein L1 is required for the stability of newly synthesized 5S rRNA and the assembly of 60S ribosomal subunits. Mol Cell Biol 13:2835–2845
Dez C, Houseley J, Tollervey D (2006) Surveillance of nuclear-restricted pre-ribosomes within a subnucleolar region of Saccharomyces cerevisiae. EMBO J 25:1534–1546. doi:10.1038/sj.emboj.7601035
Dragon F, Gallagher JE, Compagnone-Post PA, Mitchell BM, Porwancher KA, Wehner KA, Wormsley S, Settlage RE, Shabanowitz J, Osheim Y, Beyer AL, Hunt DF, Baserga SJ (2002) A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 417:967–970. doi:10.1038/nature00769
elBaradi TT, van der Sande CA, Mager WH, Raue HA, Planta RJ (1986) The cellular level of yeast ribosomal protein L25 is controlled principally by rapid degradation of excess protein. Curr Genet 10:733–739
Fermi B, Bosio MC, Dieci G (2017) Multiple roles of the general regulatory factor Abf1 in yeast ribosome biogenesis. Curr Genet 63:65–68. doi:10.1007/s00294-016-0621-3
Fernandez-Pevida A, Kressler D, de la Cruz J (2015) Processing of preribosomal RNA in Saccharomyces cerevisiae. Wiley Interdiscip Rev RNA 6:191–209. doi:10.1002/wrna.1267
Fewell SW, Woolford JL Jr (1999) Ribosomal protein S14 of Saccharomyces cerevisiae regulates its expression by binding to RPS14B pre-mRNA and to 18S rRNA. Mol Cell Biol 19:826–834
Fingerman I, Nagaraj V, Norris D, Vershon AK (2003) Sfp1 plays a key role in yeast ribosome biogenesis. Eukaryot Cell 2:1061–1068
Freckleton G, Lippman SI, Broach JR, Tavazoie S (2009) Microarray profiling of phage-display selections for rapid mapping of transcription factor-DNA interactions. PLoS Genet 5:e1000449. doi:10.1371/journal.pgen.1000449
French SL, Osheim YN, Cioci F, Nomura M, Beyer AL (2003) In exponentially growing Saccharomyces cerevisiae cells, rRNA synthesis is determined by the summed RNA polymerase I loading rate rather than by the number of active genes. Mol Cell Biol 23:1558–1568
Gallagher JE, Dunbar DA, Granneman S, Mitchell BM, Osheim Y, Beyer AL, Baserga SJ (2004) RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components. Genes Dev 18:2506–2517. doi:10.1101/gad.1226604
Garcia-Martinez J, Delgado-Ramos L, Ayala G, Pelechano V, Medina DA, Carrasco F, Gonzalez R, Andres-Leon E, Steinmetz L, Warringer J, Chavez S, Perez-Ortin JE (2016) The cellular growth rate controls overall mRNA turnover, and modulates either transcription or degradation rates of particular gene regulons. Nucleic Acids Res 44:3643–3658. doi:10.1093/nar/gkv1512
Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO (2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11:4241–4257
Gomez-Herreros F, de Miguel-Jimenez L, Morillo-Huesca M, Delgado-Ramos L, Munoz-Centeno MC, Chavez S (2012) TFIIS is required for the balanced expression of the genes encoding ribosomal components under transcriptional stress. Nucleic Acids Res 40:6508–6519. doi:10.1093/nar/gks340
Gomez-Herreros F, Rodriguez-Galan O, Morillo-Huesca M, Maya D, Arista-Romero M, de la Cruz J, Chavez S, Munoz-Centeno MC (2013) Balanced production of ribosome components is required for proper G1/S transition in Saccharomyces cerevisiae. J Biol Chem 288:31689–31700. doi:10.1074/jbc.M113.500488
Gomez-Herreros F, Margaritis T, Rodriguez-Galan O, Pelechano V, Begley V, Millan-Zambrano G, Morillo-Huesca M, Munoz-Centeno MC, Perez-Ortin JE, de la Cruz J, Holstege FCP, Chavez S (2017) The ribosome assembly gene network is controlled by the feedback regulation of transcription elongation. Nucleic Acids Res 45:9302–9318. doi:10.1093/nar/gkx529
