
Abstract
Rhizoctonia solani is an important necrotrophic fungal pathogen which causes disease on diverse plant species. It has been classified into 14 genetically distinct anastomosis groups (AGs), however, very little is known about their genomic diversity. AG1-IA causes sheath blight disease in rice and controlling this disease remains a challenge for sustainable rice cultivation. Recently the draft genome sequences of AG1-IA (rice isolate) and AG1-IB (lettuce isolate) had become publicly available. In this study, using comparative genomics, we report identification of 3,942 R. solani genes that are uniquely present in AG1-IA. Many of these genes encode important biological, molecular functions and exhibit dynamic expression during in-planta growth of the pathogen in rice. Based upon sequence similarity with genes that are required for plant and human/zoonotic diseases, we identified several putative virulence/pathogenicity determinants amongst AG1-IA specific genes. While studying the expression of 19 randomly selected genes, we identified three genes highly up-regulated during in-planta growth. The detailed in silico characterization of these genes and extent of their up-regulation in different rice genotypes, having variable degree of disease susceptibility, suggests their importance in rice–Rhizoctonia interactions. In summary, the present study reports identification, functional characterization of AG1-IA specific genes and predicts important virulence determinants that might enable the pathogen to grow inside hostile plant environment. Further characterization of these genes would shed useful insights about the pathogenicity mechanism of AG1-IA on rice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rhizoctonia solani is a non-obligate, necrotrophic, phytopathogenic Basidiomycetous fungus, known for its highly destructive lifestyle (Anderson 1982; Gonzalez Garcia et al. 2006; Sinclair 1970). It has wide host range including species in Asteraceae, Brassicaceae, Fabaceae, Poaceae, Solanaceae and as well as some ornamental plants and forest trees (Lee and Rush 1983; Pan et al. 1999; Rush and Lee 1992; Zou et al. 2000). Based upon hyphal fusion affinity (known as anastomosis), isolates of R. solani has been classified into 14 anastomosis groups, namely AG-1 to AG-13 and AG-BI (Gonzalez Garcia et al. 2006; Guillemaut et al. 2003; Ogoshi 1987; Taheri et al. 2007). Based on characteristic features like colony morphology, biochemical, genetic and pathogenicity, R. solani AG-1 has been further divided into six intraspecific groups i.e. IA, IB, IC, ID, IE and IF (Ogoshi 1987; Wibberg et al. 2013; Yang and Li 2012). Polymorphism in ITS region sequences had been explored in classifying R. solani strains into different anastomosis groups (Ahvenniemi et al. 2009; Guillemaut et al. 2003; Keijer et al. 1996; Kuninaga et al. 1997). They also produce different karyotype banding pattern when analysed through pulse field gel electrophoresis (Keijer et al. 1996). This suggests that R. solani strains demonstrate significant divergence in their gene contents even though they belong to the same taxonomic classification.
Recently genome sequences of two isolates (AG1-IA and AG1-IB) of R. solani have become publicly available (Wibberg et al. 2013; Zheng et al. 2013). The sequenced AG1-IA strain is a rice isolate whereas the sequenced AG1-IB is a lettuce isolate. The AG1-IA is well known to cause disease like sheath blight, bottom rot, brown patch, foliar blight, head-rot, leaf blight, web-blight on varieties of hosts (Yang and Li 2012). Its host includes important crops including rice, corn, barley, potato, sorghum, millet, soybean, peanut, lima bean cabbage, lettuce, etc. (Fenille et al. 2002; Sneh and Ichielevich-Auster 1998; Yang and Li 2012). The AG1-IB is causal of sheath blight, bottom rot, damping off, leaf blight, foliar blight, root rot, head-rot etc. diseases on diverse hosts including corn, sugar beet, beans, cabbage, apple and many other plants (Sneh and Ichielevich-Auster 1998; Wibberg et al. 2013; Yang and Li 2012).
The sheath blight disease (SBD) is one of major disease of rice, reported from almost all rice cultivating country in the world. During favourable conditions SBD causes up to 50 % reduction in rice yield with an annual loss of ~6 million tons of rice grains (Zheng et al. 2013). In spite of enormous global efforts till date not even a single disease resistance gene has been identified against this disease. However, some QTLs (quantitative trait loci) associated with SBD resistance have been reported (Channamallikarjuna et al. 2009; Prasad and Eizenga 2008; Wang et al. 2011). The disease is generally controlled by fungicides; however, because of high mammalian, non-target and environmental toxic side effects; there has been growing concern about indiscriminate usages of fungicides. Thus controlling sheath blight disease remains a challenge for global rice cultivation and ensuring food security. Rice is an important staple food crop and to meet the demand of global food security, its productivity has to be increased by ~1.7 % every year and to 50 % by the year 2050 (Khush 2005). This challenge has to be met in spite of all antagonistic effects of climate change and growing competition for land, water and energy resources. Understanding virulence of the pathogen and identifying key genes/molecules involved in its interactions with host would facilitate developing better strategies to control this important plant pathogen.
In the present study, we report comparative genomics of AG1-IA and AG1-IB genomes, to identify and characterize AG1-IA group specific genes with an emphasis to identify potential virulence/pathogenicity determinants of R. solani on rice. We hypothesize that strain/anastomosis group specific genes might have been acquired during evolution to facilitate pathogenicity/virulence of this pathogen. The detailed in silico analysis revealed many of AG1-IA specific genes to encode important cellular, molecular as well as virulence functions. Digital gene expression analysis reflected several of them to exhibit differential and dynamic gene expression during rice infections. We report three of them to be highly induced during in-planta growth of R. solani on different rice genotypes. We further discuss how up-regulation of these genes could assist pathogen growth in hostile plant environment to cause sheath blight disease in rice.
Materials and methods
Sequence analysis
Genomic data of AG1-IA (rice isolate) and AG1-IB (lettuce isolate) R. solani strains were collected from previously assembled sequences and annotations available at UniProt database (Wibberg et al. 2013; Zheng et al. 2013). Amongst total genes obtained for both the strains, 48.68 and 53.37 % were annotated as uncharacterized/hypothetical proteins in AG1-IA and AG1-IB respectively. The sequences of length ≤40 amino acids were filtered out to remove potentially truncated protein. Reciprocal BLASTP searches were performed to identify significant homologs between these two strains of R. solani (E value <1e−15). The proteins that do not show any significant hits in AG1-IB genome were classified as unique for AG1-IA (n = 4,053).
These genes were searched in available genome assembly (contigs) of AG1-IB at different stringency (E value 1e−15, 1e−10 and 1e−05). Further to identify genes sharing low sequence similarities between AG1-IA and AG1-IB genomes, we performed orthology search by pairwise comparison and clustering using reciprocal best match and bootstrapping procedure implemented in InParanoid (O’Brien et al. 2005; Remm et al. 2001). After removing these putative orthologous genes, we considered 3,942 genes as AG1-IA specific for further functional analysis.
