
Abstract
Sheath blight disease is one of the predominant diseases of rice and it is caused by the necrotrophic fungal pathogen Rhizoctonia solani. The mechanistic insight about its widespread success as a broad host range pathogen is limited. In this study, we endeavor to identify pathogenicity determinants of R. solani during infection process in rice. Through RNAseq analysis, we identified a total of 65 and 232 R. solani (strain BRS1) genes to be commonly upregulated in three different rice genotypes (PB1, Tetep, and TP309) at establishment and necrotrophic phase, respectively. The induction of genes encoding extracellular protease, ABC transporter, and transcription factors were notable during establishment phase. While during necrotrophic phase, several CAZymes, sugar transporters, cellular metabolism, and protein degradation-related genes were prominently induced. We have also identified few putative secreted effector encoding genes that were upregulated during pathogenesis. The qPCR analysis further validated the phase-specific expression dynamics of some selected putative effectors and pathogenicity-associated genes. Overall, the present study reports identification of key genes and processes that might be crucial for R. solani pathogenesis. The ability to effectively damage host cell wall and survive in hostile plant environment by managing oxidative stress, cytotoxic compounds, etc. is being proposed to be important for pathogenesis of R. solani in rice. The functional characterization of these genes would provide key insights about this important pathosystem and facilitate development of strategies to control this devastating disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The sheath blight disease is one of the most economically important fungal diseases of rice. It is caused by the basidiomycete necrotrophic pathogen Rhizoctonia solani Kuhn (telemorph; Thanatephorus cucumeris) AG1-IA. In general, R. solani strains demonstrate broad host range and cause disease on diverse plants including cereals, potato, bean, cotton, sugar beet, lettuce, melon, forest trees, and ornamental plants, etc (Anderson 1982; Sneh et al. 1991). They also demonstrate considerable variability in terms of their morphological and pathological attributes (Wang et al. 2013). R. solani has been classified into 14 different anastomosis groups, i.e., AG-1 to AG-13 and AG-BI (Kuninaga et al. 1997; Guillemaut et al. 2003; Ahvenniemi et al. 2009). Beside hyphal fusion characteristics and sequence polymorphisms in ITS region, karyotype banding patterns have also been used to classify strains into different anastomosis groups (Keijer et al. 1996). The strains belonging to AG1-IA anastomosis group cause disease in rice as well as other agriculturally important crops like corn, barley, sorghum, potato, millet, soybean, and peanut etc (Sneh and Ichielevich-Auster 1998; Fenille et al. 2002). Moreover, different AG1-IA strains demonstrate hypervariable pathological attributes such as number of disease lesions, size of the lesion, RVSC (relative vertical sheath colonization), disease score, relative lesion length, etc, on a particular host (Taheri et al. 2007; Das et al. 2013).
The sclerotia formed by R. solani serve as a major source of primary inoculum. They can survive for long durations in the soil and infect rice plants during its growing season. Under favorable conditions of high temperature (~ 28 °C) and humidity (~ 95%), infection is known to spread rapidly on rice tillers (Lee and Rush 1983). It is worth noting that inspite of extensive global efforts, no source of complete disease resistance has been identified yet. However, some major and minor QTLs providing quantitative resistance against R. solani are known (Prasad and Eizenga 2008; Channamallikarjuna et al. 2009; Wang et al. 2011).
With advent of next-generation sequencing platform, it has become easier to perform functional genomic studies involving phytopathogens (Jeon et al. 2007; Rhind et al. 2011; Laabei and Massey 2016; Lee et al. 2016). Also an arduous task of research on filamentous fungi like R. solani has gained an impetus in recent years. The draft genome sequences of AG1-IA (36.94 Mb encoding 10,489 ORFs) along with some other R. solani strains of different anastomosis groups (AG1-IB, AG2-2IIIB, AG3, and AG8) are now publicly available (Wibberg et al. 2013, 2016; Zheng et al. 2013; Cubeta et al. 2014; Hane et al. 2014). These studies have generated a plethora of genomic information which can be a propitious resource to understand the pathogenicity mechanism of R. solani. However due to multinucleate nature of the pathogen (Sinclair 1970), it is difficult to functionally characterize the R. solani genes, making it a challenging pathosystem to study.
In our previous study, we observed PB1 and TP309 rice cultivars to be susceptible whereas Tetep to be partially resistant against R. solani infections (Ghosh et al. 2014). During the early stage of its pathogenesis (1 dpi; days post inoculation), mycelia growing parallel to rice veins without any anatomical alteration or physiological alteration of host tissues were observed (Ghosh et al. 2017). However, during 3 dpi of pathogenesis, we observed severe anatomical alterations, disintegration of chloroplast integrity along with cell death of infected tissues. Thus, we have considered 1 dpi of R. solani pathogenesis as its establishment phase while 3 dpi as its necrotrophic phase. Recently, we have summarized the molecular and physiological alterations in the host, during pathogenesis of R. solani (Ghosh et al. 2017). In this study, we are focusing on characterizing the transcriptional dynamics of R. solani genes that were commonly expressed during its establishment and necrotrophic phases of pathogenesis on different rice genotypes (PB1, TP309, and Tetep). Several candidate pathogenicity determinants including putative effectors were predicted and their roles during R. solani pathogenesis are being postulated in this study.
Materials and methods
Biological materials and pathological assays
Oryza sativa ssp. indica (cv. PB1), O. sativa ssp. japonica (cv. TP309), O. sativa ssp. indica (cv. Tetep), and R. solani AG1-IA strain BRS1 were used in this study. BRS1 sclerotia were cultured on PDA (39 g/L; Potato Dextrose Agar; Himedia, Mumbai, India) plates at 28 °C. The freshly prepared equal-sized sclerotia were used for inoculation. Single sclerotium was used to infect each rice tillers by inserting them into the second sheath. Rice was grown in a PGV36 conviron plant growth chamber at 28 °C temperature, 80% relative humidity, and 12/12 h of day/night cycle. 60-day-old rice sheaths were infected with R. solani sclerotia, following the procedures described in (Ghosh et al. 2014). After 1, 2, and 3 day post-inoculation, the infected tissues (including 1 cm up and down from the site of infection) were harvested for transcriptome and expression analysis.