Gorenstein C, Warner JR (1977) Synthesis and turnover of ribosomal proteins in the absence of 60S subunit assembly in Saccharomyces cerevisiae. Mol Gen Genet MGG 157:327–332
Gupta I, Villanyi Z, Kassem S, Hughes C, Panasenko OO, Steinmetz LM, Collart MA (2016) Translational capacity of a cell is determined during transcription elongation via the Ccr4-not complex. Cell Rep 15:1782–1794. doi:10.1016/j.celrep.2016.04.055
Haimovich G, Medina DA, Causse SZ, Garber M, Millan-Zambrano G, Barkai O, Chavez S, Perez-Ortin JE, Darzacq X, Choder M (2013) Gene expression is circular: factors for mRNA degradation also foster mRNA synthesis. Cell 153:1000–1011. doi:10.1016/j.cell.2013.05.012
Hall DB, Wade JT, Struhl K (2006) An HMG protein, Hmo1, associates with promoters of many ribosomal protein genes and throughout the rRNA gene locus in Saccharomyces cerevisiae. Mol Cell Biol 26:3672–3679. doi:10.1128/MCB.26.9.3672-3679.2006
Henras AK, Soudet J, Gerus M, Lebaron S, Caizergues-Ferrer M, Mougin A, Henry Y (2008) The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci CMLS 65:2334–2359. doi:10.1007/s00018-008-8027-0
Ho YH, Gasch AP (2015) Exploiting the yeast stress-activated signaling network to inform on stress biology and disease signaling. Curr Genet 61:503–511. doi:10.1007/s00294-015-0491-0
Honma Y, Kitamura A, Shioda R, Maruyama H, Ozaki K, Oda Y, Mini T, Jeno P, Maki Y, Yonezawa K, Hurt E, Ueno M, Uritani M, Hall MN, Ushimaru T (2006) TOR regulates late steps of ribosome maturation in the nucleoplasm via Nog1 in response to nutrients. EMBO J 25:3832–3842. doi:10.1038/sj.emboj.7601262
Huber A, French SL, Tekotte H, Yerlikaya S, Stahl M, Perepelkina MP, Tyers M, Rougemont J, Beyer AL, Loewith R (2011a) Sch9 regulates ribosome biogenesis via Stb3, Dot6 and Tod6 and the histone deacetylase complex RPD3L. EMBO J 30:3052–3064. doi:10.1038/emboj.2011.221
Huber A, French SL, Tekotte H, Yerlikaya S, Stahl M, Perepelkina MP, Tyers M, Rougemont J, Beyer AL, Loewith R (2011b) Sch9 regulates ribosome biogenesis via Stb3, Dot6 and Tod6 and the histone deacetylase complex RPD3L. EMBO J 30:3052–3064. doi:10.1038/emboj.2011.221
Hughes JD, Estep PW, Tavazoie S, Church GM (2000) Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol 296:1205–1214. doi:10.1006/jmbi.2000.3519
Jakel S, Mingot JM, Schwarzmaier P, Hartmann E, Gorlich D (2002) Importins fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed basic domains. EMBO J 21:377–386. doi:10.1093/emboj/21.3.377
Johnson JM, French SL, Osheim YN, Li M, Hall L, Beyer AL, Smith JS (2013) Rpd3- and spt16-mediated nucleosome assembly and transcriptional regulation on yeast ribosomal DNA genes. Mol Cell Biol 33:2748–2759. doi:10.1128/MCB.00112-13
Jorgensen P, Nishikawa JL, Breitkreutz BJ, Tyers M (2002) Systematic identification of pathways that couple cell growth and division in yeast. Science 297:395–352
Jorgensen P, Rupes I, Sharom JR, Schneper L, Broach JR, Tyers M (2004) A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev 18:2491–2505. doi:10.1101/gad.1228804
Kos M, Tollervey D (2010) Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol Cell 37:809–820. doi:10.1016/j.molcel.2010.02.024
Kos-Braun IC, Kos M (2017) Post-transcriptional regulation of ribosome biogenesis in yeast. Microb Cell 4:179–181. doi:10.15698/mic2017.05.575
