
Abstract
We developed a reporter gene system that enables precise analysis of promoter activity in Thermus thermophilus HB27. The reporter vector employs a promoterless β-galactosidase gene of Thermus spp. strain T2. However, T. thermophilus HB27 strain has three genes (TTP0042, TTP0220 and TTP0222) whose products have β-galactosidase activity, which would interfere with correct measurements of promoter activities. Thus, to eliminate this background activity, we disrupted all three of these genes to generate a host strain for measuring promoter expression as β-galactosidase activity. In addition, T. thermophilus strains also produce carotenoids called thermoxanthins that are yellow pigments. To avoid the influence of these carotenoids on the β-galactosidase assay, we also disrupted the phytoene synthase gene (crtB). The reporter gene system developed here is a powerful tool for studying transcriptional activity and the mechanisms that regulate gene expression in T. thermophilus HB27. We also showed that the crtB gene cassette could be used in repeated gene-disruption experiments to screen transformants by colony colour, thus eliminating the need for antibiotic resistance markers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Extreme thermophiles are of great biological interest because of the biotechnological potential of the thermostable enzymes they produce (Cava et al. 2009; Taylor et al. 2011). Thermus spp. are an important source of such enzymes, and these bacteria, especially Thermus thermophilus HB27, are experimentally manipulable owing to their natural transformation properties (Koyama et al. 1986). Various tools for the genetic manipulation of T. thermophilus had been developed (de Grado et al. 1999; Hashimoto et al. 2001; Brouns et al. 2005; Nakamura et al. 2005; Cava et al. 2009; Fujita et al. 2012, 2013; Carr et al. 2015). However, an efficient reporter gene system is still needed for the studies of various transcriptional regulation. Plasmid vectors containing promoterless reporter genes have been used to analyse gene expression in many systems. In Escherichia coli, the β-galactosidase gene has been widely used to monitor in vivo gene expression because β-galactosidase activity can be easily and precisely quantified in liquid assays (Miller 1972). Several attempts have been made to use a β-galactosidase-based assay systems to detect promoter activities in T. thermophilus HB27 (Moreno et al. 2002; Park and Kilbane 2004). However, these previously reported systems showed high background levels of β-galactosidase due to the existence of three β-galactosidase-encoding genes, TTP0042, TTP0220 and TTP0222. In a previous study, the β-galactosidase activity of the TTP0042-encoded protein was shown biochemically (Park and Kilbane 2004), and TTP0220 and TTP0222 were predicted to encode β-galactosidases based on their amino acid sequence similarities to known β-galactosidases (Henne et al. 2004). Thus, β-galactosidase-based reporter systems might not be suitable for precise measurement of promoter activity in T. thermophilus.
In this paper, we describe the construction of a promoter reporter plasmid incorporating a thermostable β-galactosidase gene from Thermus spp. strain T2, and the construction of a suitable host strain that has no β-galactosidase activity (generated by disrupting all three genes encoding proteins with β-galactosidase activity) and produces no carotenoid (by disrupting carotenoid synthesis). This reporter gene system will be very useful for understanding the regulation of gene expressions in T. thermophilus.
In addition, we showed that overexpression of carotenoid 1 (ocr1) mutant strains and a cassette expressing the phytoene synthase gene (crtB) from the slpA promoter (PRslpA-crtB) can be utilized for multiple gene disruptions without the use of antibiotic resistance markers.
Materials and methods
Genetic methods, medium, strains and transformation
Growth media and all genetic manipulations of Escherichia coli DH5α were as previously described (Maniatis et al. 1982). All T. thermophilus strains including HB27 and derivative strains used in this study are listed in Table 1. T. thermophilus was grown at 68 °C in TM medium (Koyama et al. 1986). To select transformants, transformed T. thermophilus HB27 cells were grown on TM agar plates containing 40 μg/mL kanamycin (Km) or 40 μg/mL hygromycin (Hyg) and were grown for 24 h at 68 °C.