RNAseq library preparation and sequencing
RNAseq were performed for three different R. solani infected rice genotypes (PB1, TP309, and Tetep). RNA from R. solani infected rice sheath (five infected sheath pooled up for each sample) at 1 and 3 day post-infections (dpi) were isolated using RNeasy Plant Mini Kit (Qiagen). In addition, RNA from sclerotia grown in PDB broth (laboratory media) for 1 and 3 days at 28 °C were also harvested. The transcriptome sequencing was performed using paired end (PE) 2 × 100 bp library on Illumina HiSeq 2000 for each of these samples. The library was prepared using TrueSeq stranded mRNA HT sample preparation kit. TrueSeq PE cluster kit v3-cBot-HS was used for cluster generation, while the TrueSeq SBS kit v3-HS (200 cycles) was used for RNA sequencing. All these procedures were performed using standard protocols recommended by the manufacturer (Illumina Inc.).
Transcriptome assembly, annotation, and differential expression analysis
Trimmomatic version-0.32 was used for pre-processing of reads to remove TruSeq3 pair-end adapter sequences (Bolger et al. 2014). The reads obtained from 1 and 3 dpi laboratory grown R. solani strain BRS1 samples were used as bonafide fungal reads. These reads were assembled using Trinity version r20140717 (Haas et al. 2013) to obtain fungal transcripts. These transcripts were utilized to filter out the fungal transcripts from R. solani infected rice samples (Online Resource 1). Transdecoder version r20140704 (Haas et al. 2013) was used to detect coding regions. Annotation was performed using blastx, blastp, hmmscan, and rnammer tools. An e-value cutoff of 1e−6 was applied for all blast searches. RSEM v1.2.19 was used for differential gene expression analysis (Zhao et al. 2011). This program aligns input reads against a reference transcriptome using Bowtie2 and calculates expression values using the alignments. EBSeq program was used to perform differential expression analysis. All genes/transcripts and their associated statistics are reported as differentially expressed only if the FDR is less than 0.05.
Selection of phase-specific in-planta upregulated genes
The log fold change of genes expressed during in-planta infection at 1 and 3 dpi was calculated with respect to their expression in laboratory media and transcripts having fold change ≥ 2 were selected. To determine the in-planta upregulated phase-specific genes, only transcripts showing upregulation at a particular phase in each of the three rice genotypes (PB1, TP309, and Tetep) were selected. This led us to select core genes that were expressed uniquely during establishment, necrotrophic phase, and those that were commonly expressed at both these phases. A flowchart representing steps involved in selection of pathogenesis genes of R. solani is depicted in Online Resource 1. Morpheus software was used to plot the heat map of differentially expressed genes of R. solani during pathogenesis in three rice genotypes (Online Resource 2).
Functional annotation and metabolic pathway analysis
Physiological relevance and functional categorization of differentially expressed genes were carried out by consolidating genes that share a certain function or participate in a common process. The Gene Ontology terms (GO) were assigned to these genes using the Blast2GO software. GO terms showing high abundance (> 4%) were classified as predominant categories during respective phases of infection. The CAZymes encoding genes were predicted using the CAT software (CAZy Analysis Toolkit). The sugar transporters were predicted from the KEGG and KOG database. Furthermore, R. solani transcripts were searched in PHI-base (Pathogen Host Interactions Database) database with E value < 10−5 to identify putative pathogenicity determinants. The genes obtained were categorized according to their orthologs mutant phenotype (reduced virulence, loss of pathogenicity, lethal, and effector). The term reduced virulence refers to the mutant that shows lesser disease symptoms in comparison to the wild-type strain, loss of pathogenicity refers to the mutant unable to cause disease, and lethal refers to the mutant that is unable to survive. Effector prediction of R. solani was performed using SignalP, Phobius, and EffectorP software, and the common genes obtained from these softwares were designated as putative secreted effector genes of R. solani. The sequence similarities (obtained through BLASTX) in terms of percent identify along with respective E value for each of the PHI-base orthologs and effectors encoding genes across different sequenced strains of R. solani were recorded.
qPCR-based verification
To validate the expression pattern of selected pathogenicity-related genes of R. solani, qPCR-based expression analysis was carried out at three different time points (1, 2 and 3 dpi) during pathogenesis on susceptible rice cultivar (PB1). qPCR primers were designed in such a manner that they selectively amplify fungal genes but not any rice sequences (Online Resource 3). 2 µg of RNA was used for cDNA synthesis using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) as per specified protocol. 18s rDNA sequence of R. solani was used as a reference gene to normalize gene expression and relative expression was calculated using 2−ΔCt method, wherein ΔCt is the difference between Ct values of target and reference gene (Kiirika et al. 2012). However, to correlate with the RNAseq data, we also estimated fold change of expression of these genes at 1 and 3 dpi by the ΔΔCt method (Livak and Schmittgen 2001).
For some of the pathogenicity-related genes that showed higher fold induction during susceptible interactions (PB1) than in partially resistant interactions (Tetep) in RNAseq analysis, the qPCR analysis was carried out. The fold change in gene expression was calculated at 2 and 3 dpi of pathogenesis with respect to 1 dpi using ΔΔCt method. Each experiment was repeated in at least three biological replicates with three technical replicates.
Results
Transcriptome analysis during R. solani–rice interactions
Previously, we have reported that R. solani strain BRS1 is able to colonize and cause disease on different rice genotypes. Based upon disease severity index, the rice genotypes PB1 and TP309 were susceptible, while Tetep was partially resistant (Ghosh et al. 2014, 2017). During 1 dpi of pathogenesis, the pathogen remains in establishment phase, while at 3 dpi, the pathogen is in its necrotrophic phase. To obtain a robust overview of transcriptional dynamics of R. solani during host infection, the RNAseq analysis was performed at 1 dpi (establishment phase) and 3 dpi (necrotrophic phase) of R. solani pathogenesis on three different rice genotypes (PB1, TP309, and Tetep). Furthermore, the transcripts expressed during 1 and 3 days of R. solani growth in PDB broth (laboratory media) were identified through RNAseq. The differential expression of transcripts was estimated, by comparing their expression in laboratory media and during pathogenesis on each of the rice genotypes at 1 and 3 dpi (Online Resource 1). In this process, we identified a total of 1196 R. solani genes to be differentially regulated during pathogenesis and Online Resource 2 summarizes their expression dynamics (in terms of FPKM values) as heat map. The hierarchical clustering of expression data revealed three distinct clusters of genes being induced during growth in laboratory media, establishment phase, and necrotrophic phase (Online Resource 2). We selected transcripts that showed upregulation in each of the rice genotypes (PB1, TP309, and Tetep), and in this process, we identified 65 and 232 transcripts that were induced exclusively during establishment and necrotrophic phases of R. solani pathogenesis, respectively (Fig. 1a; Online Resource 4). Interestingly, 49 transcripts were found upregulated during both establishment and necrotrophic phases (Fig. 1a). It is important to note that although these genes are found expressed in laboratory media, however, they get significantly induced during pathogenesis in rice.