Kos-Braun IC, Jung I, Kos M (2017) Tor1 and CK2 kinases control a switch between alternative ribosome biogenesis pathways in a growth-dependent manner. PLoS Biol 15:e2000245. doi:10.1371/journal.pbio.2000245
Kressler D, Hurt E, Bassler J (2010) Driving ribosome assembly. Biochim Biophys Acta 1803:673–683. doi:10.1016/j.bbamcr.2009.10.009
Kressler D, Hurt E, Bassler J (2017) A puzzle of life: crafting ribosomal subunits. Trend Biochem Sci 42:640–654. doi:10.1016/j.tibs.2017.05.005
Krogan NJ, Peng WT, Cagney G, Robinson MD, Haw R, Zhong G, Guo X, Zhang X, Canadien V, Richards DP, Beattie BK, Lalev A, Zhang W, Davierwala AP, Mnaimneh S, Starostine A, Tikuisis AP, Grigull J, Datta N, Bray JE, Hughes TR, Emili A, Greenblatt JF (2004) High-definition macromolecular composition of yeast RNA-processing complexes. Mol Cell 13:225–239
Laferte A, Favry E, Sentenac A, Riva M, Carles C, Chedin S (2006) The transcriptional activity of RNA polymerase I is a key determinant for the level of all ribosome components. Genes Dev 20:2030–2040. doi:10.1101/gad.386106
Lebaron S, Segerstolpe A, French SL, Dudnakova T, de Lima Alves F, Granneman S, Rappsilber J, Beyer AL, Wieslander L, Tollervey D (2013) Rrp5 binding at multiple sites coordinates pre-rRNA processing and assembly. Mol Cell 52:707–719. doi:10.1016/j.molcel.2013.10.017
Lempiainen H, Shore D (2009) Growth control and ribosome biogenesis. Curr Opin Cell Biol 21:855–863. doi:10.1016/j.ceb.2009.09.002
Lepore N, Lafontaine DL (2011) A functional interface at the rDNA connects rRNA synthesis, pre-rRNA processing and nucleolar surveillance in budding yeast. PloS One 6:e24962. doi:10.1371/journal.pone.0024962
Li B, Nierras CR, Warner JR (1999) Transcriptional elements involved in the repression of ribosomal protein synthesis. Mol Cell Biol 19:5393–5404
Liko D, Slattery MG, Heideman W (2007) Stb3 binds to ribosomal RNA processing element motifs that control transcriptional responses to growth in Saccharomyces cerevisiae. J Biol Chem 282:26623–26628. doi:10.1074/jbc.M704762200
Lippman SI, Broach JR (2009) Protein kinase A and TORC1 activate genes for ribosomal biogenesis by inactivating repressors encoded by Dot6 and its homolog Tod6. Proceedings of the National Academy of Sciences of the United States of America 106:19928–19933 doi:10.1073/pnas.0907027106
Loewith R, Hall MN (2011) Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 189:1177–1201. doi:10.1534/genetics.111.133363
Maicas E, Pluthero FG, Friesen JD (1988) The accumulation of three yeast ribosomal proteins under conditions of excess mRNA is determined primarily by fast protein decay. Mol Cell Biol 8:169–175
Marion RM, Regev A, Segal E, Barash Y, Koller D, Friedman N, O’Shea EK (2004) Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proceedings of the National Academy of Sciences of the United States of America 101:14315–14322 doi:10.1073/pnas.0405353101
Martin DE, Soulard A, Hall MN (2004) TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL1. Cell 119:969–979. doi:10.1016/j.cell.2004.11.047
McKnight JN, Boerma JW, Breeden LL, Tsukiyama T (2015) Global promoter targeting of a conserved lysine deacetylase for transcriptional shutoff during quiescence entry. Mol Cell 59:732–743. doi:10.1016/j.molcel.2015.07.014
Medina DA, Jordan-Pla A, Millan-Zambrano G, Chavez S, Choder M, Perez-Ortin JE (2014) Cytoplasmic 5′–3′ exonuclease Xrn1p is also a genome-wide transcription factor in yeast. Front Genet 5:1. doi:10.3389/fgene.2014.00001