Vectors and plasmids
Plasmid pOS103 contains the Thermus spp. strain T2 β-galactosidase gene (bglT2) (Koyama et al. 1990). pTRK1T is an E. coli–T. thermophilus shuttle vector (Fujita et al. 2012). pTRK1-PRslpA is a vector used for overexpression (Fujita et al. 2013). pTRK1-crtBmcs-PP is a vector for promoter probe (Fujita et al. 2013). The gene cassette containing the crtB ORF (encoding phytoene synthase) fused to the slpA promoter was amplified by PCR using pTRK1-PRslpA-bglT2 as a template and primers PEV1 and PEV2 (see Table S1 for the sequence of all primer used in this study, Online Resources). The resultant PCR products were digested with EcoRV and ligated to EcoRV-digested pUC13T (Fujita et al. 2012) to generate pUC13T-PRslpA-crtB. The PRslpA-crtB gene cassette, which expresses the crtB gene from the slpA promoter, can be used for colony-colour screening of gene disruptions (Fig. 1a, b) Plasmid DNA was isolated from E. coli and T. thermophilus, using a PI-50 auto-plasmid-isolator (KURABO, Japan). To prepare plasmids, overnight liquid cultures of E. coli (1 mL grown at 37 °C) and T. thermophilus (4 mL grown at 68 °C) were used.

Schematic of the three chromosomal β-galactosidase genes in T. thermophilus HB27 (TTP0042, TTP0220 and TTP0222) and their disruptions. Yellow triangles indicate the primers used to amplify various DNA regions. a Structure of TTP0042 (green arrow) and its disruption. The DNA region containing TTP0042 was amplified by PCR and cloned into pUC13 (pUC-TTP42). The coding region of TTP0042 was digested with EcoNI and NaeI, and was replaced with a Kmr or Hygr marker (blue box), or PRslpA-crtB cassette (orange box). The constructs used for the gene disruptions were pUC-TTP42DK, pUC-TTP42DH, and pUC-TTP42DcrtB, respectively. b Deletion of the PRslpA-crtB cassette in TTP0042Δ::PRslpA-crtB region using pUC-TTP42del, a construct for the deletion of TTP0042 containing no maker. c Structure of TTP0220 (red arrow), TTP221 (grey arrow), and TTP0222 (yellow arrow), and their gene disruptions. The DNA region containing TTP0220 was amplified by PCR and cloned into pUC13 (pUC-TTP220). The coding region of TTP0220 was digested with Acc65I and was replaced with a Kmr or Hygr marker (blue box). The constructs used for the gene disruptions were pUC-TTP220DK and pUC-TTP220DH. The DNA region containing TTP0222 was amplified by PCR and cloned into pUC13 (pUC-TTP222). The coding region of TTP0222 was digested with XhoI and was replaced with a Kmr or Hygr marker (blue box). The constructs used for the gene disruptions were pUC-TTP222DK and pUC-TTP222DH. d Disruption of the region between TTP0220 and TTP0222. The coding region for the three chromosomal genes (TTP0220, TTP221, and TTP0222) was replaced with a Hygr marker (blue box). The upstream region of TT00220 and the downstream region of TTP0222 were amplified by PCR and cloned into pUC13 (pUC-TTP220-222). The construct used to delete the TTP0220-TTP0222 region was pUC-TTP220-222DH. TTP0221 was estimated to encode α-glucosidase; however, its disruption did not affect cell growth
Constructs for gene disruptions
The DNA region containing TTP0042 was amplified by PCR using T. thermophilus genomic DNA and primers P5388 and P5389 (Fig. 1a). The resultant PCR products were digested with EcoRI/HindIII and ligated to EcoRI/HindIII-digested pUC13 to generate pUC-TTP42 (Fig. 1a). pUC-TTP42 was digested with EcoNI and NaeI, and filled in, and then, a Kmr or Hygr marker was ligated between the two sites to generate pUC-TTP42DK or pUC-TTP42DH, respectively (Fig. 1a). These constructs were used to disrupt the TTP0042 region. The PRslpA-crtB gene cassette, which expresses phytoene synthase, under the slpA promoter, was also ligated into the plasmid used to disrupt the TTP0042 region for colony colour-based selection (white vs. deep-yellow). pUC-TTP42 was digested with EcoNI and NaeI, filled in and self-ligated to generate pUC-TTP42del (Fig. 1a). This plasmid was used to remove the