Representation of phase-specific genes of R. solani upregulated during pathogenesis. a Venn diagram showing the number of in-planta-induced phase-specific genes. b Representation of number of upregulated transcripts encoding CAZymes, sugar transporters, effectors, and PHI-base homologs (categories are defined based upon available mutant phenotypes in other pathosystems) during different phases of R. solani pathogenesis
Functional annotation of establishment and necrotrophic phase-associated genes of R. solani
The gene ontology annotation of the phase associated genes revealed induction of diverse functions during pathogenesis of R. solani (Online Resource 5). The enrichment of GO molecular functions, i.e., catalytic activity and binding as predominant molecular functions were observed during both establishment phase and necrotrophic phase (Fig. 2). The protein kinase activity was prominent catalytic activity during establishment phase, while the hydrolase activity involved in breakdown of glycosyl bonds was abundant during necrotrophic phase. Amongst binding GO term, heterocyclic compound along with organic cyclic compound binding was found induced during both establishment and necrotrophic phases. However, protein binding term was uniquely associated with establishment phase, while carbohydrate binding and pattern (polysaccharide/cellulose) binding GO terms were abundant during necrotrophic phase. In addition, transcripts associated with nutrient reservoir and transmembrane transporter activities were overrepresented during establishment phase.

Gene Ontology classification of R. solani transcripts being expressed during different phases of pathogenesis. Box represents percentage of GO molecular function terms associated with upregulated genes during establishment phase (a), in both establishment as well as necrotrophic phase (b), and during necrotrophic phase (c) of R. solani pathogenesis
Expression dynamics of putative pathogenesis-associated genes of R. solani
Necrotrophic fungi like R. solani secrete an array of cell wall degrading enzymes (CAZymes) to break down complex plant macromolecules like cellulose and pectin into simple sugars to facilitate host colonization (Talbot 2010; de Wit et al. 2012; Bennati-Granier et al. 2015). Successful pathogenesis involves availability and transportation of nutrients, such as sugars from infected host tissues to the pathogen (Solomon et al. 2003; Cornell et al. 2007; Talbot 2010). Fungal pathogens are known to utilize secreted effector proteins to promote pathogenesis by either suppressing host defense response or by promoting cell wall damage (De Jonge et al. 2011; Wang et al. 2014). In addition, PHI-base provides information about experimentally curated genes that are associated with reduced virulence, unaffected pathogenicity, loss of pathogenicity, lethal and effectors of different fungi, bacteria, and oomycetes pathogens (Winnenburg et al. 2006). Considering their importance, in this study, we focused on understanding the expression dynamics of in-planta upregulated R. solani genes that encode CAZymes, sugar transporters, effectors, and PHI-base orthologs (Fig. 1b).
Cell wall degrading enzymes
We observed 9 CAZymes to be induced during establishment phase, while 116 CAZymes upregulated during necrotrophic phase. The GT32 (glycosyl transferase) and CBM57 (carbohydrate binding-module) family were exclusively induced during establishment phase, while GH18 (glycosyl hydrolase) encoding chitinase, CE10 (carbohydrate esterase) encoding monooxygenase, and CBM12 encoding aminopeptidase were induced during both necrotrophic and establishment phases. The transcripts belonging to AA3|AA8 (auxillary activity family), CE5, GH1, GH10, GH105, GH43, GH5|CBM1, GH51, GH76, GH92, GT1, GT34, PL1 (polysaccharide lyase), and PL4 CAZyme family were found to be particularly induced during necrotrophic phase (Fig. 3; Online Resource 6). These transcripts were encoding important cell wall degrading enzymes like cellobiose dehydrogenase, β-xylanase, β-glucanase, β-glucosidase, arabinosidase, arabinofuranosidase, cutinase, and glucosyl transferase, etc, highlighting their importance during necrotrophy. Upregulation of a chitin deacetylase encoding gene (CL8196Contig1) which converts chitin (a primary constituent of fungal cell wall) into chitosan was also observed during necrotrophic phase. Chitosan is believed to be more elastic and resistant against action of host enzymes (such as chitinases) (Zhao et al. 2010). Thus, upregulation of chitin deacetylase might help the pathogen to protect itself from action of host cell wall degrading enzymes.

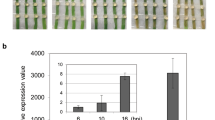
Schematic representation of genes associated with different phases of R. solani pathogenesis. The differentially expressed R. solani genes during pathogenesis on susceptible rice genotype (PB1) were classified under three categories, namely establishment phase, necrotrophic phase, and common in both these phases. The prominent functions along with name of particular upregulated genes under each category are summarized. The colored boxes represent number of upregulated paralogous genes in RNAseq analysis and color scale (red to blue in descending manner) represents extent of upregulation. The relative expression with respect to reference gene (18s rRNA) was estimated for few selected genes (highlighted in bold) through qPCR and data is represented as bar graphs. Standard error bars are represented as vertical lines and different letters represent significant difference at p value < 0.05 (estimated by one-way ANOVA). GST; refers to general substrate transporter
Interestingly, we observed ~ 24% the CAZymes induced during necrotrophic phase to show higher induction (≥ twofold) in susceptible cultivars (PB1 and TP309) than in Tetep, partially resistant cultivar (Online Resource 7). The glycoside hydrolase family, chitin deacetylase, cellobiohydrolyase, cellobiose dehydrogenase, aldose epimerase, etc. were prominent amongst them.
Sugar transporters
A total of 18 major facilitator superfamily proteins, comprising of membrane transporters putatively involved in transport of small solutes across the membrane (Cui et al. 2015; Quistgaard et al. 2016), were found induced during pathogenesis in rice (Fig. 1b; Online Resource 8). We detected 3 such transcripts being particularly induced during establishment phase while 14 of them to be induced during necrotrophic phase. Notably, the carboxylic acid transporters (CL7257Contig1 and CL8578Contig1) were upregulated at establishment phase, while genes encoding lactose permease (CL3435Contig1, CL12132Contig1 and CL12687Contig1), sugar transporter (c20529_g1_i1), hexose transporter (CL5845Contig1), and drug resistance transporter (CL7221Contig1) were induced exclusively during necrotrophic phase (Fig. 3). In addition, we observed an MFS general substrate transporter (CL3034Contig1) being upregulated at both establishment and necrotrophic phases of R. solani infection.