Functional annotation
The annotation of 3,942 AG1-IA specific genes was performed with E value cut-off of e < 1e−5 using NCBI non-redundant and UniProt databases. The putative orthologs of these genes were also searched in FungiDB (http://fungidb.org/fungidb) which is a database of 52 strains of 46 fungi species covering 571,622 genes. Functional categorization of GO terms was performed using BLAST2GO software using similarity search against NCBI non-redundant database (nr). GO terms were classified into three categories: biological process, molecular functions and cellular component and we estimated the fraction of AG1-IA specific genes involved in each of these categories. The relative abundance of GO terms in AG1-IA specific genes with respect to total AG1-IA genes was computed using custom scripts. AG1-IA specific proteins were also classified to their functional KOG categories using eggNOG application (http://eggnog.embl.de/version_3.0/overview.html). Pathway analysis was performed based on KEGG database using KAAS annotation server. The domain architecture was identified by MotifScan (http://myhits.isb-sib.ch/cgi-bin/motif_scan) and presence of post-translational modification sites were analysed by Prosite (de Castro et al. 2006). TargetP was used to predict the sub-cellular localization (http://www.cbs.dtu.dk/services/TargetP/).
Differential analysis of AG1-IA specific genes during rice infection
The expression data of genes expressed during rice infections at various time points (10, 18, 24, 32, 48 and 72 h after inoculation) were collected from published data (Zheng et al. 2013). The fragments per kilo base of transcript per million fragments (FPKM) values were extracted for AG1-IA specific genes and log2 fold ratio were computed. Average linkage hierarchical clustering method and Pearson correlation were employed to measure the similarity of expressed genes using Cluster v3.0 for preparing expression heat map (de Hoon et al. 2004; Eisen et al. 1998).
Identification of pathogenicity associated genes
The comparison of AG1-IA specific genes with CAZymes was performed using BLAST with significance e < 1e−15 to identify transcripts putatively encoding CAZymes. BLASTP analysis with e < 1e−05 cut-off against PHI-base: the pathogen–host interaction gene database (http://www.phi-base.org/) was performed to predict potential pathogenicity genes associated with loss of pathogenicity, reduced virulence, lethal, etc. (Winnenburg et al. 2006). Furthermore, we performed BLAST analysis against another pathogen–host interaction database (PHIDIAS) available online at http://www.phidias.us/index.php. The PHIDIAS is however, predominantly animal/zoonotic pathogens database (Xiang et al. 2007).
Isolation and culture condition of Rhizoctonia solani strains
The diseased rice samples were collected in sealed polythene bags and were handled with all safety precautions in laboratory condition to isolate R. solani strains used in this study. The locations of sample collection are mentioned in Table 1. The infected leaf samples were washed thoroughly with distilled water, cut into small pieces; surface sterilized with 1 % sodium hypochlorite solution and plated on 1 % Agar (Himedia, Mumbai, India) plates (Sinclair 1970). After 7 days of incubation at 28 °C, growing tips of R. solani like fungal mycelium were harvested and plated on to PDA (39 g/L; Potato Dextrose Agar; Himedia, Mumbai, India) plates. The plates were incubated at 28 °C until distinct sclerotia were observed. The identity of isolated strains was confirmed by rDNA sequencing using ITS1 and ITS4 primers (Online Resource 1), microscopic analysis and ability to cause disease on rice cultivars. The pathogens were re-isolated from artificially infected rice sheaths and were found to cause SBD when inoculated afresh onto rice cultivar.
Molecular biology techniques
Rhizoctonia solani sclerotia were grown for 3 days in potato dextrose broth (PDB; Himedia, Mumbai, India) at 28 °C and 200 rpm. DNA was isolated from 100 mg of fungal mycelium using CTAB (Cetyl Tri methyl Ammonium Bromide) method as per Doyle and Doyle (1990). PCR was performed with ~100 ng of DNA using NEB Taq polymerase and ITS1 and ITS4 primer pairs (White et al. 1990). The PCR reaction conditions were: 95 °C–30 s; 34 cycles of 95 °C–30 s; 56 °C–30 s; 68 °C–30 s with final extension at 68 °C–10 min. The PCR amplicons were sequence verified by Sanger dideoxy chain termination method using ABI3730XL DNA analyser (Applied Biosystems, Foster City, USA). Homology searches were performed in NCBI database by using BLAST algorithm (Altschul et al. 1997).
Pathogenicity bioassay
The rice seedlings of indica cultivar (IR64, PB1 and Tetep) and japonica cultivar (TP309) were grown in a PGV15 conviron plant growth chamber at 28 °C temperature, 80 % relative humidity, and 16/8 h of day/night with 250 µmol light intensity. 45 days old tillers of rice cultivars were inoculated with freshly prepared equal sized sclerotium of BRS-1 (AG1-IA R. solani isolate). The inoculation was performed on second topmost collar of the tiller, just below leaf collar on the inner side of sheath. The plants were incubated in growth chamber having above mentioned growth conditions and routinely monitored for appearance of disease symptoms. Manual water spraying was also carried out to keep high humidity that is conducive for sheath blight disease. The phenotypic observations were recorded at 3, 6 and 9 days post pathogen inoculation. The following disease variables were measured for each tiller: number of lesions; length of each lesion; distance between inoculum and tip of the farthest lesion; and vertical sheath colonization (VSC), distance between tips of the lowest and highest lesions of the infected sheath. Sheath length, the distance from base of tiller to topmost leaf collar was also measured as per methodology described (Willocquet et al. 2011). The relative vertical sheath colonization (RVSC) was calculated by formula VSC/sheath length × 100. The disease response was classified into 0–9 disease scale as described (Jia et al. 2013; Park et al. 2008). Each experiment was repeated at least three times with at least 15 tillers (3 pots; 5 tillers in each pot) of each cultivar inoculated with BRS-1 sclerotia. In each experiment similar number of un-inoculated tillers was used as negative control. As characteristic disease symptoms first appear at 3 DPI (days post inoculations) and significantly spread at 6 DPI, we used these time points for expression studies. The diseased zone from each samples were harvested in liquid N2 and were stored at −80 °C till further used for RNA isolation.
RNA isolation and cDNA synthesis
Total RNA was extracted from 200 mg of infected plant samples using Qiagen RNA Extraction Kit as per manufacturer’s instructions. Similarly, the total RNA was isolated from 100 mg of PDB grown fungal mycelium. The DNase treated 5 µg of total RNA from infected rice tissues and 3 µg of total RNA obtained from in vitro grown fungal culture were individually used for cDNA synthesis by using Verso cDNA Synthesis Kit (Thermo Fisher Scientific Inc.) following protocol provided by the manufacturer.
Normalization of cDNA for expression studies
Majority of biomass in AG1-IA infected rice sheaths is of rice origin and only a limited proportion is of fungus. It is expected that total RNA isolated from infected rice tissues would carry higher proportion of plant RNA and lesser proportion of pathogen RNA. Hence, we had to normalize the concentration of cDNA to estimate expression of pathogenic genes during in-planta infection and compare them with that expressed during its growth in laboratory media. cDNA was prepared using 2, 3 and 5 µg of total RNA isolated from laboratory grown BRS-1 culture and 3 and 5 µg of total RNA isolated from BRS-1 infected rice sheaths. A semi-quantitative PCR with specially designed 18S R. solani ribosomal DNA specific PCR primers (18S-3F and 18S-3R) which can amplify R. solani 18S rDNA but not amplify rice 18S rDNA was performed. Different dilutions of laboratory grown BRS-1 cDNA (5×, 10× and 20× dilutions) and BRS-1 infected rice (1× and 5× dilutions) were used for semi-quantitative PCR. The thermal cycling conditions were initial denaturation at 95 °C for 10 min followed by 34 cycles of denaturation at 95 °C for 30 s, annealing at 56 °C for 1 min, and extension at 68 °C for 1 min in a BIORAD (MyCycler) thermo cycler. Final extension was not provided during semi-quantitative PCR. The PCR products were resolved by 1.5 % agarose gel electrophoresis at 90 V for 30 min and gel was analysed in UVP Gel Doc-IT imaging system. It was observed that band intensity of 10× diluted laboratory grown BRS-1 cDNA and undiluted BRS-1 infected rice cDNA was comparable.