Metzl-Raz E, Kafri M, Yaakov G, Soifer I, Gurvich Y, Barkai N (2017) Principles of cellular resource allocation revealed by condition-dependent proteome profiling. eLife 6:e28034. doi:10.7554/eLife.28034
Miyoshi K, Shirai C, Mizuta K (2003) Transcription of genes encoding trans-acting factors required for rRNA maturation/ribosomal subunit assembly is coordinately regulated with ribosomal protein genes and involves Rap1 in Saccharomyces cerevisiae. Nucleic Acid Res 31:1969–1973
Mizuta K, Warner JR (1994) Continued functioning of the secretory pathway is essential for ribosome synthesis. Mol Cell Biol 14:2493–2502
Mougey EB, O’Reilly M, Osheim Y, Miller OL Jr, Beyer A, Sollner-Webb B (1993) The terminal balls characteristic of eukaryotic rRNA transcription units in chromatin spreads are rRNA processing complexes. Genes Dev 7:1609–1619
Nerurkar P, Altvater M, Gerhardy S, Schutz S, Fischer U, Weirich C, Panse VG (2015) Eukaryotic ribosome assembly and nuclear export. Int Rev Cell Mol Biol 319:107–140. doi:10.1016/bs.ircmb.2015.07.002
Osheim YN, French SL, Keck KM, Champion EA, Spasov K, Dragon F, Baserga SJ, Beyer AL (2004) Pre-18S ribosomal RNA is structurally compacted into the SSU processome prior to being cleaved from nascent transcripts in Saccharomyces cerevisiae. Mol Cell 16:943–954. doi:10.1016/j.molcel.2004.11.031
Pelava A, Schneider C, Watkins NJ (2016) The importance of ribosome production, and the 5S RNP-MDM2 pathway, in health and disease. Biochem Soc Trans 44:1086–1090. doi:10.1042/BST20160106
Pelechano V, Perez-Ortin JE (2010) There is a steady-state transcriptome in exponentially growing yeast cells. Yeast 27:413–422. doi:10.1002/yea.1768
Pelechano V, Jimeno-Gonzalez S, Rodriguez-Gil A, Garcia-Martinez J, Perez-Ortin JE, Chavez S (2009) Regulon-specific control of transcription elongation across the yeast genome. PLoS Genet 5:e1000614. doi:10.1371/journal.pgen.1000614
Perez-Fernandez J, Roman A, De Las Rivas J, Bustelo XR, Dosil M (2007) The 90S preribosome is a multimodular structure that is assembled through a hierarchical mechanism. Mol Cell Biol 27:5414–5429. doi:10.1128/MCB.00380-07
Pestov DG, Shcherbik N (2012) Rapid cytoplasmic turnover of yeast ribosomes in response to rapamycin inhibition of TOR. Mol Cell Biol 32:2135–2144. doi:10.1128/MCB.06763-11
Philippi A, Steinbauer R, Reiter A, Fath S, Leger-Silvestre I, Milkereit P, Griesenbeck J, Tschochner H (2010) TOR-dependent reduction in the expression level of Rrn3p lowers the activity of the yeast RNA Pol I machinery, but does not account for the strong inhibition of rRNA production. Nucleic Acids Res 38:5315–5326. doi:10.1093/nar/gkq264
Powers T, Walter P (1999) Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signaling pathway in Saccharomyces cerevisiae. Mol Biol Cell 10:987–1000
Reiter A, Steinbauer R, Philippi A, Gerber J, Tschochner H, Milkereit P, Griesenbeck J (2011) Reduction in ribosomal protein synthesis is sufficient to explain major effects on ribosome production after short-term TOR inactivation in Saccharomyces cerevisiae. Mol Cell Biol 31:803–817. doi:10.1128/MCB.01227-10
Rosado IV, Kressler D, de la Cruz J (2007) Functional analysis of Saccharomyces cerevisiae ribosomal protein Rpl3p in ribosome synthesis. Nucleic Acids Res 35:4203–4213. doi:10.1093/nar/gkm388
Rudra D, Zhao Y, Warner JR (2005) Central role of Ifh1p-Fhl1p interaction in the synthesis of yeast ribosomal proteins. EMBO J 24:533–542. doi:10.1038/sj.emboj.7600553