Effectors
The putative secreted effector proteins of R. solani induced during pathogenesis in rice were predicted by different computational tools (SignalP, Phobius, and EffectorP). The common (n = 12) amongst them were selected and most of them were uncharacterized (Online Resource 9). We also investigated the conservation of these genes across different sequenced strains of R. solani. In general, most of them were having low sequence similarity; however, a few of the necrotrophic phase-specific effectors encoding putative cell wall degrading enzymes were relatively more conserved (Online Resource 9). Furthermore, the expression profiles of five selected effector encoding genes of R. solani were studied through qPCR and their expression patterns were found to be comparable to that obtained through RNAseq analysis (Fig. 4).

Expression dynamics of putative secreted effectors of R. solani during different phases of pathogenesis in PB1 rice. The relative expression of selected secreted effector encoding genes with respect to reference gene (18s rRNA) was quantified through qPCR and are summarized as bar graphs. Standard error bars are represented as vertical lines and different letters represent significant difference at p value < 0.05 (estimated by one-way ANOVA)
PHI-base homologs
We identified 63 PHI-base orthologs to be upregulated during R. solani infection process (Online Resource 10). Several of them showed homology to virulence determinants of important fungal pathogens such as Fusarium graminearum, Magnaporthe oryzae, Aspergillus fumigatus, Candida albicans, and Cryptococcus neoformans. Nine of PHI-base orthologs were found induced during establishment phase of R. solani pathogenesis. Amongst them, seven were associated with reduced virulence, while two were related to loss of pathogenicity phenotype (Fig. 1). We also detected 44 PHI-base orthologs being induced during necrotrophic phase wherein 33 genes were associated with reduced virulence, 2 genes with loss of pathogenicity, 5 genes having lethal phenotype, and 4 genes to encode potential effectors (Online Resource 10). 10 genes having reduced virulence phenotype were found to be upregulated during both establishment and necrotrophic phases of R. solani pathogenesis on rice (Fig. 1). We studied their conservation across different R. solani strains and found most of them to have high sequence similarity with i strains belonging to different anastomosis groups (Online Resource 10).
Validation of expression dynamics of putative pathogenicity determinants of R. solani through qPCR
We selected few establishment and necrotrophic-specific genes to validate their expression dynamics through qPCR. Reduced virulence phenotype-associated genes, i.e., Mpr1 (c22975_g3_i1), transcription factor GzZC236 (CL7543Contig1), and loss of pathogenicity phenotype-associated ABC3 transporter (c19012_g1_i2) were selected amongst establishment phase specific genes. Similarly, GH43 family (CL10447Contig1), GT34 family (CL16Contig3), MFS general substrate transporter (CL978Contig1) along with reduced virulence phenotype AOX1 (CL4725Contig1), MEP1 (CL8460Contig1), and SPM1 (c23749_g6_i2) were selected amongst necrotrophic phase-specific genes. We could observe strong correlation between RNAseq and qPCR based expression data (Fig. 3; Online Resource 11).
Interestingly, several genes were depicting higher induction (> twofold change difference) during pathogenesis in susceptible rice genotypes (PB1 and TP309) than in partially resistant rice genotype (Tetep) (Online Resource 7). The qPCR-based expression analysis of few such genes like CAZymes (chitin deacetylase; CL8196Contig1 and endoglucanase; CL6520Contig1), PHI-base category encoding potential effector (c26016_g1_i1; CL3589Contig1 and CL5400Contig1), and transcription factors (c18866_g1_i2) were carried out in R. solani infected PB1 and Tetep rice genotypes (Fig. 5). Both RNAseq and qPCR analyses correlated higher fold induction of these genes during pathogenesis on PB1 than on Tetep (Fig. 5; Online Resource 12).

qPCR-based expression profile of R. solani genes during pathogenesis in PB1 (susceptible) and Tetep (partially resistant) rice genotype. The fold change in the expression of CL8196Contig1 (chitin deacetylase), CL6520Contig1 (endoglucanase), c18866_g1_i2 (transcription factor), and c26016_g1_i1, CL3589Contig1, CL5400Contig1 (orthologs of MoCDIP4; effector) genes at 2 and 3 dpi with respect to 1 dpi of pathogenesis is represented as bar graphs. 18s rRNA gene of R. solani was used as reference for normalization. The Y-axis represents mean fold change observed in three biological replicates. Standard error bars are represented as vertical lines. Different letters indicate significant difference at p value < 0.05 (estimated by one-way ANOVA)
Discussion
In this study, using an Indian isolate of R. solani AG1-IA (strain BRS1), we identify various candidate pathogenicity determinants during establishment and necrotrophic phases of its pathogenesis on rice. The transcriptome analysis suggested that BRS1 is quite diverse from the other R. solani strains whose genome sequences are publically available. Only 47% of the BRS1 transcripts showed best sequence similarity to AG1-IA strain (Chinese isolate), while 23 and 28% of them showed BLASTX hits with AG1-IB and AG3 strains, respectively. Previously, gene expression studies in R. solani were explored by detached leaf assay using mycelial plugs or inoculum toothpicks (Zhao et al. 2008; Zheng et al. 2013). However, in this study, we have used sclerotia (natural infection propagules of R. solani) of BRS1 to infect intact rice tillers. Furthermore, we particularly selected R. solani transcripts that were found upregulated in three different rice genotypes (having variable degree of disease susceptibility) and identified those which were associated with establishment and necrotrophic phase of R. solani pathogenesis. The upregulation of these R. solani genes in each of the rice genotypes highlights their ubiquitous and indispensable role during sheath blight disease in rice. Many of them showed sequence homology with PHI-base genes that were associated with reduced virulence, loss of pathogenicity, lethal, and effectors in various other pathosystems. Based upon in-planta induction pattern and conservation amongst different R. solani strains (Online Resource 8), it is tempting to speculate that these genes might be playing crucial role during pathogenesis of R. solani. We observed ABC (ATP-binding cassette) transporter, transcription factors, and extracellular metalloprotease (Mpr1) to be induced exclusively during establishment phase. The ABC3 transporters of M. oryzae are known to be involved in the efflux of cytotoxic compounds such as phytoalexins produced by the host (Sun et al. 2006; Patkar et al. 2012). The Mpr1 orthologs in F. verticillioides encode fungalysins (zinc metalloproteases) and they protect the pathogen from the action of host chitinases produced as a defense strategy (Naumann et al. 2011; Vu et al. 2014). Taken together, this suggests that during establishment phase, R. solani upregulates its arsenals to protect itself from cell wall degrading enzymes and cytotoxic compounds produced by the host (Fig. 3). Furthermore, upregulation of transcriptional factor encoding genes such as CRZ1 (“CRaZy,” calcineurin-responsive zinc finger transcription factor) and GzZC236 highlights their importance in regulating expression of pathogenicity-associated genes during host colonization. It is noteworthy that the CRZ1 homolog in M. oryzae is involved in modulating virulence functions and regulating delivery of effectors (Schumacher et al. 2008; Kim et al. 2010).