Expression studies
The nucleotide sequences of 19 selected candidates R. solani AG1-IA specific genes were downloaded from NCBI and primers were designed using PRIMER 3 software (http://frodo.wi.mit.edu/primer3/) and synthesized from Sigma-Aldrich chemicals private limited (Bangalore, India). The Online Resource 1 enlists primers used in this study. The tenfold diluted cDNA from 3 and 6 days PDB grown BRS-1 culture and undiluted cDNA obtained from 3 and 6 days of BRS-1 growth in susceptible (IR64) and partially resistant (Tetep) rice were used for semi-quantitative PCR based expression analysis. The PCR conditions were same to that used during normalization of different cDNA samples. For three selected genes which demonstrate in-planta up-regulation, expression was quantified using Brilliant III Ultra-Fast SYBR Green Master Mix (Agilent Technologies) on ABI Step One real time PCR (Applied Biosystems). The thermal cycling conditions were initial denaturation of 95 °C for 3 min, 40 cycles of 95 °C for 0.05 min, annealing and extension at 60 °C for 0.15 min. Dissociation curve analysis was performed by fluorescence reading at 2° intervals between 60 and 95 °C to ensure that only one PCR product was amplified. All experiments were performed with at least three biological replicates and two technical replicates. The expression data were normalized with 18S rRNA gene and the relative fold change was calculated by using \(2^{{ - \Delta \Delta C_{t} }}\) method (Livak and Schmittgen 2001).
Results
Identification of AG1-IA anastomosis group specific genes of R. solani
A detailed comparative genomics study was performed to identify AG1-IA specific genes. We retrieved 10,482 and 12,198 gene sequences of AG1-IA and AG1-IB R. solani strains, respectively from UniProt database. The gene content of R. solani AG1-IA and AG1-IB was compared using BLASTP based sequence similarity searches (E value <1e−15). The proteins that do not show significant hits in AG1-IB were classified as unique for AG1-IA anastomosis group. This process identified 756 annotated and 3,383 uncharacterized proteins (total: 4,139 proteins) that are specific to AG1-IA. Sequences <40 amino acid lengths were excluded from further analysis to remove potentially truncated/non-functional proteins. In this process we identified 4,053 proteins; however, 10 of them (encoding hypothetical proteins) were found to be also present in the AG1-IB genomic contigs at low stringent (1e−5, % identity >95) search conditions (Online Resource 2). Further, InParanoid database search (O’Brien et al. 2005) revealed 101 out of 4,053 AG1-IA proteins to be orthologous to AG1-IB proteins. After removing these putative 111 AG1-IB orthologs, 3,942 proteins were considered to be AG1-IA specific (Online Resource 3).
Out of 6,452 contigs reported in the draft genome, AG1-IA specific genes were distributed across 1,879 contigs (Fig. 1). 25 contigs had 10 or more AG1-IA specific genes and two of them namely AFRT01000802 and AFRT01000985 were encompassing 22 and 16 genes, respectively (Online Resource 4). Several of AG1-IA specific genes were localized in continuous manner on the genomic contigs. This suggests role of gene transfer events in shaping evolution of these genes in AG1-IA strains. We could assign GO terms to limited set of genes that were present together in a cluster and few of them encoded similar functions (Online Resource 5).

Frequency distribution of AG1-IA specific R. solani genes across different genomic contigs. The AG1-IA specific genes (n = 3,942) were mapped onto the 6,452 available genomic contigs, reported in the available draft genome of R. solani AG1-IA and these genes were found to be distributed across 1,879 contigs. The frequency of the occurrence across different AG1-IA genomic contigs is plotted on the Y-axis
Annotation and functional classifications of AG1-IA specific proteins
The predicted AG1-IA specific genes were further annotated using blast searches against NCBI non-redundant protein database (nr) and FungiDB (http://fungidb.org/fungidb), a database of 52 strains of 48 fungal species. Out of 3,942 genes, 1,061 (27 %) demonstrated significant hits (e < 1e−5) to nr database and 862 demonstrated homology in FungiDB. Interestingly, majority of AG1-IA specific genes demonstrated homology with either a mushroom Coprinopsis cinerea strain Okayama 7 (#130) (42 %) or with a lignin degrading white rot fungi Phanerochaete chrysosporium strain RP78 (31 %) (Online Resource 6).
The hits associated with gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) were analysed to functionally characterize AG1-IA specific genes. GO terms for 501 proteins were identified and top 10 functions related to each GO categories i.e. cellular component, molecular function and biological process are summarized in Fig. 2. The metabolic process (44 proteins), translation (26), oxidation–reduction process (20), transport (19), transcription related functions etc. were predominant biological process. Binding (zinc ion, metal ion, nucleotide, nucleic acid, ATP, DNA) as well as catalytic or transferase activities were predominant molecular function. Integral to membrane, intracellular, nucleus, cytoplasm, ribosome as well as mitochondria were abundant cellular component. The relative abundance of GO terms associated with AG1-IA specific R. solani genes was computed by considering total number of such genes present in the sequenced genome. The cellular process, methylation, lipid metabolic process, vesicle mediated transport, transcription DNA dependent, transcription from RNA polymerase II promoter, translation were enriched biological process (Online Resource 7A). The methyltransferase activity, structural constituent of ribosome, zinc ion binding and electron carrier activity were enriched molecular function of AG1-IA specific genes (Online Resource 7B). As shown in Online Resource 7A and 7B, each of these functions constitute >20 % of total such functions encoded by AG1-IA strain.

Gene ontology (GO) classification of AG1-IA specific genes. GO classification was carried out based on similarity searches against NCBI non-redundant database using BLAST2GO software. The top 10 terms under each GO category (cellular component, molecular function and biological process) are displayed
The KOG analysis reflected significant proportion of these genes to be involved in post-translational modifications, protein turnover, translation and intracellular trafficking, secretion, vesicular transport and several other important cellular functions (Online Resource 8A). KEGG analysis classified 201 AG1-IA specific genes across metabolic pathways (54 genes), ribosome (20), biosynthesis of secondary metabolites (13) etc. (Online Resource 8B).
AGI-1A anastomosis group specific genes encode potential virulence determinants
To predict potential pathogenicity genes amongst AG1-IA specific genes, BLASTP searches against PHI-base database was performed. Out of 74 AG1-IA specific genes that demonstrate homology (1e−05) in PHI-base database, 31 % were associated with reduced virulence and 12 % were related to loss of pathogenicity (Fig. 3a). GO classification of PHI-base orthologs revealed zinc ion binding and nucleic acid as major molecular function (Fig. 3c). Regulation of transcription, metabolic process etc. was predominant biological process (Fig. 3b). The sub-cellular localization pattern of these genes is summarized in Fig. 3d. We further identified AG1-IA specific genes that show homology with PHIDIAS (http://www.phidias.us/index.php), a web based pathogen–host interactions (PHI) database. PHIDIAS encompasses literature curated information and gene expression data of several human and zoonotic pathogenic bacteria, viruses, parasites (Xiang et al. 2007). BLAST search against PHIDIAS identified 180 hits with E value <1e−4 and is summarized in Online Resource 9. KEGG annotation of PHIDIAS orthologs of AG1-IA specific genes revealed significant proportion of them to be potentially involved in metabolic pathways or biosynthesis of secondary metabolites (Online Resource 10).