Rudra D, Mallick J, Zhao Y, Warner JR (2007) Potential interface between ribosomal protein production and pre-rRNA processing. Mol Cell Biol 27:4815–4824. doi:10.1128/MCB.02062-06
Schawalder SB, Kabani M, Howald I, Choudhury U, Werner M, Shore D (2004) Growth-regulated recruitment of the essential yeast ribosomal protein gene activator Ifh1. Nature 432:1058–1061. doi:10.1038/nature03200
Schneider DA, Michel A, Sikes ML, Vu L, Dodd JA, Salgia S, Osheim YN, Beyer AL, Nomura M (2007) Transcription elongation by RNA polymerase I is linked to efficient rRNA processing and ribosome assembly. Mol Cell 26:217–229. doi:10.1016/j.molcel.2007.04.007
Sharma S, Lafontaine DL (2015) ‘View from a bridge’: a new perspective on eukaryotic rRNA base modification. Trend Biochem Sci 40:560–575. doi:10.1016/j.tibs.2015.07.008
Sung MK, Porras-Yakushi TR, Reitsma JM, Huber FM, Sweredoski MJ, Hoelz A, Hess S, Deshaies RJ (2016a) A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. eLife 5:e19105. doi:10.7554/eLife.19105
Sung MK, Reitsma JM, Sweredoski MJ, Hess S, Deshaies RJ (2016b) Ribosomal proteins produced in excess are degraded by the ubiquitin–proteasome system. Mol Biol Cell 27:2642–2652. doi:10.1091/mbc.E16-05-0290
Talkish J, Biedka S, Jakovljevic J, Zhang J, Tang L, Strahler JR, Andrews PC, Maddock JR, Woolford JL Jr (2016) Disruption of ribosome assembly in yeast blocks cotranscriptional pre-rRNA processing and affects the global hierarchy of ribosome biogenesis. RNA 22:852–866. doi:10.1261/rna.055780.115
Teng T, Thomas G, Mercer CA (2013) Growth control and ribosomopathies. Curr Opin Genet Dev 23:63–71. doi:10.1016/j.gde.2013.02.001
Tsay YF, Thompson JR, Rotenberg MO, Larkin JC, Woolford JL Jr (1988) Ribosomal protein synthesis is not regulated at the translational level in Saccharomyces cerevisiae: balanced accumulation of ribosomal proteins L16 and rp59 is mediated by turnover of excess protein. Genes Dev 2:664–676
Turowski TW, Tollervey D (2015) Cotranscriptional events in eukaryotic ribosome synthesis. Wiley Interdiscip Rev RNA 6:129–139. doi:10.1002/wrna.1263
Vanrobays E, Leplus A, Osheim YN, Beyer AL, Wacheul L, Lafontaine DL (2008) TOR regulates the subcellular distribution of DIM2, a KH domain protein required for cotranscriptional ribosome assembly and pre-40S ribosome export. RNA 14:2061–2073. doi:10.1261/rna.1176708
Vilardell J, Warner JR (1994) Regulation of splicing at an intermediate step in the formation of the spliceosome. Genes Dev 8:211–220
Vilardell J, Warner JR (1997) Ribosomal protein L32 of Saccharomyces cerevisiae influences both the splicing of its own transcript and the processing of rRNA. Mol Cell Biol 17:1959–1965
Vilardell J, Chartrand P, Singer RH, Warner JR (2000) The odyssey of a regulated transcript. RNA 6:1773–1780
von der Haar T (2008) A quantitative estimation of the global translational activity in logarithmically growing yeast cells. BMC Syst Biol 2:87. doi:10.1186/1752-0509-2-87
Wade C, Shea KA, Jensen RV, McAlear MA (2001) EBP2 is a member of the yeast RRB regulon, a transcriptionally coregulated set of genes that are required for ribosome and rRNA biosynthesis. Mol Cell Biol 21:8638–8650. doi:10.1128/MCB.21.24.8638-8650.2001
Wade CH, Umbarger MA, McAlear MA (2006) The budding yeast rRNA and ribosome biosynthesis (RRB) regulon contains over 200 genes. Yeast 23:293–306. doi:10.1002/yea.1353