PRslpA-crtB cassette from TTP0042Δ::PRslpA-crtB by colony-colour selection (deep-yellow vs. white, Fig. 1b). The DNA region containing TTP0220 was amplified by PCR using T. thermophilus genomic DNA and primers P5990 and P5991 (Fig. 1c). The resultant PCR products were digested with EcoRI/SalI and ligated to EcoRI/SalI-digested pUC13 to generate pUC-TTP220 (Fig. 1c). pUC-TTP220 was digested with Acc65I and filled in, and then, a Kmr or the Hygr marker was ligated into the site to generate pUC-TTP220DK or pUC-TTP220DH, respectively (Fig. 1c). These constructs were used to disrupt the TTP0220 region. The DNA region containing TTP0222 was amplified by PCR using T. thermophilus genomic DNA and primers P5992 and P5993 (Fig. 1c). The resultant PCR products were digested with EcoRI/SalI and ligated to EcoRI/SalI-digested pUC13 to generate pUC-TTP222 (Fig. 1c). pUC-TTP222 was digested with XhoI and filled in, and then, a Kmr or the Hygr marker was ligated into the site to generate pUC-TTP222DK or pUC-TTP222DH, respectively (Fig. 1c). These constructs were used to disrupt the TTP0222 region. The upstream region of TTP0220 was amplified by PCR using T. thermophilus genomic DNA and primers P6328 and P6329 (Fig. 1d). The resultant PCR products were digested with EcoRI/EcoRV (Fig. 1d). The downstream region of TTP0222 was amplified by PCR using T. thermophilus genomic DNA and primers P6330 and P6331 (Fig. 1d). The resultant PCR products were digested with EcoRV/SalI and ligated to EcoRV/SalI-digested pUC13T (Fujita et al. 2012) to generate pUC13T-TTP222DOWN. pUC13T-TTP222DOWN was digested with EcoRI and EcoRV, and the upstream region of TTP0220, which was digested with EcoRI/EcoRV was ligated between these two sites to generate pUC-TTP220-222. pUCTTP220-222 was digested with EcoRV, and the Hygr marker was ligated into the digested plasmid to generate pUC-TTP220-222DH (Fig. 1d). This construct was used to delete the TTP0220-TTP0221-TTP0222 region.
Construction of the pTRK-bglT2mcsHEstop reporter vector
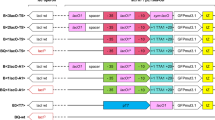
Previously we had developed an expression vector (pTRK1-PRslpA) (Fig. 2a) for overproducing a gene of interest from the slpA promoter (Fujita et al. 2013). First, the bglT2 ORF was amplified by PCR using pOS103 as a template with primers the P5946 and P5328. The resultant PCR products were digested with EcoRI/HindIII and were ligated into EcoRI/HindIII-digested pTRK1-PRslpA (Fig. 2a) to generate pTRK1-PRslpA-bglT2 (Fig. 2b). This construct could be used to overexpress the bglT2 gene. To convert this construct into a reporter vector, a BsrGI/EcoRI-digested DNA fragment containing the multiple cloning site (MCS) of pTRK1-crtBmcs-PP (Fig. 2c) (Fujita et al. 2013) was cloned into the BsrGI/EcoRI-digested pTRK1-PRslpA-bglT2 (Fig. 2b) to generate pTRK1-bglT2mcsHEH (Fig. 2d). This plasmid has an MCS instead of the slpA promoter. However, since this construct also had two HindIII sites, we eliminated the HindIII site downstream of bglT2. The MCS and bglT2 were amplified by PCR with primers P6543 and P6548 using pTRK1-bglT2mcsHEH (Fig. 2d) as a template. The resultant PCR products were digested with BalI and were ligated into the PvuII-digested shuttle vector, pTRK1T (Fig. 2e) (Fujita et al. 2012) to generate pTRK1-bglT2mcsHE (Fig. 2f). The MCS in this vector can be used to clone various DNA fragments. However, when some DNA fragments are cloned into this vector, there is a possibility that peptide-β-galactosidase fusions are expressed. Thus, we introduced a stop codon just upstream of bglT2 ORF. The DNA fragment containing the stop codon was amplified from pTRK1-bglT2mcsHE (Fig. 2f) using primers P6582 and P6548. The resultant PCR products were digested with EcoRI/AvrII, and cloned into EcoRI/AvrII-digested pTRK1-bglT2mcsHE (Fig. 2f) to generate pTRK-bglT2mcsHEstop (Fig. 2g). The sequence of pTRK-bglT2mcsHEstop has been deposited in the DDBJ Nucleotide Sequence Database (Accession Number: LC021516).