Orthologs of AG1-IA specific genes in pathogen–host interactions (PHI-base) database. a Summary of different phenotypic categories of AG1-IA group specific genes in PHI-base database. b–d Represent the associated GO terms under biological process, molecular function and cellular component, respectively
Apart from genes homologous to PHI-base and PHIDIAS database entries, we also identified carbohydrate-active enzymes (CAZYmes) that are encoded by AG1-IA specific genes (Table 2). The presence of 12 genes across 11 different families suggests that some of them might be deployed by the pathogen for depolymerisation of host cell wall for successful colonization/pathogenesis. Previously hydrolase like transcripts were found enriched in the ESTs obtained from AG1-IA infected rice tissues (Rioux et al. 2011). Such activity can be potentially associated with host cell wall degradation.
The AG1-IA specific genes are conserved across different AG1-IA strains of R. solani
In order to analyse whether genes identified in this study demonstrate AG1-IA strain specificity, we tested the presence of 19 randomly selected genes (including five PHIDIAS orthologs; Table 3) in different field isolates of R. solani that were isolated from geographically different part of India. Online Resource 11 depicts the microscopic image of actively growing mycelium of each of these isolates. On the basis of ITS rDNA sequencing, blast and phylogenetic analysis, four of the field isolates were found to be AG1-IA strain and another one was R. zeae strain (Online Resource 12). PCR analysis revealed majority of the 19 selected genes to be present in different field isolates of AG1-IA (Online Resource 13). At first glance, presence of a few genes (marked as dashed circles; Online Resource 13) was not very clear in some of the AG1-IA strains. However, when tested individually, these genes were found to be present in all AG1-IA strains analysed, RS_L6 seems to be an exception (Online Resource 14). The presence of these genes was further analysed in BRS-4 (R. zeae strain), isolated from sheath blight disease like symptomatic zone of PB1, a susceptible indica rice cultivar. BRS-4 showed distinct PCR banding pattern when compared to R. solani AG1-IA strains (Online Resource 13E).
Pathogenicity of BRS-1, the AG1-IA R. solani strain on different rice genotypes
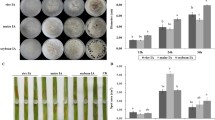
In order to understand pathogenicity of R. solani strain BRS-1 (AG1-IA rice isolate) different rice genotypes [IR64, PB1, Tetep (indica cultivars) and TP309 (japonica cultivar)] were inoculated with freshly grown sclerotia. The BRS-1 imparted characteristic sheath blight disease symptoms (i.e. water soaked, green grey, ellipsoid, necrotic lesions). Distinct symptoms appeared at 3DPI in IR64 and PB1 rice cultivars but at a later stage in TP309 and Tetep. The characteristic disease symptoms on IR64 and Tetep at 3 and 6 days post inoculation (DPI) are depicted in Fig. 4a and relative vertical sheath colonization data [calculated by the formula 100 × VSC (vertical sheath colonization)/sheath length] are summarized in Fig. 4b. Based upon various disease parameters, IR64 and PB1 were found to be susceptible while Tetep and TP309 were moderately resistant during initial stage of infection i.e. 3DPI. However, at later stage (6 DPI), the disease index of TP309 and Tetep was found similar to that exhibited by IR64 and PB1. Our result coincided with earlier reports which suggested IR64 and PB1 to be susceptible while Tetep and TP309 as moderately sheath blight disease resistant rice cultivars (Channamallikarjuna et al. 2009; Sato et al. 2004; Singh and Singh 2012; Willocquet et al. 2011; Xiaoxing et al. 2013).

Pathogenesis of R. solani AG1-IA isolate (BRS-1) on different rice genotypes. a Sheath blight disease symptoms at 3 and 6 days upon BRS-1 inoculation on IR64 (susceptible) and Tetep (partially resistant) indica rice cultivars. b Relative vertical sheath colonization (RVSC) index with error bar at 95 % confidence interval. The data are representative of at least three biological replicates with 15 infected tillers at each time points
Expression analysis of AGI-1A anastomosis group specific genes
The gene expression data (FPKM values) of AG1-IA anastomosis group specific genes were obtained from NCBI database and were used to calculate log2 fold change during different stages of rice infections (Online Resource 15). Majority of these genes were dynamically expressed during rice infection and their expression varied across different time intervals (Fig. 5a). Several of AG1-IA specific genes exhibited differential expression during in-planta growth of the pathogen (Fig. 5b). 129 genes were up-regulated while 150 genes were down-regulated at 10 h compared to 18 h of rice infections. Limited overlaps amongst up-regulated genes across different time intervals were observed. Out of 129 up-regulated genes at 10 vs 18 h; only 5 genes at 18 vs 24 h, 17 genes at 24 vs 32 h and 10 genes at 32 vs 48 h of infections were found to be similarly regulated (Fig. 5c). Similar trends were also observed for down-regulated genes. Comparative account of differentially regulated genes at different time intervals of R. solani infections on rice are summarized in Online Resource 16.

Differential regulation of AG1-IA specific genes during in-planta growth of the pathogen. a Heat map clustering pattern of FKPM based gene expression profile of AG1-IA specific genes, across different time intervals of pathogen infection in rice. b Bar graph representing number of up and down-regulated AG1-IA specific genes. c Venn diagram representing number of up-regulated and down-regulated genes across different time points
Semi-quantitative PCR based expression analysis
The expression dynamics of 19 selected AG1-IA specific genes (Table 3) were analysed during 3 and 6 DPI of in-planta growth of BRS-1 on IR64 and Tetep rice cultivars. Also the expression of these genes was studied during pathogen’s growth in PDB media. PCR primers were designed for each of these genes in such a manner that they do not amplify any rice sequences. Majority of AG1-IA specific genes were expressed during BRS-1 growth in PDB media and no major difference in gene expression was detected at 3 and 6 DPI of its growth (Online Resource 17). However, expression of many of them was not observed during 3 and 6 DPI of in-planta growth. We speculate that these genes might be down-regulated during in-planta growth of the pathogen. Possibility of these genes to be expressed at earlier time points or have very low expression to be detected by semi-quantitative PCR could not be ruled out. It was noteworthy that RS_P1, RS_P3 and RS_P4, PHIDIAS orthologous AG1-IA specific genes demonstrated induced expression during in-planta growth of the pathogen, as revealed by their corresponding band intensity on agarose gel.
