
Abstract
The promoter of the cre1 gene, encoding the glucose-dependent regulator CRE1 from the β-lactam producer Acremonium chrysogenum, carries 15 putative CRE1 binding sites (BS1 to BS15). For a detailed analysis, we fused cre1 promoter deletion derivatives with the DsRed reporter gene to perform a comparative gene expression analysis. Plate assays, Northern hybridizations, and spectrofluorometric measurements of DsRed identified the minimal D4 promoter sequence that promoted glucose-dependent expression. Truncated recombinant CRE1 interacted with D4 in electromobility shift analysis and these binding studies were further extended with two oligonucleotides, carrying putative CRE1 binding sites BS14 and BS15. Surface plasmon resonance analysis was performed using BS14 and BS15, along with four derivatives containing 2 or 4 bp substitutions within BS14 and BS15, respectively. Substitutions within BS14 abolished the high affinity interaction with CRE1, while mutations in BS15 only marginally diminished the affinity with CRE1. In vivo analysis of a modified D4 sequence with substitutions in the two binding sites confirmed the in vitro binding results and still promoted glucose-dependent gene expression. Our results will contribute to the construction of versatile expression vectors carrying a minimal cre1 promoter sequence that still confers glucose-dependent induction of gene expression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucose is a readily metabolizable carbon source, which leads to enhanced growth rates during fermentation processes. Glucose also causes repression of several enzymes and secondary metabolites by a phenomenon called carbon catabolite repression (CCR) that has been observed in diverse prokaryotic and eukaryotic microorganisms (Martín et al. 1999; Warner and Lolkema 2003). In the yeast Saccharomyces cerevisiae, CCR is mediated by the zinc-finger protein MIG1 that, via binding a consensus sequence (5′-SYGGRG-3′) in several glucose-repressible promoters, interacts with the TUP1/SSN6 general repressor complex to form an active complex and represses gene expression (reviewed by Gancedo 1998). In filamentous fungi, the homologue of the MIG1 repressor (CREA or CRE1) was first isolated from Aspergillus nidulans (Dowzer and Kelly 1989) and later shown to function in diverse pathways, e.g. regulation of proline, ethanol, xylan, and arabinan utilization (reviewed by Ruijter and Visser 1997). In Trichoderma reesei, the industrially important producer of cellulases and hemicellulases, the CRE1 homologue is partially responsible for the inhibition of cellulase expression and also represses the expression of the xylanase I (xyn1) gene (Ilmén et al. 1996; Mach et al. 1996). The CRE proteins function either by directly competing for binding sites of activator proteins or by an indirect mechanism that prevents the action of an activator (Ronne 1995). Characterization of the promoters of many glucose-repressible target genes led to the identification of CRE binding consensus sequences (5′-SYGGRG-3′) (e.g. Kulmburg et al. 1993; Cubero and Scazzocchio 1994; Mathieu et al. 2000; Rauscher et al. 2006). Furthermore, CRE1 does not always directly regulate transcription by interacting with binding sites in promoter sequences, but also indirectly regulates transcription by affecting nucleosome positioning, such as in the 5′-regulatory sequence of the cbh2 gene (Zeilinger et al. 2003). CRE binding sites are not only found in the promoters of target genes but also in the promoters of cre genes from diverse filamentous fungi. The best molecularly characterized creA promoter is the A. nidulans promoter; which contains four CREA binding sequences and large blocks of CT-rich sequences, including long stretches of polydT sequence (Dowzer and Kelly 1989; Kulmburg et al. 1993; Cubero and Scazzocchio 1994). Two of these CREA consensus binding sequences were previously characterized in A. nidulans and shown to function as an autoregulation motif in vivo. The authors showed that the glucose pulse-mediated downregulation of creA transcript levels is abolished when the creA gene was fused with a promoter carrying mutated CREA recognition sequences (Strauss et al. 1999).
In this study, we provide a functional characterization of the cre1 promoter from the β-lactam antibiotic producing fungus Acremonium chrysogenum, in which antibiotic biosynthesis is repressed by glucose (reviewed by Schmitt et al. 2004a). Detailed transcript analysis revealed that the transcript levels of pcbC and cefEF, encoding isopenicillin N synthase and deacetoxycephalosporin C/deacetylcephalosporin C synthetase, respectively, were reduced in the wild type but not in a producer strain (Jekosch and Kück 2000b). The cre1 gene from A. chrysogenum was previously isolated in order to analyze glucose-dependent gene expression, and the CRE1 protein sequence shows high similarity to other glucose repressor proteins from filamentous fungi, suggesting a similar regulation and related role in CCR. Analysis of the cre1 transcript levels in two A. chrysogenum strains showing different production rates of the antibiotic cephalosporin C displayed a different glucose regulation of the cre1 transcript in both strains. The cre1 transcript level in the wild type strain was increased sixfold during growth in glucose-containing, pH-buffered medium, whereas the transcript of the producer strain was unaffected, indicating a positive autoregulation of cre1 in the wild type strain and an absence of glucose regulation in the producer strain.
In order to measure the cre1 promoter-driven gene expression in A. chrysogenum, we used the DsRed reporter gene encoding the red fluorescent protein from Discosoma spec. We recently have shown that the DsRed gene is a valuable reporter not only for quantifying promoter strengths but also for the phenotypic detection of robustly expressed genes on Petri dishes (Janus et al. 2007). Previous sequence analysis of the 1,030 bp cre1 promoter sequence identified 15 putative CRE1 binding sites (BS1 to BS15) (Jekosch and Kück 2000a) that are further characterized in this investigation. This includes in vivo expression studies with different cre1 promoter derivatives as well as in vitro binding studies of CRE1 with putative CRE1 binding sites using electromobility shift analysis (EMSA) and surface plasmon resonance (SPR) analysis. Our experiments are complemented by investigating transformants that carry modified cre1 promoters with mutated CRE1 binding sites. This study identifies a 114 bp minimal promoter with two CRE1 binding sites, the first minimal regulatory promoter sequence for the β-lactam antibiotic producing fungus.