Overview of the construction of the β-galactosidase reporter vector used in this study. Details of the construction are provided in the “Materials and methods”. Arrows repA from pTT8 (Aoki and Itoh 2007) (green); Ampr (red); E.coli lacZ, (grey); thermostable Kmr (blue); Thermus T2 bglT2, (orange), and slpA promoter: PRslpA (pink). The positions of the primers used for PCR are shown as yellow triangles. The changed nucleotide sequence in the vicinity of the EcoRI site is shown in the box above pTRK1-bglT2mcsHEstop
DNA construct for disruption of the litR-crtB region
pUC-LRCB is a pUC13 construct that contains the litR-crtB region (Fujita et al. 2013). pUC-LRCB was digested with Eco47III and BamHI, filled in, and self-ligated to generate pUC-LRCBdel. pUC-LRCBdel was used to disrupt the litR-crtB region, and transformants with litR-crtB disrupted were selected by the change in colony colour (Supplementary Fig. S1, Online Resources).
Cloning of the promoter regions of three T. thermophilus genes
The promoter regions of three genes (TTC0189, TTC0549 and TTC1172) were cloned by PCR using T. thermophilus genomic DNA. The promoter region of TTC0189 was amplified using primers P6597 and P6598; the promoter region of TTC0549 was amplified using primers P6591 and P6592; the promoter region of TTC1172 was amplified using primers P6593 and P6594. These resultant PCR products for TTC0189, TTC0549 and TTC1172 were digested with EcoRI/HindIII, and ligated into the EcoRI/HindIII-digested pTRK-bgl-T2mcsHEstop to generate plasmids, pTRK-PRTTC189-bglT2mcsHEstop, pTRK-PRTTC549-bglT2mcsHEstop and pTRK-PRTTC1172-bglT2mcsHEstop, respectively.
β-Galactosidase assays
Cells were grown in TM medium (a complex medium) (Koyama et al. 1986). β-Galactosidase activity was measured when the cells were in exponential growth phase. The cell samples were assayed for β-galactosidase activities at 68 °C as described previously (Ulrich et al. 1972; Miller 1972; http://biochemistry.ucsf.edu/labs/herskowitz/bgal2.html) (Supplementary “Materials and methods”, Online Resources).
Results and discussion
β-Galactosidase activity in T. thermophilus HB27
An efficient reporter gene system is a very important tool for studying the regulation of gene expression. In thermophilic species, a thermostable β-galactosidase gene can be used as a reporter to analyse expression in vivo (Koyama et al. 1990). Several attempts have been made to use β-galactosidase-based assay systems to detect promoter activity in T. thermophilus HB27 (Moreno et al. 2002; Park and Kilbane 2004). However, Park and Kilbane showed that the gene product encoded by TTP0042 has β-galactosidase activity, and that a TTP0042-disrupted strain still had a high background level of β-galactosidase activity. This residual activity might be due to the existence of two other genes TTP0220 and TTP0222 which were predicted to encode β-galactosidases in the analysis of the complete genome sequence of T. thermophilus HB27 (Henne et al. 2004; Ohta et al. 2006). The T. thermophilus HB27 genome is divided between a 1.89 Mb chromosome and a 0.23 Mb megaplasmid called pTT27 (Henne et al. 2004). These three genes that encode proteins with β-galactosidase activities are located on pTT27.
Host strains used for β-galactosidase-based reporter systems should have no endogenous β-galactosidase activity. Therefore, we first constructed single-, double- and triple-gene-disrupted strains using a gene replacement technique with a Kmr or Hygr marker (Fig. 1; Table 1) (Hoseki et al. 1999; Ooga et al. 2009). We then measured the β-galactosidase activities of the wild-type and these disrupted strains (Fig. 3). The TTP0042-disrupted strain (TAF193) still had a β-galactosidase activity as was previously reported. Furthermore, the double-gene-disrupted strains (TAF201, TAF200 and TAF199) also had residual β-galactosidase activity. Only the triple-gene-disrupted strain (TAF236) had no β-galactosidase activity. Thus, this triple disruptnat was a suitable host strain for our reporter system.

β-Galactosidase activities of wild-type and various β-galactosidase-disrupted strains (Table 1). The data shown are the mean ± standard deviations (SD) of three independent experiments
Construction of a β-galactosidase reporter vector for T. thermophilus HB27
For a promoter reporter vector, we used a β-galactosidase gene (bglT2) from Thermus spp. strain T2 (Koyama et al. 1990; Vian et al. 1998). We compared nucleotide sequence of bglT2 to the that of the chromosome and megaplasmid (pTT27) of T. thermophilus HB27 using the BLAST program. No significant similarities were found in these sequences. Thus, we determined that it was an adequate reporter gene since we could eliminate the possibility of homologous recombination with the genome. The construction of the vector pTRK-bglT2mcsHEstop for our β-galactosidase-based reporter system is described in the “Materials and methods”.