The PHI-base orthologs encoding cytochrome P450s (CYP52X1), appressorial penetration-associated proteins (GAS1), and phosphate utilization-related proteins (PHO84) were found induced during both establishment and necrotrophic phases of pathogenesis (Fig. 3). Interestingly, the GAS1 (encoding β-1,3-glucanosyltransferase) mutants of M. oryzae and F. oxysporum were defective in appressorium penetration and causing disease (Xue et al. 2002; Caracuel et al. 2005). As during pathogenesis, R. solani forms infection cushions or lobate appressoria (Basu et al. 2016; Ghosh et al. 2017), it would be interesting to test the potential involvement of the GAS1 gene (CL4124Contig1 and CL5792Contig1) in this context.
Our study suggests that progression of R. solani into necrotrophic phase is associated with induction of large suite of cell wall degrading enzymes, effector proteins, and ability to manage oxidative stress. The induction of two orthologs of AOX1 (encoding alternate oxidase) genes was observed during necrotrophic phase. AOX1 is involved in alternative oxidative pathway, and it provides resistance against oxidative stress during pathogenesis of Cladosporium fulvum and C. neoformans, etc (Segers et al. 2001; Akhter et al. 2003). We also observed induction of two orthologs of SidH (Anoyl-CoA hydratase) and an ortholog of DHOD (fumarate reductase) during necrotrophic phase of R. solani. SidH is known to be involved in production of siderophore in A. fumigatus, which in turn assists the pathogen to survive under iron starved oxidative stress conditions. On the other hand, DHOD is involved in maintaining cellular redox homeostasis in Trypanosoma cruzi (human pathogen) to survive in anaerobic host conditions (Annoura et al. 2005; Yasmin et al. 2012). This affirms that R. solani has adapted its arsenals to survive under hostile oxidative and anaerobic stress conditions inside host plants (Fig. 3). It is worth noting that recent transcriptome studies had also suggested that R. solani adopts strategies to combat oxidative stress to infect diverse host plants such as soybean, turf grass, rice, maize, and lettuce etc (Zheng et al. 2013; Zhu et al. 2016; Copley et al. 2017).
A few secreted effector encoding genes of R. solani were found induced during pathogenesis on rice (Online Resource 9). The limited sequence similarities of these effectors amongst various R. solani strains suggest that they might be evolving rapidly. It is known that phytopathogens diversify effector proteins to escape from host recognition (Spanu et al. 2010; De Jonge et al. 2011; de Wit et al. 2012), although we observed some of the necrotrophy-specific effector genes encoding plant cell wall damaging enzymes to be relatively more conserved amongst different R. solani strains. It is worth noting that effectors involved in necrosis-associated host cell death have been often described in other pathosystems (Oliver et al. 2012; Lo Presti et al. 2015). For example, M. oryzae utilizes MoCDIP4, encoding secreted effector proteins to induce necrotrophic phase-specific cell death response in rice (Chen et al. 2013; Shirke et al. 2016). Notably, three orthologs of MoCDIP4 in R. solani were showing more than sixfold higher induction in susceptible rice genotypes (PB1 and TP309) than in partially resistant (Tetep) rice (Fig. 5). Not only MoCDIP4 orthologs, several other necrotrophy associated cell wall degrading enzymes were also having several fold induction in susceptible than partially resistant interactions. Overall, this suggests that induction level of these genes might be associated with extent of host cell wall damage which in turn might influence the disease severity of R. solani on different rice genotypes. It is possible that the expression level of these genes can be explored as markers of disease susceptibility or resistance in the conventional breeding programs. In general, rapid induction of host cell wall damage is known to activate plant defense response (Jha et al. 2007; Schwessinger and Zipfel 2008; Dodds and Rathjen 2010; Sinha et al. 2013; Tayi et al. 2016). As Tetep is known to harbor several major disease resistance QTLs against various pathogens (Barman et al. 2004; Channamallikarjuna et al. 2009), thus to avoid induction of plant defense, it is possible that R. solani might be restricting higher fold induction of these genes during pathogenesis on Tetep.
In conclusion, our study has identified candidate pathogenicity determinants of R. solani that might play a crucial role during establishment of rice sheath blight disease. In general, homologous recombination, insertional mutagenesis, RNAi-based gene silencing, etc had been explored to functionally characterize pathogenicity-associated genes of fungi (Jeon et al. 2007; Li et al. 2015; Zhang et al. 2015). However, as it is difficult to genetically transform R. solani, establishing the role of identified pathogenicity determinants remains a challenge. Exploring host induced gene silencing approach (Nowara et al. 2010; Jahan et al. 2015) to knock-down the R. solani genes during its pathogenesis process might turn out to be an alternate strategy.