Materials and methods
Strains and culture conditions
Escherichia coli strain K-12 XL1-blue (Stratagene) was used for plasmid construction and maintenance (Bullock et al. 1987). As already described, liquid cultures of A. chrysogenum strains were grown at 27°C and 180 rpm for 3–5 days in CCM medium, CCM complemented with 100 mM HEPES (CCMH), or CCMH with the addition of 6.3% (w/v) glucose (CCMHS) (Walz and Kück 1991; Jekosch and Kück 2000a).
Transformation of A. chrysogenum strain ATCC 14553 was done according to conventional transformation procedures (Walz and Kück 1993; Radzio and Kück 1997). Resulting transformants were selected on media containing hygromycin B at concentrations as previously reported (Walz and Kück 1993; Radzio and Kück 1997). All strains used in this study are listed in Table 1 and their genotypes were verified by Southern blot analysis to determine the copy number of the transformed DsRed gene (data not shown). Copy number determination was done with the Scion Image program (Scion Corporation) to quantify signals from autoradiographs that were obtained from hybridization with the labeled DsRed probe. For reference, the membranes were reprobed with a labeled cre1 fragment that detects the single copy cre1 gene in A. chrysogenum. DsRed copy numbers of individual transformants are given in Table 1.
Construction of the cre1 promoter derivatives
For amplifying the cre1 promoter from plasmid pCRE13/2 (Jekosch and Kück 2000a) primers 3148 and 3149 were used. The PCR fragment was ligated into plasmid pDrive (Qiagen) and subsequently sequenced. The resulting plasmid was hydrolyzed with AgeI and partially digested with NcoI. The corresponding DNA fragment was used for ligation with the AgeI-NcoI-restricted fragment of plasmid pRHN1. The reporter gene plasmid pRHN1 carries the full-size DsRed gene (DsRed-Express; BD Biosciences Clontech, Heidelberg, Germany) under the control of the gpd promoter and trpC terminator of A. nidulans and an hph-resistance cassette (Godehardt and Kück, unpublished data). The resulting recombinant plasmid was designated pRPcre1 carrying the DsRed gene under control of the cre1 promoter (Pcre1).
The construction of the cre1 promoter deletion derivatives was performed by conventional PCR or fusion PCR based on modified protocols described previously (Shevchuk et al. 2004). The cre1 promoter fragments for generating deletion derivatives D1, D3, and D4 were amplified by using primers Pcre1-D1/Pcre1-D3/Pcre1-D4 and 3149, respectively, and plasmid pCRE13/2 as template. Using primers 3148 and 3149 and plasmid pCRECEFD2, promoter deletion fragment D2 was amplified to generate plasmid pRPcre1-D2. pCRECEFD2 carries the full-size A. chrysogenum cefEF gene controlled by a truncated version of the cre1 promoter (Δbp 488–687). cre1 promoter derivatives D5, D6 and D7 harbor internal deletions and were therefore made by a three step fusion PCR. In the first step, both fragments which should be fused, were amplified using A. chrysogenum genomic DNA as template and primers cre1ko5 and D5-anti/D6-anti/D7-anti, and cre1-as and D5-sense/D6-sense/D7-sense, respectively. For deletion derivative D4*, primers cre1-as and Pcre1-D4*-s, and Pcre1-D3 and Pcre1-D4*-as were used to amplify the first two PCR fragments of the promoter. In the next step the two fragments that correspond to one deletion fragment were fused by a further PCR step. In the final step, the cre1 promoter deletion fragments were amplified using nested primers 3148 and 3149 (deletion derivatives D5, D6, and D7), and primers 3149 and Pcre1-D4 (deletion derivative D4*) to attach restriction sites AgeI and NcoI. The corresponding cre1 promoter fragments were ligated into pDrive (Qiagen) and sequenced. In the next step, all obtained fragments were digested and inserted into AgeI–NcoI restriction sites in plasmid pRHN1 to generate the different promoter deletion constructs (Table 2). The sequences of all the recombinant plasmids and oligonucleotides are given in Tables 2 and 3, respectively.
Preparation and analysis of RNA
RNA was isolated as described by Jekosch and Kück (2000a), and the integrity of all RNAs was verified by agarose gel electrophoresis and Northern blot analyses (Sambrook and Russell 2001). The blots were hybridized with 32P-radiolabeled DNA probes as specified in “Results”.
Protein purification
The purification of fungal proteins was carried out as described previously (Schmitt et al. 2004b), with the modification that the mycelium was resuspended in 2 ml of buffer A [0.1 M morpholinepropanesulfonic acid (MOPS, pH 7.5), 0.2 M KCl, 10 mM MgCl2, 1 mM EDTA, 10 mM dithiothreitol, 4.2 mM phenylmethylsulfonyl fluoride, 40% (w/v) glycerol].
Spectrofluorometric measurements
The fluorescence of the DsRed protein was measured at 576 nm with the excitation set at 554 nm using a JASCO FP-6500 spectrofluorometer (JASCO, Tokyo, Japan) as previously described (Janus et al. 2007). A calibration curve using purified DsRed protein (BD Biosciences) was produced and served as control to quantify DsRed levels in A. chrysogenum protein extracts.
Synthesis of recombinant CRE1 polypeptide in E. coli
GST and GST fusion proteins were purified from E. coli BL21 (DE3) cells (Stratagene). The gene sequence for the first 183 amino acids from CRE1 was ligated in frame to the coding sequence of GST by using restriction sites EcoRI and NotI of pGEX-4T1 (Amersham Bioscience). The resulting plasmid named pGEXcre1_183 was transformed into E. coli expression host BL21 (DE3). Gene expression was induced at midlog phase by adding 1 mM isopropyl β-d-thiogalactoside and the cells were incubated for 1 h at 30°C. GST and GST fusion proteins were purified by affinity chromatography using a 50% slurry of Glutathione Sepharose beads according to the supplier’s manual (Amersham Biosciences). On-column cleavage has been performed with 80 U thrombin according to the supplier’s manual (Amersham Biosciences). Purified proteins were stored at −80°C until used for electrophoretic mobility shift assays.