Construction of a host strain without β-galactosidase and phytoene synthase activities
Thermus thermophilus HB27 synthesizes carotenoids called thermozeaxanthins, which are yellow pigments that result in the formation of light-yellow colonies. These yellow pigments produced by the host strain might affect β-galactosidase activity measurements. Thus, we constructed a host strain that cannot produce both carotenoids or β-galactosidases. Previously, we had isolated a mutant, TAF189 (ocr1), which overproduced carotenoids (Table 1) (Fujita et al. 2013). TAF189 cells formed deep-yellow colonies. The T. thermophilus crtB gene encodes phytoene synthase, which is required for carotenoid synthesis. First, we constructed a plasmid, pUC-LRCBdel (Fig. S1, Online Resources) to delete the litR-crtB region in TAF189 without an antibiotic resistance marker. pUC-LRCBdel was digested with EcoRI. The resultant DNA was transformed into TAF189 cells to delete the litR-crtB region. Cells were plated on non-selective medium, and one white colony was picked among the many deep-yellow colonies. Deletion of the litR-crtB region was confirmed by PCR. We named this strain as TAF221. Next, we disrupted TTP0042 using colony-colour selection. Using the gene PRslpA-crtB gene cassette, we constructed pUC-TTP42DcrtB (Fig. 1a). The SalI-digested pUC-TTP42DcrtB DNA was transformed into TAF221 to delete TTP0042. Cells were plated on non-selective medium, and one deep-yellow colony was picked among the many white colonies. Disruption of TTP0042 was confirmed by PCR. We named this strain TAF227. Next, we deleted the TTP0220-TTP0221-TTP0222 region using pUC-TTP220-222DH (Fig. 1d). pUC-TTP220-222DH was digested with SalI, and the resultant DNA was transformed into TAF227. One Hygr-colony was picked, and deletion of TTP0220-TTP0222 was confirmed by PCR. We named this strain TAF232. Finally we deleted the PRslpA-crtB cassette of TAF232. EcoRI-digested pUC-TTP42del DNA (Fig. 1b) was transformed into TAF232 cells. Then, the cells were plated on non-selective medium, and one white colony among the many deep-yellow colonies was picked. Deletion of the PRslpA-crtB cassette was confirmed by PCR. We named this strain TAF233, and used it as a host strain for our β-galactosidase-based reporter system. It should be noted that ocr1 (overexpression of carotenoid 1) mutant strains, and a gene cassette expressing phytoene synthase, such as PRslpA-crtB, can be utilized for repeated of gene disruptions with colony colour-based selection instead of antibiotic resistance-based selection (Fig. 2).
Application of a β-galactosidase-based reporter system
We tested our reporter gene system in T. thermophilus HB27 using three newly cloned three promoters of TTC0549 (encoding glyceraldehyde 3-phosphate dehydrogenase, one of the enzymes in glycolysis and gluconeogenesis), TTC1172 (encoding isocitrate dehydrogenase, one of the enzymes in the tricarboxylic acid cycle), and TTC0189 (encoding superoxide dismutase, an enzyme which catalyses the dismutation of the toxic superoxide radical). We also cloned the promoter region of a gene encoding S-layer protein (slpAp, TTC1532) as a strong promoter (positive control) (Fujita et al. 2013). The four plasmids containing these promoters were constructed in pTRK-bglT2mcsHEstop (pTRK-PRTTC189-bglT2mcsHEstop, pTRK-PRTTC549-bglT2mcsHEstop, pTRK-PRTTC1172-bglT2mcsHEstop, and pTRK-PRslpA-bglT2mcsHEstop), and these plasmid were transformed into TAF233 cells. We measured the β-galactosidase activity in these transformed cells (Fig. 4). We could precisely measure the promoter activities, as there was no background activity. As expected, the activity of the slpA promoter was very strong.