References
Ahvenniemi P, Wolf M, Lehtonen MJ et al (2009) Evolutionary diversification indicated by compensatory base changes in ITS2 secondary structures in a complex fungal species, Rhizoctonia solani. J Mol Evol 69:150–163. https://doi.org/10.1007/s00239-009-9260-3
Akhter S, Mcdade HC, Gorlach JM et al (2003) Role of alternative oxidase gene in pathogenesis of Cryptococcus neoformans role of alternative oxidase gene in pathogenesis of Cryptococcus neoformans. Infect Immun 71:5794–5802. https://doi.org/10.1128/IAI.71.10.5794
Anderson NA (1982) The genetics and pathology of Rhizoctonia solani. Annu Rev Phytopathol 20:329–347. https://doi.org/10.1146/annurev.py.20.090182.001553
Annoura T, Nara T, Makiuchi T et al (2005) The origin of dihydroorotate dehydrogenase genes of kinetoplastids, with special reference to their biological significance and adaptation to anaerobic, parasitic conditions. J Mol Evol 60:113–127. https://doi.org/10.1007/s00239-004-0078-8
Barman SR, Gowda M, Venu RC, Chattoo BB (2004) Identification of a major blast resistance gene in the rice cultivar “Tetep”. Plant Breed 123:300–302. https://doi.org/10.1111/j.1439-0523.2004.00982.x
Basu A, Chowdhury S, Ray Chaudhuri T, Kundu S (2016) Differential behaviour of sheath blight pathogen Rhizoctonia solani in tolerant and susceptible rice varieties before and during infection. Plant Pathol 65:1333–1346. https://doi.org/10.1111/ppa.12502
Bennati-Granier C, Garajova S, Champion C et al (2015) Substrate specificity and regioselectivity of fungal AA9 lytic polysaccharide monooxygenases secreted by Podospora anserina. Biotechnol Biofuels 8:90. https://doi.org/10.1186/s13068-015-0274-3
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Caracuel Z, Martínez-rocha AL, Pietro A, Di et al (2005) Fusarium oxysporum gas1 encodes a putative β-1,3-glucanosyltransferase required for virulence on tomato plants. Mol Plant Microbe Interact 18:1140–1147. https://doi.org/10.1094/MPMI-18-1140
Channamallikarjuna V, Sonah H, Prasad M et al (2009) Identification of major quantitative trait loci qSBR11-1 for sheath blight resistance in rice. Mol Breed 25:155–166. https://doi.org/10.1007/s11032-009-9316-5
Chen S, Songkumarn P, Venu RC et al (2013) Identification and characterization of in planta-expressed secreted effector proteins from Magnaporthe oryzae that induce cell death in rice. Mol Plant Microbe Interact 26:191–202. https://doi.org/10.1094/MPMI-05-12-0117-R
Copley TR, Duggavathi R, Jabaji S et al (2017) The transcriptional landscape of Rhizoctonia solani AG1-IA during infection of soybean as defined by RNA-seq. PLoS One 12:e0184095. https://doi.org/10.1371/journal.pone.0184095
Cornell MJ, Alam I, Soanes DM et al (2007) Comparative genome analysis across a kingdom of eukaryotic organisms: specialization and diversification in the fungi. Genome Res 17:1809–1822. https://doi.org/10.1101/gr.6531807
Cubeta MA, Thomas E, Dean RA et al (2014) Draft genome sequence of the plant–pathogenic soil fungus Rhizoctonia solani anastomosis group 3 strain Rhs1AP. Genome Announc. 2(5):e1072-14 https://doi.org/10.1128/genomeA.01072-14
Cui Z, Gao N, Wang Q et al (2015) BcMctA, a putative monocarboxylate transporter, is required for pathogenicity in Botrytis cinerea. Curr Genet 61:545–553. https://doi.org/10.1007/s00294-015-0474-1
Das S, Shah FA, Butler RC et al (2013) Genetic variability and pathogenicity of Rhizoctonia solan i associated with black scurf of potato in New Zealand. Plant Pathol 63:651–666. https://doi.org/10.1111/ppa.12139
De Jonge R, Bolton MD, Thomma BPHJ. (2011) How filamentous pathogens co-opt plants: the ins and outs of fungal effectors. Curr Opin Plant Biol 14:400–406. https://doi.org/10.1016/j.pbi.2011.03.005
de Wit PJGM., van der Burgt A, Ökmen B et al (2012) The Genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet 8(11): e1003088. https://doi.org/10.1371/journal.pgen.1003088
Dodds PN, Rathjen JP (2010) Plant immunity: towards an integrated view of plant–pathogen interactions. Nat Rev Genet 11:539–548. https://doi.org/10.1038/nrg2812
Fenille RC, de Souza NL, Kuramae EE (2002) Characterization of Rhizoctonia solani associated with soybean in Brazil. Eur J Plant Pathol 108:783–792. https://doi.org/10.1023/A:1020811019189
Ghosh S, Gupta SK, Jha G (2014) Identification and functional analysis of AG1-IA specific genes of Rhizoctonia solani. Curr Genet. 60: 327-341. https://doi.org/10.1007/s00294-014-0438-x
Ghosh S, Kanwar P, Jha G (2017) Alterations in rice chloroplast integrity, photosynthesis and metabolome associated with pathogenesis of Rhizoctonia solani. Sci Rep 7:41610. https://doi.org/10.1038/srep41610
Guillemaut C, Edel-Hermann V, Camporota P et al (2003) Typing of anastomosis groups of Rhizoctonia solani by restriction analysis of ribosomal DNA. Can J Microbiol 49:556–568. https://doi.org/10.1139/w03-066
Haas BJ, Papanicolaou A, Yassour M et al (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512. https://doi.org/10.1038/nprot.2013.084
Hane JK, Anderson JP, Williams AH et al (2014) Genome sequencing and comparative genomics of the broad host-range pathogen Rhizoctonia solani AG8. PLoS Genet 10(5): e1004281. https://doi.org/10.1371/journal.pgen.1004281
Jahan SN, Asman AKM, Corcoran P et al (2015) Plant-mediated gene silencing restricts growth of the potato late blight pathogen Phytophthora infestans. J Exp Bot 66:2785–2794. https://doi.org/10.1093/jxb/erv094
Jeon J, Park S-Y, Chi M-H et al (2007) Genome-wide functional analysis of pathogenicity genes in the rice blast fungus. Nat Genet 39:561–565. https://doi.org/10.1038/ng2002
Jha G, Rajeshwari R, Sonti RV (2007) Functional interplay between two Xanthomonas oryzae pv,. oryzae secretion systems in modulating virulence on rice. Mol Plant Microbe Interact 20:31–40. https://doi.org/10.1094/MPMI-20-0031
Keijer J, Houterman PM, Dullemans AM, Korsman MG (1996) Heterogeneity in electrophoretic karyotype within and between anastomosis groups of Rhizoctonia solani. Mycological Research 100(7):789–797