Real time PCR based validation of in-planta up-regulated AG1-IA specific genes
SYBR green based real time PCR was performed to understand the differential expression of RS_P1, RS_P3 and RS_P4 AG1-IA specific genes. The expression was studied during growth of R. solani on different rice genotypes having variable degree of disease susceptibility [IR64 and PB1 (susceptible), TP309 and Tetep (moderately resistant)] and also during its growth in PDB broth at 3 and 6 DPI. The 18S fungal rRNA was used as house keeping gene for normalization of cDNA across different data points. The analysis revealed RS_P1, RS_P3 and RS_P4 genes to be up-regulated during in-planta growth of the pathogen when compared to their expression in laboratory media (Fig. 6a). However, degree of up-regulation varied during in-planta growth of the pathogen in different rice genotypes. Each of them were up-regulated at 6 DPI of in-planta growth; however, Tetep and TP309 induced higher degree of up-regulation compared to their expression during growth in IR64 and PB1 (Fig. 6b). This correlates with disease severity index of Tetep and TP309 (Fig. 4b). We also observed each of these genes being up-regulated in susceptible maize variety (Maharaja Bio 9637) (Fig. 6b). Notably the extent of up-regulation was significantly less in maize compared to that in different rice genotypes analysed in this study.

In-planta induction of R. solani AG1-IA specific genes. a SYBR green based Real Time PCR was used to quantify the relative expression analysis of three candidates AG1-IA specific genes (RS_P1, RS_P3 and RS_P4) during pathogenesis on IR64 and PB1 (susceptible); TP309 (moderately susceptible) and Tetep (partially resistant) rice cultivars at 3 (dark shaded bars) and 6 (light shaded bars) DPI following R. solani inoculation. The comparative expression was calculated with that of PDB grown R. solani strain, harvested at the same time point. b The in-planta expression of these genes was compared at 3 and 6 DPI. The R. solani specific 18S primers were used as normalization control. Similar expression profile of each of these genes was observed in at least three independent experiments
In silico characterization of in-planta induced AG1-IA specific genes
Various databases were analysed to predict presence of specific sequence motifs, post-translational modification sites and biological functions of each of these proteins (Table 4). The RS_P1 gene encodes a parvulin subfamily of peptide-prolyl cis/trans isomerase (PPiC) protein having Rotamase domain (Online Resource 18A). The RS_P1 orthologs of this protein (named Ess1) have previously been shown to be required for growth of yeast and candida strains (Devasahayam et al. 2002; Gemmill et al. 2005). RS_P3 encodes NifU like protein that serves as a scaffold during iron–sulphur biogenesis (Online Resource 18B). Such proteins are responsible for maintaining iron homeostasis in a cell, particularly mitochondria (Lill 2009) and can serve as sensor for molecular oxygen, stress response and iron concentration (Crack et al. 2012; Rouault and Tong 2005). Another in-planta up-regulated gene (RS_P4) encodes V-SNARE like protein (Online Resource 18C), the orthologs of which is required for translocation of pathogenic effector proteins into host (Frei dit Frey and Robatzek 2009; Giraldo et al. 2013; Yang et al. 2013).
Interestingly, Prosite analysis revealed each of these in-planta up-regulated AG1-IA specific genes to contain post-translational modification sites that are absent in their nearest orthologs (Table 4). The RS_P1 protein had a distinct 25 aa long residue with a protein kinase C (5–7 aa) and cAMP/cGMP dependent protein kinase (6–9 aa) phosphorylation sites (Online Resource 18A). Similarly alignment of RS_P3 and its orthologs revealed that AG1-IA protein particularly contain a PkC phosphorylation site (10–12 aa) at N-terminal region and N-Myristoylation (260–265 aa) and Asn glycosylation site (267–270 aa) at its C-terminal end (Online Resource 18B). Multiple sequence alignment of RS_P4 and its nearest orthologs identified a stretch of 69 aa residue with two potential N-myristoylation sites (at 11–16 and 26–31 aa) present exclusively in AG1-IA protein (Online Resource 18C).
Discussion
In this study, we report identification and detailed in silico characterization of R. solani genes that are specific to AG1-IA, the causal of sheath blight disease of rice. The GO, KEGG and KOG based functional annotation of these genes revealed several of them to encode important cellular and molecular functions. Significant proportion of these genes demonstrates homology with either a mushroom Coprinopsis cinerea strain Okayama 7 (#130) or a lignin degrading white rot fungi Phanerochaete chrysosporium strain RP78. Previously phylogenetic analysis has also suggested close relationship of AG1-IA to C. cinerea (Zheng et al. 2013). Interestingly, many of AG1-IA specific genes were found localized in a sequential manner on particular genomic contigs of available draft genome, suggesting the role of gene transfer events in their evolution.
Many of the AG1-IA specific genes demonstrated homology in PHI-base database and were orthologs of genes that are involved in loss of pathogenicity or reduced virulence. This suggests that they can serve as potential pathogenicity determinants of AG1-IA in rice. Considering the close relationship of fungal pathogens of plants and animals and the fact that in some cases they exhibit similar pathogenicity mechanisms imparting similar symptoms (Ortoneda et al. 2004; Sexton and Howlett 2006; Sharon and Shlezinger 2013), we analysed the homology of AG1-IA specific genes in PHIDIAS database. We could identify 180 PHIDIAS orthologs and KEGG annotation suggested significant proportions of them to be related to genes involved in biosynthesis of secondary metabolites. The secondary metabolites play pivotal role during pathogenesis of necrotrophic plant pathogens (Howlett 2006; Panstruga 2010; Tsuge et al. 2013). For example, modification of a gene encoding secondary metabolite of Alternaria alternata (a necrotrophic pathogen) altered its host range (Ito et al. 2004). Some small molecular weight secondary metabolite acts as host specific and nonspecific toxins (Howlett 2006; Tsuge et al. 2013; Vidhyasekaran et al. 1997). Interestingly, R. solani strains are known to produce host specific toxins which impart characteristic disease like symptoms on host plants (Brooks 2007; Vidhyasekaran et al. 1997). Considering the importance of secondary metabolism in toxin biosynthesis/modification and fungal virulence, we hypothesize that some of the PHIDIAS orthologous may be involved in biosynthesis, modification or secretion of AG1-IA specific R. solani toxins, thereby rendering them to act in a host genotype specific manner.
Majority of AG1-IA specific genes identified in this study were expressed and exhibited dynamic expression during in-planta growth of the pathogen. The expression analysis of 19 randomly selected genes revealed most of them to be expressed during growth of the pathogen in PDB broth. We identified and validated three of them (namely RS_P1, RS-P2 and RS_P3) to be up-regulated during in-planta growth of the pathogen when compared to their expression in laboratory media. The up-regulation was also observed during growth of the pathogen in different rice genotypes having variable degree of disease susceptibility and also in susceptible maize. Each of them showed higher up-regulation at 6DPI compared with 3 DPI of in-planta growth. However, extent of up-regulation of these genes was much higher in TP309 and Tetep (partially resistant) when compared to susceptible rice cultivars IR64 and PB1. Interestingly, up-regulation was limited in maize compared to that in rice genotypes. We hypothesize R. solani might regulate its gene expression in host genotype specific manner to use right set of arsenals and regulate their expression in different hosts. This in turn might facilitate the pathogen to make the environment conducive for its growth and establishment of disease. Based upon functions of orthologs of these in-planta induced genes, we propose a model to suggest their role during rice–Rhizoctonia interactions (Fig. 7). We speculate enhanced expression of RS_P1 gene encoding parvulin like protein might promote growth of R. solani during plant pathogenesis and protect the pathogen from fungal cell wall degrading enzymes secreted by the host plant. Indeed Cryptococcus neoformans and Magnaporthe oryzae orthologs of this protein are required for pathogenesis on their respective host (Ren et al. 2005; Soanes et al. 2012). The up-regulation of RS_P3 that encodes NifU like protein might ensure proper functioning of iron–sulphur synthesis machinery and assist the pathogen to tackle enhanced reactive oxygen species (ROS) that are produced as a first line of plant defence upon pathogen attack (Heller and Tudzynski 2011). Pathogens deliver small molecular weight proteinaceous effectors inside the host to suppress plant defence and alter host cellular environment to conquer and establish disease (Ciuffetti et al. 2010; Tan et al. 2010; Liu et al. 2012; Oliver et al. 2012). The up-regulation of RS_P4 that encodes V-SNARE like protein might be required to transport R. solani effectors into host plant to facilitate establishment of sheath blight disease.