β-Galactosidase activities of several T. thermophilus promoters. The data shown are the mean ± SD of three independent experiments
Isolation of DNA fragments that have strong promoter activity by colony-colour screening
In E. coli, promoter-probe experiments are easily performed as plate assays due to the change in colony colour in the presence of the chromogenic substrate for β-galactosidase, X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactoside) (Casadaban et al. 1980; Vieira and Messing 1982). In T. thermophilus, when random DNA fragments were cloned into pTRK-bglT2mcsHEstop some of them showed promoter activity. Cells containing such plasmids exhibit bluish colony colour on medium containing X-gal that is proportional to promoter activity. We tried to isolate DNA fragments with relatively strong promoter activity by colony-colour screening. We constructed a DNA library for a promoter-probe experiment using Saccharomyces cerevisiae genomic DNA. Since the AT-content of S. cerevisiae (62 %) (Goffeau et al. 1996) is much higher than that of T. thermophilus (31 %) (Henne et al. 2004), it has many EcoRI and HindIII sites. S. cerevisiae genomic DNA was digested with EcoRI and HindIII, and cloned into the EcoRI–HindIII-digested pTRK-bglT2mcsHEstop. A pool of clones was transformed into TAF233 cells. As shown in Fig. 5a, unexpectedly, many colonies on medium containing X-gal were light blue, blue or deep-blue. The percentage of bluish colonies was about 15 %, and that of the deep-blue colonies was about 0.5 %. Although the GC-content of S. cerevisiae DNA (38 %) (Goffeau et al. 1996) is very low compared to that of T. thermophilus (69 %) (Henne et al. 2004), many DNA fragments from S. cerevisiae are capable of driving gene expression in T. thermophilus HB27. We picked five deep-blue colonies and rescued plasmids from these transformants (pXG3, pXG8, pXG9, PXG11 and pXG13), and the β-galactosidase activities of these transformants were measured. Their β-galactosidase activities were about 30–70 MU (Fig. 5b). The nucleotide sequences of the inserted DNA fragments and their positions in the S. cerevisiae genome were determined (Table S2, Online Resources). We compared these nucleotide sequences to the consensus E. coli and Thermus-35 and -10 sequences (Hawley and McClure 1983; Maseda and Hoshino 1995); however, we could not detect any similarity. It should be noted that transformation efficiencies are sometimes considerably lower on X-gal-containing mediums probably because of the accumulation of indoxyl derivatives that might inhibit the growth of T. thermophilus (Angelov et al. 2013). Although we obtained some DNA fragments with strong promoter activity, this system is not suitable for large-scale screening of promoter-probe experiments.

Identification of DNA fragments with strong promoter activities by colony-colour screening, and the β-galactosidase activities of the obtained clones. a The S. cerevisiae genomic library in pTRK1-bglT2mcsHEstop was transformed into the TAF233 cells. Transformants were plated on TM medium containing X-gal. The yellow arrow shows one deep-blue transformant that might have strong β-galactosidase activity. b β-Galactosidase activities of five clones obtained (Table S2, Online Resources). The data shown are the mean ± SD of three independent experiments
Conclusions
Here, we constructed a reporter plasmid containing the β-galactosidase gene from Thermus spp. T2. We also constructed a host strain without β-galactosidase activity by disrupting all three genes predicted to encode proteins with β-galactosidase activity. We also disrupted the crtB gene to prevent the production of yellow pigments. The host–vector system developed here is a powerful tool for investigating the regulatory mechanisms of gene expression in T. thermophilus. In addition, we showed that the utilization of carotenoid synthesis genes enables repeated gene disruptions without using antibiotic resistance markers. The frequency of gene disruption by colony-colour screening is higher than the frequency of spontaneous carotenoid-less mutants. One disruptnat was found in approximately every 500–1000 colonies. Carotenoid-less mutants were not found at such a high frequency. Thus, this colony colour-based strategy is usable for repeated disruptions of several genes.
It should be noted that, using of X-gal for the promoter-probe experiments does not always work because T. thermophilus growth is inhibited by the toxicity of indoxyl derivative accumulation resulting from the cleavage of X-gal (Angelov et al. 2013). Moreover, we recently deleted a Hygr maker of TAF233 to construct an antibiotic-sensitive strain (TAF250; Table 1) by colony-colour screening. This strain is also usable in our β-galactosidase-based reporter system.