Kiirika LM, Bergmann HF, Schikowsky C et al (2012) Silencing of the Rac1 GTPase MtROP9 in Medicago truncatula stimulates early mycorrhizal and oomycete root colonizations but negatively affects rhizobial infection. Plant Physiol 159:501–516. https://doi.org/10.1104/pp.112.193706
Kim S, Hu J, Oh Y et al (2010) Combining chip-chip and expression profiling to model the MoCRZ1 mediated circuit for Ca2+/calcineurin signaling in the rice blast fungus. PLoS Pathog 6(5): e1000909. https://doi.org/10.1371/journal.ppat.1000909
Kuninaga S, Natsuaki T, Takeuchi T, Yokosawa R (1997) Sequence variation of the rDNA ITS regions within and between anastomosis groups in Rhizoctonia solani. Curr Genet 32:237–243. https://doi.org/10.1007/s002940050272
Laabei M, Massey R (2016) Using functional genomics to decipher the complexity of microbial pathogenicity. Curr Genet 62:523–525. https://doi.org/10.1007/s00294-016-0576-4
Lee FN, Rush MC (1983) Rice sheath blight: a major rice disease. Plant Dis 67:829–832. https://doi.org/10.1094/PD-67-829
Lee HH, Park J, Kim J et al (2016) Understanding the direction of evolution in Burkholderia glumae through comparative genomics. Curr Genet 62:115–123. https://doi.org/10.1007/s00294-015-0523-9
Li Y, Que Y, Liu Y et al (2015) The putative Gγ subunit gene MGG1 is required for conidiation, appressorium formation, mating and pathogenicity in Magnaporthe oryzae. Curr Genet 61:641–651. https://doi.org/10.1007/s00294-015-0490-1
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Lo Presti L, Lanver D, Schweizer G et al (2015) Fungal effectors and plant susceptibility. Annu Rev Plant Biol 66:513–545. https://doi.org/10.1146/annurev-arplant-043014-114623
Naumann TA, Wicklow DT, Price NPJ (2011) Identification of a chitinase-modifying protein from Fsarium verticillioides: truncation of a host resistance protein by a fungalysin metalloprotease. J Biol Chem 286:35358–35366. https://doi.org/10.1074/jbc.M111.279646
Nowara D, Gay A, Lacomme C et al (2010) HIGS: Host-induced gene silencing in the obligate biotrophic fungal pathogen Blumeria graminis. Plant Cell 22:3130–3141. https://doi.org/10.1105/tpc.110.077040
Oliver RP, Friesen TL, Faris JD, Solomon PS (2012) Stagonospora nodorum: from pathology to genomics and host resistance. Annu Rev Phytopathol 50:23–43. https://doi.org/10.1146/annurev-phyto-081211-173019
Patkar RN, Xue YK, Shui G et al (2012) Abc3-mediated efflux of an endogenous digoxin-like steroidal glycoside by Magnaporthe oryzae is necessary for host invasion during blast disease. PLoS Pathog. 8(8): e1002888. https://doi.org/10.1371/journal.ppat.1002888
Prasad B, Eizenga GC (2008) Rice sheath blight disease resistance identified in Oryza spp. accessions. Plant Dis 92:1503–1509. https://doi.org/10.1094/PDIS-92-11-1503
Quistgaard EM, Löw C, Guettou F, Nordlund P (2016) Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat Publ Gr 17:1–10. https://doi.org/10.1038/nrm.2015.25
Rhind N, Chen Z, Yassour M et al (2011) Comparative functional genomics of the fission yeasts. Science 332:930–936. https://doi.org/10.1126/science.1203357
Schumacher J, De Larrinoa IF, Tudzynski B (2008) Calcineurin-responsive zinc finger transcription factor CRZ1 of Botrytis cinerea is required for growth, development, and full virulence on bean plants. Eukaryot Cell 7:584–601. https://doi.org/10.1128/EC.00426-07
Schwessinger B, Zipfel C (2008) News from the frontline: recent insights into PAMP-triggered immunity in plants. Curr Opin Plant Biol 11:389–395. https://doi.org/10.1016/j.pbi.2008.06.001
Segers G, Bradshaw N, Archer D et al (2001) Alcohol oxidase is a novel pathogenicity factor for Cladosporium fulvum, but aldehyde dehydrogenase is dispensable. Mol Plant Microbe Interact 14:367–377. https://doi.org/10.1094/MPMI.2001.14.3.367
Shirke MD, Mahesh HB, Gowda M (2016) Genome-wide comparison of Magnaporthe species reveals a host-specific pattern of secretory proteins and transposable elements. PLoS One 11(9): e0162458. https://doi.org/10.1371/journal.pone.0162458
Sinclair JB (1970) Rhizoctonia solani: biology and pathology. In: Parmeter J (ed) Rhizoctonia solani: biology and pathology. University of California Press, Berkeley, pp 199–217
Sinha D, Gupta MK, Patel HK et al (2013) Cell wall degrading enzyme induced rice innate immune responses are suppressed by the type 3 secretion system effectors XopN, XopQ, XopX and XopZ of Xanthomonas oryzae pv. oryzae. PLoS One 8(9): e75867. https://doi.org/10.1371/journal.pone.0075867
Sneh B, Ichielevich-Auster M (1998) Induced resistance of cucumber seedlings caused by some non-pathogenicRhizoctonia (np-R) isolates. Phytoparasitica 26:27–38. https://doi.org/10.1007/BF02981263
Sneh B, Burpee L, Ogoshi A (1991) Identification of Rhizoctonia species. APS Press, St. Paul
Solomon PS, Tan KC, Oliver RP (2003) The nutrient supply of pathogenic fungi; a fertile field for study. Mol Plant Pathol 4:203–210. https://doi.org/10.1046/j.1364-3703.2003.00161.x
Spanu PD, Abbott JC, Amselem J et al (2010) Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science 330:1543–1546. https://doi.org/10.1126/science.1194573
Sun CB, Suresh A, Deng YZ, Naqvi NI (2006) A multidrug resistance transporter in Magnaporthe is required for host penetration and for survival during oxidative stress. Plant Cell 18:3686–3705. https://doi.org/10.1105/tpc.105.037861
Taheri P, Gnanamanickam S, Höfte M (2007) Characterization, genetic structure, and pathogenicity of Rhizoctonia spp. associated with rice sheath diseases in India. Phytopathology 97:373–383. https://doi.org/10.1094/PHYTO-97-3-0373
Talbot NJ (2010) Living the sweet life: How does a plant pathogenic fungus acquire sugar from plants? PLoS Biol 8(2):e1000308. https://doi.org/10.1371/journal.pbio.1000308
Tayi L, Maku R, Patel HK, Sonti R (2016) Action of multiple cell wall degrading enzymes is required for elicitation of innate immune responses during Xanthomonas oryzae pv. oryzae infection in rice. Mol Plant Microbe Interact 29:599–608. https://doi.org/10.1094/MPMI-02-16-0039-R
Vu K, Tham R, Uhrig JP et al (2014) Invasion of the central nervous system by Cryptococcus neoformans requires a secreted fungal metalloprotease. MBio 5(3):e01101–e01114 https://doi.org/10.1128/mBio.01101-01114