Model illustrating the role of in-planta upregulated AG1-IA specific genes during rice–Rhizoctonia interaction. The R. solani sclerotia germinate to form infection cushion and hyphae emerge out to penetrate into the host cell. Upon sensing the attack rice plant may activate its basal defence responses and may secrete PR proteins (pathogenesis related), β-1,3 glucanase, chitinase, CWDE (cell wall degrading enzymes) etc. Rice may also produce ROS (reactive oxygen species) to prevent the growth of pathogen. The R. solani inturn counteracts by inducing RS_P1, RS_P3 and RS_P4 genes. The RS_P1 might assist fortification of fungal cell wall and RS_P3 may prepare the pathogen to tackle the enhanced oxidative stress due to increased ROS production. Also they might assist in activating stress responsive genes of the pathogen. The RS_P4, encoding V-SNARE protein might be involved in secreting fungal effector molecules into the host cell mediated via vesicular trafficking. The fungal effectors generally function to make the host environment conducive for its growth and pathogenesis. In this model rice cell is depicted in green while R. solani is depicted in brown. The secreted plant PR and CWDE proteins are depicted in green (empty and filled circle) while ROS production is highlighted as green star. The V-SNARE mediated vesicular transport of fungal effectors is depicted as brown stars, while the fungal effectors are represented as brown dots
Overall our analysis identified AG1-IA specific important genes/functions, which could be required for its pathogenesis on rice. Future availability of genome sequences of R. solani strains belonging to other anastomosis group would assist our analysis to ascertain AG1-IA specific nature of these genes. We further report identification of three in-planta up-regulated AG1-IA specific genes and propose a model to support their role during rice–Rhizoctonia interactions. The knock-down/overexpression studies are in progress to create loss and gain of function allele of some of these genes to verify their role during rice–Rhizoctonia interactions. This study would facilitate further research to understand the biology and pathogenicity mechanism of AG1-IA on rice, which in turn would make possible to evolve novel strategies to develop sheath blight disease resistant rice as well as several other crops of agricultural importance.
References
Ahvenniemi P, Wolf M, Lehtonen MJ, Wilson P, German-Kinnari M, Valkonen JPT (2009) Evolutionary diversification indicated by compensatory base changes in ITS2 secondary structures in a complex fungal species, Rhizoctonia solani. J Mol Evol 69:150–163. doi:10.1007/s00239-009-9260-3
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Anderson NA (1982) The genetics and pathology of Rhizoctonia solani. Annu Rev Phytopathol 20:329–347
Brooks SA (2007) Sensitivity to a phytotoxin from Rhizoctonia solani correlates with sheath blight susceptibility in rice. Phytopathology 97:1207–1212. doi:10.1094/PHYTO-97-10-1207
Channamallikarjuna V, Sonah H, Prasad M, Rao GJN, Chand S, Upreti HC, Singh NK, Sharma TR (2009) Identification of major quantitative trait loci qSBR11-1 for sheath blight resistance in rice. Mol Breed 25:155–166. doi:10.1007/s11032-009-9316-5
Ciuffetti LM, Manning VA, Pandelova I, Betts MF, Martinez JP (2010) Host-selective toxins, Ptr ToxA and Ptr ToxB, as necrotrophic effectors in the Pyrenophora tritici-repentis–wheat interaction. New Phytol 187:911–919. doi:10.1111/j.1469-8137.2010.03362.x
Crack JC, Green J, Thomson AJ, Le Brun NE (2012) Iron–sulfur cluster sensor-regulators. Curr Opin Chem Biol 16:35–44. doi:10.1016/j.cbpa.2012.02.009
De Castro E, Sigrist CJA, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res 34:W362–W365. doi:10.1093/nar/gkl124
De Hoon MJL, Imoto S, Nolan J, Miyano S (2004) Open source clustering software. Bioinformatics 20:1453–1454. doi:10.1093/bioinformatics/bth078
Devasahayam G, Chaturvedi V, Hanes SD (2002) The Ess1 prolyl isomerase is required for growth and morphogenetic switching in Candida albicans. Genetics 160:37–48
Doyle JJ, Doyle JL (1990) A rapid total DNA preparation procedure for fresh plant tissue. Focus (Madison) 12:13–15
Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 95:14863–14868. doi:10.1073/pnas.95.25.14863
Fenille RC, de Souza NL, Kuramae EE (2002) Characterization of Rhizoctonia solani associated with soybean in Brazil. Eur J Plant Pathol 108:783–792
Frei dit Frey N, Robatzek S (2009) Trafficking vesicles: pro or contra pathogens? Curr Opin Plant Biol 12:437–443. doi:10.1016/j.pbi.2009.06.002
Gemmill TR, Wu X, Hanes SD (2005) Vanishingly low levels of Ess1 prolyl-isomerase activity are sufficient for growth in Saccharomyces cerevisiae. J Biol Chem 280:15510–15517. doi:10.1074/jbc.M412172200
Giraldo MC, Dagdas YF, Gupta YK, Mentlak Ta, Yi M, Martinez-Rocha AL, Saitoh H, Terauchi R, Talbot NJ, Valent B (2013) Two distinct secretion systems facilitate tissue invasion by the rice blast fungus Magnaporthe oryzae. Nat Commun 4:1996. doi:10.1038/ncomms2996
Gonzalez Garcia V, Portal Onco MA, Rubio Susan V (2006) Review. Biology and systematics of the form genus Rhizoctonia. Span J Agric Res 4:55–79
Guillemaut C, Edel-Hermann V, Camporota P, Alabouvette C, Richard-Molard M, Steinberg C (2003) Typing of anastomosis groups of Rhizoctonia solani by restriction analysis of ribosomal DNA. Can J Microbiol 49:556–568. doi:10.1139/w03-066
Heller J, Tudzynski P (2011) Reactive oxygen species in phytopathogenic fungi: signaling, development, and disease. Annu Rev Phytopathol 49:369–390. doi:10.1146/annurev-phyto-072910-095355
Howlett BJ (2006) Secondary metabolite toxins and nutrition of plant pathogenic fungi. Curr Opin Plant Biol 9:371–375. doi:10.1016/j.pbi.2006.05.004
Ito K, Tanaka T, Hatta R, Yamamoto M, Akimitsu K, Tsuge T (2004) Dissection of the host range of the fungal plant pathogen Alternaria alternata by modification of secondary metabolism. Mol Microbiol 52:399–411. doi:10.1111/j.1365-2958.2004.04004.x
Jia Y, Liu G, Park DS, Yang Y (2013) Inoculation and scoring methods for rice sheath blight disease. Methods Mol Biol 956:257–268
Keijer J, Houterman PM, Dullemans AM, Korsman MG (1996) Heterogeneity in electrophoretic karyotype within and between anastomosis groups of Rhizoctonia solani. Mycol Res 100:789–797. doi:10.1016/S0953-7562(96)80023-2