Electromobility shift assays
Gel shift experiments were performed using either radiolabeled PCR fragments or annealed oligonucleotides from the cre1 promoter. The promoter fragments were amplified with oligonucleotides listed in Table 3 and radiolabeled during PCR as described previously (Schmitt et al. 2004b). The PCR mixture contained 90 ng of pCRE13/2 as template, 40 ng of each primer, 4 μM dATP, 4 μM dTTP, 4 μM dGTP, 0.2 μM dCTP, 8.33 × 10−12 mol of [α-32P] dCTP, and 1 U Taq polymerase (Eppendorf) in a volume of 50 μl. To remove free nucleotides from the samples, an electrophoresis was performed in a 5% polyacrylamid gel with 0.5× Tris–borate–EDTA buffer. Single bands were extracted, incubated with elution buffer (0.5 M ammonium acetate, 1 mM EDTA, 0.1% SDS) and precipitated with ethanol. The precipitate was finally dissolved in 100 μl H2O. Labeling of annealed oligonucleotides was achieved by 5′-end labeling using polynucleotide kinase (Roche) and [γ-32P]-dATP. To investigate the binding of the CRE1 polypeptide, 10–30 fmol of various radiolabeled promoter fragments or oligonucleotides were incubated with varying protein concentrations (indicated in the corresponding figures) in the presence of 2 μl binding buffer (250 mM Tris/HCl pH 8.0, 1 M KCl, 50% glycerin) and 1 μg of poly(dI–dC) · poly(dI–dC) in a total volume of 20 μl. After 20-min incubation at room temperature, the samples were electrophoresed in a 5% polyacrylamide gel at 4°C with Tris/glycine buffer.
Protein purification of CRE1 for Biacore measurements
GST-tagged CRE1 (1–183) was produced by autoinduction in E. coli Rosetta 2 (DE3) cells grown at 30°C in 1 l Overnight Express Instant TB Medium (Novagen) (Studier 2005). Fifteen grams of cells were collected by centrifugation, resuspended in 200 ml lysis buffer (20 mM NaH2PO4, 150 mM NaCl, 1 mM PMSF, pH 7.4) and homogenized using an Emulsiflex C5 high-pressure homogenizer (Avestin). Clarified cellular extract was loaded onto a 20 ml Glutathione Sepharose 4 FF column (GE Healthcare) followed by an overnight on-column cleavage at room temperature using 320 U human thrombin (Sigma). CRE1 was eluted with 50 mM Tris/HCl, 10 mM reduced glutathione, pH 8.0, transferred to 50 mM NaH2PO4, 150 mM NaCl, pH 7.4 and applied to a 5 ml Heparin Sepharose HP column (GE Healthcare), followed by elution with a salt gradient up to 2 M NaCl. CRE1 containing fractions were concentrated using a Vivaspin 10 kD concentrator (Vivascience) and purified to homogeneity by gel filtration chromatography on a Superdex PG 200 column (GE Healthcare) with 50 mM NaH2PO4, 150 mM NaCl, 1 mM DTT, pH 7.4 as running buffer.
Binding analysis by SPR
Real-time analysis of protein–DNA interaction was performed on a Biacore 2000 system at 25°C. A commercially available SA sensor chip (Biacore) was used having a layer of a thin gold film coated with carboxymethyldextran hydrogel matrix to which streptavidin was cross-linked. Data were processed with the BIAevaluation software version 4.1 (Biacore). The running buffer used for DNA immobilization and SPR assay was 10 mM HEPES pH 7.4, 0.15 M NaCl, 1 mM DTT, 0.005% (v/v) surfactant P20. The buffer was freshly prepared, filtered through a 0.22 μm membrane and degassed prior to use. Refractive index errors due to bulk solvent effects were corrected by substracting responses from the non-coated flow cell 1. 5′-Biotinylated DNA duplexes (5 nM) containing CRE1 wild type and mutant binding sites were immobilized on a SA sensor chip on flow cells 2, 3 and 4 by injection at a flow rate of 10 μl/min until 80 response units (RU) were bound. The purified CRE1 protein was diluted to nanomolar concentrations in running buffer and a concentration series from 12.5 to 400 nM was examined for each DNA duplex. Sample injection and dissociation time was set to 2.5 min at a flow rate of 20 μl/min. The chip surface was regenerated with an injection of running buffer containing 0.5 M NaCl for one minute. Dissociation constants were calculated from the concentration-dependent steady-state binding using the 1:1 steady-state affinity model.
Results
cre1 promoter directed DsRed expression
Transcriptional expression of the cre1 gene is up regulated in the presence of glucose in the A. chrysogenum wild type strain (Jekosch and Kück 2000a). We examined the 5′-upstream region of the cre1 gene and detected several putative CRE1 consensus binding sites (5′-SYGGRG-3′), suggesting a positive autoregulation of cre1 expression by its own gene product. We discovered 15 CRE1 consensus sequences (BS1 to BS15) within the 1,030 bp upstream of the cre1 transcription start site, which is preceded by an 18 bp T-stretch as well as a CT-rich region. In order to study cre1 promoter-driven expression in vivo, we used a recently developed DsRed gene-based reporter system. Expression of the gene encoding the red fluorescent protein (DsRed) results in fungal transformants displaying a red colored phenotype on solid medium. Moreover, variations in DsRed synthesis can be quantified with spectrofluorometric measurements (Janus et al. 2007).
For our expression studies, we constructed plasmid pRPcre1, which contains the DsRed gene under control of the full-length cre1 promoter (Pcre1). Transformation of pRPcre1 into A. chrysogenum resulted in several differently colored transformants obtained on agar plates. Of the total 19 transformants, 7 showed a red or pink colored phenotype and were further analyzed for plasmid copy number and glucose-induced gene expression. We identified low (T2) and multi copy number transformants (T13, T17), and glucose-induction was further analyzed when the strains were grown in the presence (+G) or absence (−G) of glucose. From these various strains in different growth conditions, RNA was isolated and subjected to Northern blot hybridizations. As presented in Fig. 1a, the DsRed transcript markedly increases in all transformants when grown in glucose-containing medium. Transcript levels are barely detectable when the strains were grown in the absence of glucose. Only T17, which is a multi copy strain, showed detectable levels of DsRed in the absence of glucose.