Wang Y, Pinson SRM, Fjellstrom RG, Tabien RE (2011) Phenotypic gain from introgression of two QTL, qSB9-2 and qSB12-1, for rice sheath blight resistance. Mol Breed 30:293–303. https://doi.org/10.1007/s11032-011-9619-1
Wang L, Liu LM, Wang ZG, Huang SW (2013) Genetic structure and aggressiveness of Rhizoctonia solani AG1-IA, the cause of sheath blight of rice in Southern China. J Phytopathol 161:753–762. https://doi.org/10.1111/jph.12127
Wang X, Jiang N, Liu J et al (2014) The role of effectors and host immunity in plant–necrotrophic fungal interactions. Virulence 5:722–732. https://doi.org/10.4161/viru.29798
Wibberg D, Jelonek L, Rupp O et al (2013) Establishment and interpretation of the genome sequence of the phytopathogenic fungus Rhizoctonia solani AG1-IB isolate 7/3/14. J Biotechnol 167:142–155. https://doi.org/10.1016/j.jbiotec.2012.12.010
Wibberg D, Andersson L, Tzelepis G et al (2016) Genome analysis of the sugar beet pathogen Rhizoctonia solani AG2-2IIIB revealed high numbers in secreted proteins and cell wall degrading enzymes. BMC Genom 17:245. https://doi.org/10.1186/s12864-016-2561-1
Winnenburg R, Baldwin TK, Urban M et al (2006) PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. https://doi.org/10.1093/nar/gkj047
Xue C, Park G, Choi W et al (2002) Two novel fungal virulence genes specifically expressed in appressoria of the rice blast fungus. Plant Cell 14:2107–2119. https://doi.org/10.1105/tpc.003426
Yasmin S, Alcazar-Fuoli L, Grundlinger M et al (2012) PNAS Plus: mevalonate governs interdependency of ergosterol and siderophore biosyntheses in the fungal pathogen Aspergillus fumigatus. Proc Natl Acad Sci 109:E497–E504. https://doi.org/10.1073/pnas.1106399108
Zhang YL, Li ZF, Feng ZL et al (2015) Isolation and functional analysis of the pathogenicity-related gene VdPR3 from Verticillium dahliae on cotton. Curr Genet 61:555–566. https://doi.org/10.1007/s00294-015-0476-z
Zhao CJ, Wang AR, Shi YJ et al (2008) Identification of defense-related genes in rice responding to challenge by Rhizoctonia solani. Theor Appl Genet 116:501–516. https://doi.org/10.1007/s00122-007-0686-y
Zhao Y, Park R-D, Muzzarelli RAA (2010) Chitin deacetylases: properties and applications. Mar Drugs 8:24–46. https://doi.org/10.3390/md8010024
Zhao Q, Hautamäki V, Fränti P (2011) RSEM: an accelerated algorithm on repeated EM. In: Proceedings—6th International conference on image and graphics, ICIG 2011, pp 135–140
Zheng A, Lin R, Zhang D et al (2013) The evolution and pathogenic mechanisms of the rice sheath blight pathogen. Nat Commun 4:1424. https://doi.org/10.1038/ncomms2427
Zhu C, Ai L, Wang L et al (2016) De novo transcriptome analysis of Rhizoctonia solani AG1 IA strain early invasion in Zoysia japonica root. Front Microbiol 7:1–12. https://doi.org/10.3389/fmicb.2016.00708
Acknowledgements
SG is supported by SPM fellowship from Council of Scientific and Industrial Research (Govt. of India). PK is supported by Post-Doctoral Research fellowship from Department of Biotechnology (DBT, Govt. of India). We acknowledge Nucleome Informatics Pvt. Ltd for assistance in RNAseq. The assistance of central instrumentation facilities of NIPGR for qPCR is acknowledged. This work was supported by DBT, Government of India as well as core research Grant from National Institute of Plant Genome Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
Accession Number: The Illumina sequence data from this study have been submitted as BioProject ID: PRJNA298635 to the NCBI Sequence read archive.
Electronic supplementary material
Below is the link to the electronic supplementary material.
294_2017_791_MOESM1_ESM.tif
Online Resource 1: Pipeline used to delineate in-planta upregulated phase-specific transcripts of R. solani during its pathogenesis in rice (TIF 113 KB)
294_2017_791_MOESM2_ESM.tif
Online Resource 2: Heat map of differentially regulated genes of R. solani showing distinct induction patterns (TIF 1016 KB)
294_2017_791_MOESM4_ESM.xlsx
Online Resource 4: R. solani transcripts expressed during infection in three different rice genotypes (PB1, Tetep and TP309) at establishment, necrotrophic and common in both the phases (XLSX 68 KB)
294_2017_791_MOESM5_ESM.xlsx
Online Resource 5: GO annotation of in-planta upregulated genes of R. solani during different phases of pathogenesis (XLSX 20 KB)
294_2017_791_MOESM6_ESM.xlsx
Online Resource 6: Induction pattern of R. solani transcripts encoding CAZymes during different phases of pathogenesis (XLSX 34 KB)
294_2017_791_MOESM7_ESM.xlsx
Online Resource 7: R. solani genes having >2 log fold difference between susceptible (PB1 and TP309) and partially resistant (Tetep) interactions (XLSX 14 KB)
294_2017_791_MOESM8_ESM.xlsx
Online Resource 8: Induction pattern of R. solani transcripts encoding sugar transporters during different phases of pathogenesis (XLSX 14 KB)
294_2017_791_MOESM9_ESM.xlsx
Online Resource 9: Induction pattern of R. solani transcripts encoding putative secreted effectors during different phases of pathogenesis (XLSX 16 KB)
294_2017_791_MOESM10_ESM.xlsx
Online Resource 10: Induction pattern of R. solani transcripts showing homology to PHI-base during different phases of pathogenesis. The homology of R. solani strain BRS1 genes with other R. solani strains belonging to different anastomosis groups are summarized (XLSX 31 KB)
294_2017_791_MOESM11_ESM.docx
Online Resource 11: Correlation of qPCR and RNAseq based differential expression quantification of a few R. solani genes induced during pathogenesis on PB1 (susceptible) rice genotype (DOCX 16 KB)
294_2017_791_MOESM12_ESM.docx
Online Resource 12: Correlation of qPCR and RNAseq based differential expression quantification of a few R. solani genes induced during pathogenesis on susceptible (PB1) and partially resistant (Tetep) rice genotype (DOCX 15 KB)
Rights and permissions
About this article

Cite this article
Ghosh, S., Kanwar, P. & Jha, G. Identification of candidate pathogenicity determinants of Rhizoctonia solani AG1-IA, which causes sheath blight disease in rice. Curr Genet 64, 729–740 (2018). https://doi.org/10.1007/s00294-017-0791-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-017-0791-7