Khush GS (2005) What it will take to feed 5.0 billion rice consumers in 2030. Plant Mol Biol 59:1–6. doi:10.1007/s11103-005-2159-5
Kuninaga S, Natsuaki T, Takeuchi T, Yokosawa R (1997) Sequence variation of the rDNA ITS regions within and between anastomosis groups in Rhizoctonia solani. Curr Genet 32:237–243. doi:10.1007/s002940050272
Lee FN, Rush MC (1983) Rice sheath blight: a major rice disease. Plant Dis 67:829–832
Lill R (2009) Function and biogenesis of iron–sulphur proteins. Nature 460:831–838. doi:10.1038/nature08301
Liu Z, Zhang Z, Faris JD, Oliver RP, Syme R, McDonald MC, McDonald BA, Solomon PS, Lu S, Shelver WL, Xu S, Friesen TL (2012) The cysteine rich necrotrophic effector SnTox1 produced by Stagonospora nodorum triggers susceptibility of wheat lines harboring Snn1. PLoS Pathog 8:e1002467. doi:10.1371/journal.ppat.1002467
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi:10.1006/meth.2001.1262
O’Brien KP, Remm M, Sonnhammer ELL (2005) Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. doi:10.1093/nar/gki107
Ogoshi A (1987) Ecology and pathogenicity of anastomosis and intraspecific groups of Rhizoctonia solani Kuhn. Annu Rev Phytopathol 25:125–143
Oliver RP, Friesen TL, Faris JD, Solomon PS (2012) From pathology to genomics and host resistance. Annu Rev Phytopathol 50:23–43. doi:10.1146/annurev-phyto-081211-173019
Ortoneda M, Guarro J, Madrid MP, Caracuel Z, Roncero MIG, Mayayo E, Di Pietro A (2004) Fusarium oxysporum as a multihost model for the genetic dissection of fungal virulence in plants and mammals. Infect Immun 72:1760–1766. doi:10.1128/IAI.72.3.1760-1766.2004
Pan XB, Rush MC, Sha XY, Xie QJ, Liscombe SD, Stenina SR, Orad JH (1999) Major gene non-allelic sheath blight resistance from the rice cultivars Jasmine 85 and Teqing. Crop Sci 39:338–346
Panstruga R (2010) Introduction to a virtual special issue on pathogenic plant–fungus interactions. New Phytol 188:907–910. doi:10.1111/j.1469-8137.2010.03530.x
Park D-S, Sayler RJ, Hong Y-G, Nam M-H, Yang Y (2008) A method for inoculation and evaluation of rice sheath blight disease. Plant Dis 92:25–29. doi:10.1094/PDIS-92-1-0025
Prasad B, Eizenga GC (2008) Rice sheath blight disease resistance identified in Oryza spp. accessions. Plant Dis 92:1503–1509. doi:10.1094/PDIS-92-11-1503
Remm M, Storm CE, Sonnhammer EL (2001) Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol 314:1041–1052. doi:10.1006/jmbi.2000.5197
Ren P, Rossettini A, Chaturvedi V, Hanes SD (2005) The Ess1 prolyl isomerase is dispensable for growth but required for virulence in Cryptococcus neoformans. Microbiology 151:1593–1605. doi:10.1099/mic.0.27786-0
Rioux R, Manmathan H, Singh P, los Reyes B, Jia Y, Tavantzis S (2011) Comparative analysis of putative pathogenesis-related gene expression in two Rhizoctonia solani pathosystems. Curr Genet 57:391–408. doi:10.1007/s00294-011-0353-3
Rouault TA, Tong W-H (2005) Iron–sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat Rev Mol Cell Biol 6:345–351
Rush MC, Lee F (1992) Sheath blight. In: Webster F, Gunnell P (eds) Compendium of Rice Diseases. APS Press, St. Paul, pp 22–23
Sato H, Ideta IO, Ando I, Kunihiro Y, Hirabayashi H, Iwano M, Miyasaka A, Nemoto H, Imbe T (2004) Mapping QTLs for sheath blight resistance in the rice line WSS2. Breed Sci 54:265–271
Sexton AC, Howlett BJ (2006) Parallels in fungal pathogenesis on plant and animal hosts. Eukaryot Cell 5:1941–1949. doi:10.1128/EC.00277-06
Sharon A, Shlezinger N (2013) Fungi infecting plants and animals: killers, non-killers, and cell death. PLoS Pathog 9:e1003517
Sinclair JB (1970) Rhizoctonia solani: special methods of study. In: Parmeter J (ed) Rhizoctonia solani biology and pathology. University of California Press, Berkeley, Los Angeles and London, pp 199–217
Singh HK, Singh U (2012) Virulence diversity of Rhizoctonia solani causing sheath blight disease in rice and its host pathogen interaction. Int J Agric Environ Biotechnol 5:367–372
Sneh B, Ichielevich-Auster M (1998) Induced resistance of cucumber seedlings caused by some non-pathogenicRhizoctonia (np-R) isolates. Phytoparasitica 26:27–38. doi:10.1007/BF02981263
Soanes DM, Chakrabarti A, Paszkiewicz KH, Dawe AL, Talbot NJ (2012) Genome-wide transcriptional profiling of appressorium development by the rice blast fungus Magnaporthe oryzae. PLoS Pathog. doi:10.1371/journal.ppat.1002514
Taheri P, Gnanamanickam S, Höfte M (2007) Characterization, genetic structure, and pathogenicity of Rhizoctonia spp. associated with rice sheath diseases in India. Phytopathology 97:373–383. doi:10.1094/PHYTO-97-3-0373
Tan K-C, Oliver RP, Solomon PS, Moffat CS (2010) Proteinaceous necrotrophic effectors in fungal virulence. Funct Plant Biol 37:907. doi:10.1071/FP10067
Tsuge T, Harimoto Y, Akimitsu K, Ohtani K, Kodama M, Akagi Y, Egusa M, Yamamoto M, Otani H (2013) Host-selective toxins produced by the plant pathogenic fungus Alternaria alternata. FEMS Microbiol Rev 37:44–66. doi:10.1111/j.1574-6976.2012.00350.x
Vidhyasekaran P, Ponmalar TR, Samiyappan R, Velazhahan R, Vimala R, Ramanathan A, Paranidharan V, Muthukrishnan S (1997) Host-specific toxin production by Rhizoctonia solani, the rice sheath blight pathogen. Phytopathology 87:1258–1263. doi:10.1094/PHYTO.1997.87.12.1258
Wang Y, Pinson SRM, Fjellstrom RG, Tabien RE (2011) Phenotypic gain from introgression of two QTL, qSB9-2 and qSB12-1, for rice sheath blight resistance. Mol Breed 30:293–303. doi:10.1007/s11032-011-9619-1
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: PCR protocols, a guide to methods and applications. Academic Press Inc, London, pp 315–322
Wibberg D, Jelonek L, Rupp O, Hennig M, Eikmeyer F, Goesmann A, Hartmann A, Borriss R, Grosch R, Pühler A, Schlüter A (2013) Establishment and interpretation of the genome sequence of the phytopathogenic fungus Rhizoctonia solani AG1-IB isolate 7/3/14. J Biotechnol 167:142–155. doi:10.1016/j.jbiotec.2012.12.010
Willocquet L, Lore JS, Srinivasachary S, Savary S (2011) Quantification of the components of resistance to rice sheath blight using a detached tiller test under controlled conditions. Plant Dis 95:1507–1515. doi:10.1094/PDIS-01-11-0051
Winnenburg R, Baldwin TK, Urban M, Rawlings C, Köhler J, Hammond-Kosack KE (2006) PHI-base: a new database for pathogen host interactions. Nucleic Acids Res 34:D459–D464. doi:10.1093/nar/gkj047