Expression of the DsRed reporter gene under control of the cre1 promoter in multi (T13, T17) and low (T2) copy transformants. a Northern blot hybridization was performed using the DsRed gene as a probe. b Spectrofluorometric measurements of the DsRed protein in A. chrysogenum total protein extracts. +G and −G indicate that strains were grown in the presence or absence of glucose. The wild type strain served as a negative control
We next examined the time-course of DsRed expression. Strains were cultured for 7 days and protein extracts were obtained at specific time points. To guarantee sufficient cellular material for the quantification of DsRed, the measurement began 2 days after inoculation, and, as recently described, spectrofluorometric measurements were conducted to determine the amount of DsRed in each sample (Janus et al. 2007). As shown in Fig. 1b, all transformants showed the highest amount of DsRed in a copy-dependent manner when grown in the presence of glucose. Maximal expression was observed after 5–6 days of growth in all cases, confirming the usefulness of the DsRed reporter gene system to study regulated gene expression in A. chrysogenum.
Expression studies with cre1 promoter deletion derivatives
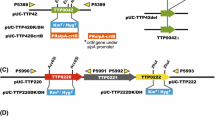
To detect critical sequences within the cre1 promoter region, we performed functional analysis of seven promoter deletion mutants that lack at least one of the putative CRE1 binding sites (Fig. 2). All promoter constructs were fused upstream of DsRed and analyzed phenotypically. Transformation of A. chrysogenum with plasmid pRPcre1 in which DsRed expression is driven by the wild type cre1 promoter resulted in a total of 19 analyzed transformants, 7 of which displayed clear red or pink color. In comparison, the panel of promoter mutants displayed a diverse range of DsRed regulation. As discussed below, ectopic integration of transformed DNAs into the genome, such as during DNA-mediated fungal transformation, leads to modification of the plasmid-derived gene expression (Janus et al. 2007). Thus, the phenotypic characterization of fungal transformants on Petri dishes is a rough estimate of promoter-driven reporter gene expression. From the data presented in Fig. 2, the T-stretch and the CT-rich region, both comprising a region of about 149 bp, are essential for gene expression, exemplified by the high frequency of reddish transformants, carrying either D1, D3, D4, or D6. In contrast, D5 and D7 showed no reddish transformants and lack the T-stretch as well as the CT-rich region. One exception seemed to be the D2 mutant that carries a deletion just upstream of the T-stretch, suggesting that the upstream region contains binding sites for further activator or repressor proteins that modulate gene expression. In separate experiments, we constructed promoter derivatives lacking the 5′-transcribed but not translated region. None of these mutants showed a detectable expression of the reporter gene (data not shown).

Schematic drawing of the wild type cre1 promoter (Pcre1) and different promoter deletion derivatives. All promoter fragments were fused to the DsRed reporter gene. Numbers on the right represent red colored transformants (first number) within the total number of analyzed transformants (second number). The putative CRE1 binding sites (bars), a T-stretch (T), a CT-rich region (CT), as well as the transcriptional start point (arrow) are indicated
To quantify the observed variations in transcriptional expression, 10–14 randomly selected transformants obtained with each promoter derivative were selected and analyzed spectrofluorometrically. After 5 days of growth, the DsRed protein content was determined compared to the total protein amount (Supplementary Data Table 1). The results of the spectrofluorometric measurements are consistent with the data shown in Fig. 2. Transformation with promoter derivatives that failed to yield any colored transformants correspondingly had barely detectable DsRed protein, while among the derivatives that generated a range of phenotypes, we observed a correlation between DsRed protein content and copy number. Notably, the D4 construct generated the most distinct protein yields (Supplementary Data Table 1).
We further analyzed DsRed expression in both, low and multi copy transformants harboring the promoter derivatives D2, D4, D5, D6, and D7, in the absence and presence of glucose. Northern blot analysis showed that cells harboring DsRed driven by the full length cre1 promoter markedly induced DsRed expression in the presence of glucose, and levels correlated well with copy number (Fig. 3a). A glucose-dependent transcriptional induction was generally observed among all transformants investigated, except D5 and D7, which acted similar to a negative control. The derivatives D4 and D6 induced strong DsRed levels similar to that obtained with the full-length promoter Pcre1. Interestingly, in cells with the full-length promoter, a basal expression was detectable in the absence of glucose, while D4 showed no detectable transcript at all. These transcriptional expression data were further verified by spectrofluorometric measurement of the DsRed protein level (Fig. 3b). Glucose-dependent protein levels were observed, with the highest yield generated by the multi copy transformant carrying the D4 derivative (D4 T9 +G). The wild type strains served as negative control to measure background fluorescence in all cases.

Impact of glucose on DsRed expression. a Northern blot hybridization analysis with RNAs from low (lc) and multi copy (mc) transformants grown with (+G) and without (−G) glucose. Radiolabeled DsRed served as hybridization probe and the rRNA served for standardization for the RNA loaded. b Spectrofluorometric measurements of the DsRed amount in the transformants as indicated. Strains were grown in the presence (black bars) or absence (gray bars) of glucose
Gel shift analysis of cre1 promoter sequences with CRE1
To determine whether the CRE1 protein could bind fragments of the cre1 promoter carrying putative CRE1 binding sites, we performed in vitro binding studies using EMSA. As detailed in “Materials and methods”, we generated a recombinant GST::CRE1 fusion protein consisting of the GST-tag and the first 183 amino acids of the CRE1 polypeptide comprising the zinc-finger DNA binding region and synthesized the fusion protein in E. coli. Different DNA fragments that were generated with individual primer pairs (Fig. 4a) were incubated with GST::CRE1, and in all these cases, almost every fragment was bound by the CRE1 protein (data not shown). As an example, incubation of two labeled DNA fragments with recombinant GST::CRE1 resulted in a high molecular weight DNA–protein complex only with fragments 21–24, which contains two putative CRE1 binding sites, while fragments 1–2, which lacks binding sites, and the GST alone were both unable to form any complex (Fig. 4b). We then tested increasing amounts (0.5–10 μg) of the GST::CRE1 fusion protein and the CRE1 protein lacking the GST-tag with fragments 21–24. In both cases, we observed an intensified binding, suggesting strong interactions between the proteins and the DNA fragment. Interestingly, multiple bands appeared when the amount of the CRE1 protein was increased, indicating consecutive binding of different sequences with the CRE1 polypeptide (Fig. 4c, d).