Waliullah TM, Yeasmin AM, Kaneko A, Koike N, Terasawa M, Totsuka T, Ushimaru T (2017) Rim15 and Sch9 kinases are involved in induction of autophagic degradation of ribosomes in budding yeast. Biosci Biotechnol Biochem 81: 307–310. doi:10.1080/09168451.2016.1234928
Wan K, Yabuki Y, Mizuta K (2015) Roles of Ebp2 and ribosomal protein L36 in ribosome biogenesis in Saccharomyces cerevisiae. Curr Genet 61:31–41. doi:10.1007/s00294-014-0442-1
Warner JR (1989) Synthesis of ribosomes in Saccharomyces cerevisiae. Microbiol Rev 53:256–271
Warner JR (1999) The economics of ribosome biosynthesis in yeast. Trend Biochem Sci 24:437–440
Warner JR, Mitra G, Schwindinger WF, Studeny M, Fried HM (1985) Saccharomyces cerevisiae coordinates accumulation of yeast ribosomal proteins by modulating mRNA splicing, translational initiation, and protein turnover. Mol Cell Biol 5:1512–1521
Watkins NJ, Bohnsack MT (2012) The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA 3:397–414. doi:10.1002/wrna.117
Wei Y, Zheng XF (2009) Sch9 partially mediates TORC1 signaling to control ribosomal RNA synthesis. Cell Cycle 8:4085–4090. doi:10.4161/cc.8.24.10170
Wery M, Ruidant S, Schillewaert S, Lepore N, Lafontaine DL (2009) The nuclear poly(A) polymerase and Exosome cofactor Trf5 is recruited cotranscriptionally to nucleolar surveillance. RNA 15:406–419. doi:10.1261/rna.1402709
Woolford JL Jr, Baserga SJ (2013) Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195:643–681. doi:10.1534/genetics.113.153197
Wu LF, Hughes TR, Davierwala AP, Robinson MD, Stoughton R, Altschuler SJ (2002a) Large-scale prediction of Saccharomyces cerevisiae gene function using overlapping transcriptional clusters. Nat Genet 31:255–265
Wu LF, Hughes TR, Davierwala AP, Robinson MD, Stoughton R, Altschuler SJ (2002b) Large-scale prediction of Saccharomyces cerevisiae gene function using overlapping transcriptional clusters. Nat Genet 31:255–265. doi:10.1038/ng906
Zhang Y, Smith ADt, Renfrow MB, Schneider DA (2010) The RNA polymerase-associated factor 1 complex (Paf1C) directly increases the elongation rate of RNA polymerase I and is required for efficient regulation of rRNA synthesis. J Biol Chem 285:14152–14159. doi:10.1074/jbc.M110.115220
Zhao Y, McIntosh KB, Rudra D, Schawalder S, Shore D, Warner JR (2006) Fine-structure analysis of ribosomal protein gene transcription. Mol Cell Biol 26:4853–4862. doi:10.1128/MCB.02367-05
Zhu C, Byers KJ, McCord RP, Shi Z, Berger MF, Newburger DE, Saulrieta K, Smith Z, Shah MV, Radhakrishnan M, Philippakis AA, Hu Y, De Masi F, Pacek M, Rolfs A, Murthy T, Labaer J, Bulyk ML (2009) High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res 19:556–566. doi:10.1101/gr.090233.108
Acknowledgements
We are grateful to M. Koš for thoughtful comments. This work was supported by Grants from the Spanish Ministry of Economy and Competitiveness (MINECO) and the European Regional Development Fund (ERDF): BFU2016-77728-C3-3-P AEI-FEDER (to S.C.), BFU2016-75352-P AEI-FEDER (to J.d.l.C.) and BFU2016-76446-P AEI-FEDER and RYC-2014-16665 (to F.G.-H.). We also acknowledge the support from the Andalusian Government and ERDF (grant P12-BIO-1938 MO to S.C.). V.B. is a recipient of a FPI fellowship from MINECO.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by M. Kupiec.
Rights and permissions
About this article

Cite this article
de la Cruz, J., Gómez-Herreros, F., Rodríguez-Galán, O. et al. Feedback regulation of ribosome assembly. Curr Genet 64, 393–404 (2018). https://doi.org/10.1007/s00294-017-0764-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-017-0764-x