Xiang Z, Tian Y, He Y (2007) PHIDIAS: a pathogen–host interaction data integration and analysis system. Genome Biol 8:R150. doi:10.1186/gb-2007-8-7-r150
Xiaoxing L, Ping L, Aiping Z (2013) In vitro selection of sheath blight resistance germplams in rice. Afr J Microbiol Res 7:4422–4429. doi:10.5897/AJMR2013.5659
Yang G, Li C (2012) General description of Rhizoctonia species complex. In: Cumagun CJ (ed) Plant Pathology, pp 41–52
Yang X, Ben S, Sun Y, Fan X, Tian C, Wang Y (2013) Genome-wide identification, phylogeny and expression profile of vesicle fusion components in Verticillium dahliae. PLoS One 8:e68681. doi:10.1371/journal.pone.0068681
Zheng A, Lin R, Zhang D, Qin P, Xu L, Ai P, Ding L, Wang Y, Chen Y, Liu Y, Sun Z, Feng H, Liang X, Fu R, Tang C, Li Q, Zhang J, Xie Z, Deng Q, Li S, Wang S, Zhu J, Wang L, Liu H, Li P (2013) The evolution and pathogenic mechanisms of the rice sheath blight pathogen. Nat Commun 4:1424. doi:10.1038/ncomms2427
Zou JH, Pan XB, Chen ZX, Xu JY, Lu JF, Zhai WX, Zhu LH (2000) Mapping quantitative trait loci controlling sheath blight resistance in two rice cultivars (Oryza sativa L.). Theory Appl Genet 101:569–573
Acknowledgments
SG was supported by SPM fellowship from Council of Scientific and Industrial Research (Govt. of India) and SKG was supported by Post-Doctoral Research fellowship from Department of Biotechnology (DBT, Govt of India). We acknowledge Praveen Raj S, CromDx Solutions Pvt. Ltd for assistance in computational analysis and central instrumentation facility at NIPGR for sequencing and qRT-PCR analysis. We also thank Dr G.S. Laha, DRR, Hyderabad for providing R. solani strains and AiPing Zheng, SICAU, China and Daniel Wibberg, CeBiTec, Bielefield University, Germany for making draft genome sequences of AG1-IA and AG1-B, R. solani strains publicly available. This work was supported by core research grant from the National Institute of Plant Genome Research, India. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
S. Ghosh and S. K. Gupta contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
294_2014_438_MOESM2_ESM.xlsx
Online Resource 2: Blast analysis against AG1-IB genomic contigs and Inparanoid database based ortholog prediction (XLSX 22 kb)
294_2014_438_MOESM7_ESM.pdf
Online Resource 7: The relative proportion of AG1-IA specific GO terms compared to that present in entire AG1-IA genome. The relative fraction of GO terms was computed using custom script and top 20 terms in Biological Process (A) and Molecular Function (B) are displayed. (PDF 667 kb)
294_2014_438_MOESM8_ESM.pdf
Online Resource 8: KOG and KEGG classification of AG1-IA specific genes. KOG and KEGG classification of AG1-IA specific genes were performed using eggNOG application and KAAS annotation server, respectively. The analysis classified 613 AG1-IA specific proteins into 23 KOG groups (A) and 187 genes across KEGG pathways. (PDF 727 kb)
294_2014_438_MOESM10_ESM.pdf
Online Resource 10: KEGG annotation of AG1-IA specific proteins that demonstrate homology in PHIDIAS database. (PDF 589 kb)
294_2014_438_MOESM11_ESM.tif
Online Resource 11: Microscopic analysis of trypan blue stained mycelia of different Rhizoctonia strains. The analysis was performed using Nikon Eclipse E100 microscope at 400X magnification. (TIFF 846 kb)
294_2014_438_MOESM13_ESM.pdf
Online Resource 13: PCR analysis of selected AG1-IA specific genes across geographically diverse strains of R. solani. The 19 selected genes were PCR amplified from different R. solani strains (A. BRS-1, B. BRS-2, C. BRS-6, D. BRS-7, E. BRS-4) analysed on 1.5 % agarose gel. The presence of these genes across different strains is summarized in F. Dark dot reflects confirmative presence of these genes, while cases having ambiguity are reflected as empty dots. The lane L2 and M2 represent the same gene, however the primer binding sites are different. M represents molecular weight marker. (PDF 1310 kb)
294_2014_438_MOESM14_ESM.pdf
Online Resource 14: PCR analysis to confirm the presence of some ambiguous AG1-IA specific genes. The genes RS_L1, RS_L5, RS_L6, and RS_M5 which showed ambiguous results in PCR (Online Resource 11) were individually PCR amplified in different AG1-1A isolates. M represents molecular weight marker. (PDF 420 kb)
294_2014_438_MOESM16_ESM.xlsx
Online Resource 16: Differential expression profile of AG1-IA specific genes across different time intervals (XLSX 1131 kb)
294_2014_438_MOESM17_ESM.pdf
Online Resource 17: Expression dynamics of selected AG1-IA specific genes. Semi-quantitative PCR based expressions of 19 selected candidate AG1-IA specific genes were analysed. The expression at 3 and 6 DPI of BRS-1 growth in susceptible rice IR64 is depicted in A and D, respectively. The expression at 3 and 6 DPI of BRS-1 growth in partially resistant rice cultivar Tetep is depicted in B and E, respectively while the expression during its growth in potato dextrose broth (PDB) at 3 and 6 DPI is reflected in C and F, respectively. The expression pattern of these genes at different stages is summarized in G. Dark dot reflects confirmative expression of these genes, while ambiguous cases are reflected as empty dots. R. solani specific 18S rDNA primers were used as a normalization control. The data is a representative of at least two biological and three technical replicates. The lane L2 and M2 represent the same gene, however the primer binding sites were different. M represents molecular weight marker. (PDF 2540 kb)
294_2014_438_MOESM18_ESM.pdf
Online Resource 18: Domain architecture of in-planta induced AG1-IA specific genes. The domain architecture of RS_P1 (A), RS_P3 (B), RS_P4 (C) proteins as revealed by Prosite and Pfam analysis. a: represents AG1-IA specific sequence while b: represents AG3 sequence, the nearest ortholog as per BLASTP analysis. Red flag in A.a represents PkC phosphorylation site and CAMP phosphorylation site is denoted by grey flag. In B.a Violet flag represents a PkC phosphorylation site while red and grey flag denotes N myristylation and Asn Glycosylation sites, respectively. Red flags in C.a represent N- Myristylation sites at N-terminal. (PDF 416 kb)
Rights and permissions
About this article

Cite this article
Ghosh, S., Gupta, S.K. & Jha, G. Identification and functional analysis of AG1-IA specific genes of Rhizoctonia solani . Curr Genet 60, 327–341 (2014). https://doi.org/10.1007/s00294-014-0438-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-014-0438-x