Gel shift analysis to determine CRE1 binding sites. a Schematic map of the cre1 promoter region as already shown in Fig. 2. PCR fragments used in electromobility shift analysis (EMSA) with the CRE1 protein are indicated with numbers that represent primers for PCR amplification. b Gel retardation analysis of the CRE1 protein together with PCR fragments 1–2 and 21–24. The recombinant GST::CRE1 (G-CRE1) protein purified from E. coli and an E. coli control protein (GST) were incubated with radiolabeled PCR fragments. c EMSA with PCR fragments 21–24 and increasing amounts of the GST::CRE1 (G-CRE1) fusion protein. d EMSA with PCR fragments 21–24 and increasing amounts of recombinant CRE1 (CRE1) protein detached from the GST-tag
To more closely analyze CRE1 binding, the two putative binding sites (BS14, BS15), part of the 114 bp DNA sequence of promoter derivative D4 triggering glucose-dependent gene expression, were used for gel shift analysis (Fig. 5a). BS14 contains the putative binding site 14 flanked by 10 base pairs on both sites, while BS15 carries binding site 15 and is flanked by 7 and 13 bp. Gel shift analysis showed that both sequences interact with CRE1 lacking the GST-tag, and that the protein binds BS15 more intensely (Fig. 5b). Increasing amounts of GST::CRE1 in the binding analysis produced only a single complex with BS14 and BS15 (Fig. 5c).

Electromobility shift analysis with primers BS14 and BS15. a 5′-Sequence of deletion derivative D4. Putative CRE1 binding sites BS14 and BS15 are underlined, the transcriptional start point is marked by an arrow and the start codon of the open reading frame is shown in italics. Shaded sequences were used for gel retardation analysis and surface plasmon resonance (SPR) assays, and substitutions of the putative binding sequence are given below the sequence. b Analysis of protein–DNA interactions with radiolabeled oligonucleotides BS14 and BS15 and the recombinant CRE1 (CRE1) protein lacking the GST-tag. c Gel retardation analysis with oligonucleotides BS14 and BS15 with increasing amounts of the recombinant GST::CRE1 protein (G-CRE1) as indicated
Interactions between CRE1 and binding sites BS14 and BS15 determined by SPR analysis
To further characterize the binding specificity of CRE1, SPR analyses of protein–DNA interactions were carried out on a streptavidin-coated Biacore SA sensor chip using oligonucleotides BS14 and BS15, as well as mutant oligonucleotides with two to four substitutions (BS14m1, BS14m2, BS15m1, BS15m2) (see Table 3) within the consensus binding sites as shown in Fig. 5a. The non-coated flow cell 1 served as a reference in order to reduce bulk solvent effects. The RU-values of different DNA loaded on each flow cell were about 80 RU. As detailed in the “Materials and methods”, a highly purified CRE1 protein detached from the GST-tag was synthesized for SPR analysis. The purified truncated CRE1 protein (CRE1 1–183) used in these studies showed a single band on SDS-polyacrylamide gels (Fig. 6a).

Surface plasmon resonance analysis. a Coomassie blue-stained protein gel with highly purified CRE1 protein (lane 1). Molecular weight markers are indicated on the site. b Analysis of steady state affinity of CRE1 binding to BS14, BS14m1 and BS14m2 determined using SPR measurements. c Steady state affinity of CRE1 binding to BS15, BS15m1 and BS15m2 determined using SPR measurements. The graphs show the response units in comparison to the protein concentration
Surface plasmon resonance sensorgrams for equilibrium binding of CRE1 to immobilized DNA-duplexes were recorded (data not shown) and their corresponding steady-state affinity analyses are shown in Fig. 6b, c. Binding of the CRE1 polypeptide to BS14 compared to the two mutant DNA-duplexes BS14m1 and BS14m2 is shown in Fig. 6b. Interaction of 400 nM CRE1 with BS14 attained 73.8 RU at equilibrium whereas mutant oligonucleotides showed no or very marginal interactions at all. SPR analyses using oligonucleotides BS15 and mutant forms (BS15m1, BS15m2) are shown in Fig. 6c and demonstrate that the protein–DNA interaction between CRE1 and BS15 had a high affinity. On the other hand, mutant oligonucleotides retained lower binding specificities but still showed a weak affinity, which is reduced by a factor of approximately 3. Consequently, substitution of 2–4 bp within the consensus binding sequence of BS15 weakened the protein–DNA complex formation but did not completely inhibit CRE1 binding.
Comparing the calculated K D-values (Table 4) supports the observation that both BS14 and BS15 are specifically bound by CRE1 but BS14 (with a K D value of 154 nM) interacts with a lower affinity than BS15 (with a K D value of 85.9 nM). In contrast, mutant oligonucleotides, BS14m1, BS14m2, BS15m1 and BS15m2, show higher K D values in comparison to BS14 and BS15, which refer to a lower binding affinity to the CRE1 polypeptide. These results are in good agreement with the data obtained with gel retardation analysis of CRE1 interaction to oligonucleotides BS14 and BS15 (Fig. 5b, c).
In vivo analysis of substitutions in binding sites BS14 and BS15
The SPR analysis indicates that BS14m2 lacks almost any binding specificity with CRE1 while BS15m2, despite four nucleotide substitutions, still retains decent interaction with the CRE1 protein. This suggests that in vivo a D4 fragment containing the BS14m2, as well as the BS15m2, substitution could still have a glucose-dependent transcriptional expression. We thus constructed plasmid pRPcre1_D4* harboring 4 bp substitutions in each of the BS14 and BS15 CRE1 consensus binding sites, and transformed it into the wild type strain of A. chrysogenum, resulting in reddish colonies on agar plates. When we tested a total of 35 transformants, we found that 17 had a pink or red color while the remaining did not show any expression of the DsRed gene on solid media. This is similar to the distribution we observed when we used the D4 construct carrying the wild type sequence for fungal transformation (see also Supplementary Material Table 1). This rough approach indicates that, despite the base pair substitutions, the modified D4* promoter sequence is still able to confer glucose-dependent gene expression. These results were further confirmed when two randomly selected D4* transformants, a low copy and a multi copy strain, were subjected to Northern blot analysis and compared to transformants carrying either the full-length cre1 promoter or the D4 promoter fragment. As shown in Fig. 7, the level of the transcript clearly increased in all strains investigated under glucose-inducing growth conditions, and this was further confirmed by spectrofluorometric measurements of DsRed protein (data not shown). Thus, these data are in good agreement with the results obtained with the SPR measurements showing a sufficient affinity of CRE1 to the BS15m2 sequence harboring four T-substitutions, being capable of driving glucose-induced gene expression. The binding affinity to BS15 seemed to be stronger than interaction with BS14, as demonstrated by SPR analysis, and this is in line with the in vivo analysis with deletion derivative D4* still showing strong gene expression and induction by glucose.

Impact of nucleotide substitutions within CRE1 binding sites on DsRed expression. Northern blot hybridization with RNA from the wild type strain and from selected transformants, harboring plasmids pRPcre1 (Pcre), pRPcre1_D4 (D4) or pRPcre1_D4* (D4*). RNA was isolated from strains that were grown in the presence (+G) or absence of glucose (−G). Hybridization was done with a radiolabeled DsRed probe
Discussion
So far, in A. chrysogenum a locus-dependent integration system to study reproducible expression rates, as in other filamentous fungi (e.g. A. niger) (van Hartingsveldt et al. 1987) has not been established. Therefore, a larger number of transformants carrying ectopically integrated plasmids with variable gene expression patterns must be investigated in separate experiments. For this purpose, the DsRed gene is an ideal reporter system for a statistical approach to distinguish transformants showing high, medium, and low expression rates.
The identification of a 114 bp CT-rich sequences in the non-transcribed cre1 upstream region suggests that it is required to trigger glucose-dependent gene expression. CT-rich regions are frequently found in promoters of strongly transcribed genes in filamentous fungi, mostly in genes lacking conventional TATA and CAAT boxes (Kapoor et al. 1995). This indicates that CT-rich regions are putative activator sequences in close proximity to transcription start sites (Gurr et al. 1987; Yamazaki et al. 2000; Sirand-Pugnet et al. 2003; Fei et al. 2006; Neveu et al. 2007). For example, deletion of the CT-rich region upstream of the transcription start point in different Aspergillus gpdA and oliC genes, encoding glyceraldehyde-3-phosphate dehydrogenase and oligomycin-resistant ATP synthase subunit 9, respectively, resulted in an altered transcription initiation. This supported the involvement of this region in correct transcription initiation and determining the frequency of initiation (Ballance 1986; Ward and Turner 1986; Ward et al. 1988; Punt et al. 1990). In plants, the CT-rich regions flanking the 5′-end of plant genes function similarly, since mutagenesis of this sequence resulted in a significant decrease of transgene expression (Bolle et al. 1994).
In A. chrysogenum, the CT-rich region contains two closely spaced binding sites (BS14, BS15) that according to our gel shift analysis cooperatively bind to CRE1. Similarly, two closely spaced putative CREA binding sites were found in the creA promoter from A. nidulans, mediating autoregulation. Gel shift analysis with a recombinant GST::CREA protein and cell-free extracts showed direct interaction of the CREA protein to binding sequences in the creA promoter (Strauss et al. 1999).
The binding affinities of A. chrysogenum CRE1 with two binding sequences were further quantified using SPR analysis. To the best of our knowledge, this is the first example where the CRE1 affinity to consensus binding sites was measured using this technique. SPR measurements have, e.g. been used to determine the interaction of two proteins or protein complexes with specific DNA sequences (e.g. Hortschansky et al. 2007; Shi et al. 2007). Recently, the interaction of the S. cerevisiae alcohol dehydrogenase 1 protein with sequences of the alcohol dehydrogenase II gene was analyzed by SPR. The determination of dissociation constants for the interaction of this zinc-finger protein with the corresponding DNA has been determined with two-digit nanomolar affinities (Schaufler and Klevit 2003), similar to the results in our investigation. Furthermore, our data reveal that SPR analysis is a convenient method to determine the interaction of a protein to different DNA sequences in real-time without labeling requirements (Myszka et al. 1998). In addition, the comparison of binding specificities of a protein to wild type and mutant binding sites can be easily performed and quantified (Hart et al. 1999).
Detailed sequence analysis of binding site BS15 (5′-AGACTGGGGGGGGGGG-3′) and mutant binding sites, as well as adjacent sequences, displayed that the consensus binding site (5′-SYGGRG-3′) in the mutant oligonucleotide shifted to the 3′-end of BS15m2 (5′-AGACTTT TT GGGGGGG-3′) with only a single nucleotide substitution. Therefore, it is possible that this incompletely truncated binding site is responsible for the glucose induction in transformants carrying deletion derivative D4*. This is also consistent with our SPR analysis using oligonucleotides that have still a significant affinity to CRE1. Deletion of flanking sequences from BS15 should be performed to prevent further binding of CRE1 to the displaced binding site and to further determine whether CRE1 is the only regulator responsible for glucose induction directed by promoter derivative D4.
Another explanation could be the interaction of other proteins and possible transcriptional modulators directed by the cre1 promoter. At present, we can only speculate whether the CRE1 protein interacts with other proteins having a specificity to the 114 bp CT-rich sequence. The in silico analysis did not reveal any significant binding sites for transcriptional activators/repressors from filamentous fungi, but detailed promoter–protein interaction studies in filamentous fungi showed that a tight interplay between different transcription activators and/or repressors is a prerequisite to control environment-specific gene expression. For example, the xyn1 promoter from T. reesei, controlling xylan-dependent gene expression, is regulated by an interplay of the CRE1 repressor and the cellulose regulator Ace1 that acts as a general and specific repressor with the xylanase regulator Xyr1 (Rauscher et al. 2006). Similarly, the iron-dependent transcriptional expression in A. nidulans is regulated by the novel HapX factor, mediating repression of iron-dependent pathways under iron-depleted conditions via interaction with the CCAAT-binding core complex (Hortschansky et al. 2007).
In conclusion: the cre1 promoter from A. chrysogenum displays a positive autoregulation upon glucose induction, and our identification of a 114 bp minimal promoter with two CRE1 binding sites is the first example of a minimal regulatory promoter sequence for this β-lactam antibiotic producing fungus. Two CRE1 consensus binding sites are located within the minimal promoter sequence that are tightly bound by the CRE1 protein, and this interaction was quantified using the Biacore system and represents the first characterization of a CRE1–DNA interaction by SPR analysis. Importantly, the minimal cre1 promoter identified here will be a functionally useful tool for the construction of compact fungal expression vectors suitable for biotechnically relevant filamentous fungi. For example, normal glucose-dependent repression of cephalosporin C biosynthesis genes could be converted into expressional activation by fusing biosynthesis genes to the minimal cre1 promoter.
References
Ballance DJ (1986) Sequences important for gene expression in filamentous fungi. Yeast 2:229–236
Bolle C, Sopory S, Lubberstedt T, Herrmann RG, Oelmuller R (1994) Segments encoding 5′-untranslated leaders of genes for thylakoid proteins contain cis-elements essential for transcription. Plant J 6:513–523
Bullock WO, Fernandez JM, Short JM (1987) XL1-Blue: a high efficiency plasmid transforming recA Escherichia coli strain with beta-galactosidase selection. BioTechniques 5:376–378
Cubero B, Scazzocchio C (1994) Two different, adjacent and divergent zinc finger binding sites are necessary for CREA-mediated carbon catabolite repression in the proline gene cluster of Aspergillus nidulans. EMBO J 13:407–415
Dowzer CEA, Kelly JM (1989) Cloning of the creA gene form Aspergillus nidulans: a gene involved in carbon catabolite repression. Curr Genet 15:457–459
Fei X, Zhao MW, Li YX (2006) Cloning and sequence analysis of a glyceraldehyde-3-phosphate dehydrogenase gene from Ganoderma lucidum. J Microbiol 44:515–522
Gancedo JM (1998) Yeast carbon catabolite repression. Microbiol Mol Biol Rev 62:334–361
Gurr SJ, Uncles SE, Kinghorn JR (1987) The structure and organization of nuclear genes in filamentous fungi. In: Kinghorn JR (ed) Gene structure in eukaryotic microbes. IRL Press, Oxford, pp 93–139
Hart DJ, Speight RE, Cooper MA, Sutherland JD, Blackburn JM (1999) The salt dependence of DNA recognition by NF-kappaB p50: a detailed kinetic analysis of the effects on affinity and specificity. Nucleic Acids Res 27:1063–1069
Hortschansky P, Eisendle M, Al-Abdallah Q, Schmidt AD, Bergmann S, Thön M, Kniemeyer O, Abt B, Seeber B, Werner ER, Kato M, Brakhage AA, Haas H (2007) Interaction of HapX with the CCAAT-binding complex—a novel mechanism of gene regulation by iron. EMBO J 26:3157–3168
Ilmén M, Thrane C, Penttilä M (1996) The glucose repressor gene cre1 of Trichoderma: isolation and expression of a full-length and a truncated mutant form. Mol Gen Genet 251:451–460
Janus D, Hoff B, Hofmann E, Kück U (2007) An efficient fungal RNA-silencing system using the DsRed reporter gene. Appl Environ Microbiol 73:962–970
Jekosch K, Kück U (2000a) Glucose dependent transcriptional expression of the cre1 gene in Acremonium chrysogenum strains showing different levels of cephalosporin C production. Curr Genet 37:388–395
Jekosch K, Kück U (2000b) Loss of glucose repression in an Acremonium chrysogenum beta-lactam producer strain and its restoration by multiple copies of the cre1 gene. Appl Microbiol Biotechnol 54:556–563
Kapoor M, Curle CA, Runham C (1995) The hsp70 gene family of Neurospora crassa: cloning, sequence analysis, expression, and genetic mapping of the major stress-inducible member. J Bacteriol 177:212–221
Kulmburg P, Mathieu M, Dowzer C, Kelly J, Felenbok B (1993) Specific binding sites in the alcR and alcA promoters of the ethanol regulon for the CREA repressor mediating carbon catabolite repression in Aspergillus nidulans. Mol Microbiol 7:847–857
Mach RL, Strauss J, Zeilinger S, Schindler M, Kubicek CP (1996) Carbon catabolite repression of xylanase I (xyn1) gene expression in Trichoderma reesei. Mol Microbiol 21:1273–1281
Martín JF, Casqueiro J, Kosalková K, Marcos AT, Gutiérrez S (1999) Penicillin and cephalosporin biosynthesis: mechanism of carbon catabolite regulation of penicillin production. Antonie Van Leeuwenhoek 75:21–31
Mathieu M, Fillinger S, Felenbok B (2000) In vivo studies of upstream regulatory cis-acting elements of the alcR gene encoding the transactivator of the ethanol regulon in Aspergillus nidulans. Mol Microbiol 36:123–131
Myszka DG, Jonsen MD, Graves BJ (1998) Equilibrium analysis of high affinity interactions using BIACORE. Anal Biochem 265:326–330
Neveu B, Belzile F, Bélanger RR (2007) Cloning of the glyceraldehyde-3-phosphate dehydrogenase gene from Pseudozyma flocculosa and functionality of its promoter in two Pseudozyma species. Antonie Van Leeuwenhoek 92(2):245–255
Punt PJ, Dingemanse MA, Kuyvenhoven A, Soede RD, Pouwels PH, van den Hondel CA (1990) Functional elements in the promoter region of the Aspergillus nidulans gpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene 93:101–109
Radzio R, Kück U (1997) Efficient synthesis of the blood-coagulation inhibitor hirudin in the filamentous fungus Acremonium chrysogenum. Appl Microbiol Biotechnol 48:58–65
Rauscher R, Wurleitner E, Wacenovsky C, Aro N, Stricker AR, Zeilinger S, Kubicek CP, Penttilä M, Mach RL (2006) Transcriptional regulation of xyn1, encoding xylanase I, in Hypocrea jecorina. Eukaryot Cell 5:447–456
Ronne H (1995) Glucose repression in fungi. Trends Genet 11:12–17
Ruijter GJ, Visser J (1997) Carbon repression in Aspergilli. FEMS Microbiol Lett 151:103–114
Sambrook J, Russell DW (eds) (2001) Molecular cloning. A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Habor, pp 7.31–7.44
Schaufler LE, Klevit RE (2003) Mechanism of DNA binding by the ADR1 zinc finger transcription factor as determined by SPR. J Mol Biol 329:931–939
Schmitt EK, Hoff B, Kück U (2004a) Regulation of cephalosporin biosynthesis. In: Brakhage AA (ed) Adv Biochem Engin/Biotechnol. Springer, Berlin, pp 1–43
Schmitt EK, Bunse A, Janus D, Hoff B, Friedlin E, Kürnsteiner H, Kück U (2004b) Winged helix transcription factor CPCR1 is involved in regulation of beta-lactam biosynthesis in the fungus Acremonium chrysogenum. Eukaryot Cell 3:121–134
Shevchuk NA, Bryksin AV, Nusinovich YA, Cabello FC, Sutherland M, Ladisch S (2004) Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucleic Acids Res 32:e19
Shi C, Kaminskyj S, Caldwell S, Loewen MC (2007) A role for a complex between activated G protein-coupled receptors in yeast cellular mating. Proc Natl Acad Sci USA 104:5395–5400
Sirand-Pugnet P, Santos C, Labarere J (2003) The Aa-Pri4 gene, specifically expressed during fruiting initiation in the Agrocybe aegerita complex, contains an unusual CT-rich leader intron within the 5′ uncoding region. Curr Genet 44:124–131
Strauss J, Horvath HK, Abdallah BM, Kindermann J, Mach RL, Kubicek CP (1999) The function of CreA, the carbon catabolite repressor of Aspergillus nidulans, is regulated at the transcriptional and post-transcriptional level. Mol Microbiol 32:169–178
Studier FW (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41:207–234
van Hartingsveldt W, Mattern IE, van Zeijl CM, Pouwels PH, van den Hondel CA (1987) Development of a homologous transformation system for Aspergillus niger based on the pyrG gene. Mol Gen Genet 206:71–75
Walz M, Kück U (1991) Polymorphic karyotypes in related Acremonium strains. Curr Genet 19:73–76
Walz M, Kück U (1993) Targeted integration into the Acremonium chrysogenum genome: disruption of the pcbC gene. Curr Genet 24:421–427
Ward M, Turner G (1986) The ATP synthase subunit 9 gene of Aspergillus nidulans: sequence and transcription. Mol Gen Genet 205:331–338
Ward M, Wilson LJ, Carmona CL, Turner G (1988) The oliC3 gene of Aspergillus niger: isolation, sequence and use as a selectable marker for transformation. Curr Genet 14:37–42
Warner JB, Lolkema JS (2003) CcpA-dependent carbon catabolite repression in bacteria. Microbiol Mol Biol Rev 67:475–490
Yamazaki T, Hasebe T, Kajiwara S, Shishido K (2000) Structure and function of a pyrimidine/purine-biased sequence from the 5′-flanking region of the basidiomycete Lentinus edodes gene priA. Mol Gen Genet 263:262–270
Zeilinger S, Schmoll M, Pail M, Mach RL, Kubicek CP (2003) Nucleosome transactions on the Hypocrea jecorina (Trichoderma reesei) cellulase promoter cbh2 associated with cellulase induction. Mol Genet Genomics 270:46–55
Acknowledgments
We thank Kerstin Kalkreuter (Bochum), Sylke Fricke (Jena), and Ingeborg Godehardt (Bochum) for excellent technical assistance, Prof. Eckhard Hofmann (Bochum) for the use of the JASCO spectrofluorometer, Dr. E. Friedlin and Dr. H. Kürnsteiner (Kundl) for fruitful discussions, and Prof. A. Brakhage (Hans-Knöll Institute, Jena) for institutional support. This work was funded by Sandoz GmbH (Austria, Kundl).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by K. Breunig.
Electronic supplementary material
Below is the link to the electronic supplementary material.
294_2007_164_MOESM1_ESM.doc
Supplementary Table 1: Data from spectrofluorometric measurements to determine the DsRed amount (ng DsRed/µg protein) in crude protein extracts from different low (lc) and multi copy (mc) transformants carrying promoter deletion derivatives as indicated. (DOC 38 kb)
Rights and permissions
About this article
Cite this article
Janus, D., Hortschansky, P. & Kück, U. Identification of a minimal cre1 promoter sequence promoting glucose-dependent gene expression in the β-lactam producer Acremonium chrysogenum . Curr Genet 53, 35–48 (2008). https://doi.org/10.1007/s00294-007-0164-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-007-0164-8