References
Angelov A, Li H, Geissler A, Leis B, Liebl W (2013) Toxicity of indoxyl derivative accumulation in bacteria and its use as a new counterselection principle. Syst Appl Microbiol 36:585–592
Aoki K, Itoh T (2007) Characterization of the ColE2-like replicon of plasmid pTT8 for Thermus thermophilus. Biochem Biophys Res Commun 353:1028–1033
Brouns SJJ, Wu H, Akeboom J, Turnbull AP, de Vos WM, van der Oost J (2005) Engineering a selectable marker for hyperthermophiles. J Biol Chem 280:11422–11431
Carr JF, Gregory ST, Dahlberg AE (2015) Transposon mutagenesis of the extremely thermophilic bacterium Thermus thermophilus HB27. Extremophiles 19:221–228
Casadaban M, Chou J, Cohen SN (1980) In-vitro gene fusions that join an enzymatically active β-galactosidase segment to amino terminal fragments of exogenous proteins: Escherichia coli plasmid vectors for the detection and cloning translational initiation signals. J Bacteriol 143:971–980
Cava F, Hidalgo A, Berenguer J (2009) Thermus thermophilus as biological model. Extremophiles 13:213–231
de Grado M, Castan P, Berenguer J (1999) A high-transformation-efficiency cloning vector for Thermus thermophilus. Plasmid 42:241–245
Fujita A, Misumi Y, Koyama Y (2012) Two versatile vectors for Thermus thermophilus-Escherichia coli containing multiple cloning sites, lacZα gene and kanamycin or hygromycin resistance marker. Plasmid 67:272–275
Fujita A, Misumi Y, Honda S, Sato T, Koyama Y (2013) Construction of a new cloning vectors that employ the phytoene synthase encoding gene for color screening of cloned DNA inserts in Thermus thermophilus. Gene 527:655–662
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG (1996) Life with 6000 genes. Science 274:563–567
Hashimoto Y, Yano T, Kuramitsu S, Kagamiyama H (2001) Disruption of Thermus thermophilus genes by homologous recombination using a thermostable kanamycin-resistant marker. FEBS Lett 506:231–234
Hawley DK, McClure WR (1983) Compilation and analysis of Escherichia coli promoter DNA sequences. Nucl Acids Res 11:2237–2245
Henne A, Bruggeman H, Raasch C, Wiezer A, Hartsch T, Liesegang H, Johann A, Lienard T, Gohl O, Martinez-Arias R, Jacobi C, Starkuviene V, Schlenczeck S, Dencker S, Huber R, Klenk HP, Kramer W, Merkl R, Gottschalk G, Fritz HJ (2004) The genome sequence of the extreme thermophile Thermus thermophilus. Nat Biotechnol 22:547–553
Hoseki J, Yano T, Koyama Y, Kuramitsu S, Kagamiyama H (1999) Directed evolution of thermostable kanamaycin-resistance gene: a convenient selection marker for Thermus thermophilus. J Biochem 126:951–956
Koyama Y, Hoshino T, Tomizuka N, Furukawa K (1986) Genetic transformation of the thermophile Thermus thermophilus and of other Thermus spp. J Bacteriol 166:338–340
Koyama Y, Okamoto S, Furukawa K (1990) Cloning of α- and β-galactosidase genes from an extreme Thermophile, Thermus strain T2, and their expression in Thermus thermophilus HB27. Appl Environ Microbiol 56:2251–2254
Maniatis T, Fritsch EF, Sambrook J (1982) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor
Maseda H, Hoshino T (1995) Screening and analysis of DNA fragments that show promoter activities in Thermus thermophilus. FEMS Microbiol Lett 128:127–134
Miller J (1972) Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor
Moreno R, Zafra O, Cava F, Berenguer J (2002) Development of a gene expression vector for Thermus thermophilus based on the promoter of the respiratory nitrate reductase. Plasmid 49:2–8
Nakamura A, Takakura Y, Kobayashi H, Hoshino T (2005) In vivo directed evolution for thermostabilization of Escherichia coli hygromycin B phosphotransferase and the use of the gene as a selection marker in the host-vector system of Thermus thermophilus. J Biosci Bioeng 100:158–163
Ohta T, Tokishita S, Imazuka R, Mori I, Okamura J, Yamagata H (2006) β-Glucosidase as a reporter for the gene expression studies in Thermus thermophilus and constitutive expression of DNA repair genes. Mutagenesis 21:255–260
Ooga T, Ohashi Y, Kuramitsu S, Koyama Y, Tomita M, Soga T, Masui R (2009) Degradation of ppGpp by nudix pyrophosphatase modulates the transition of growth phase in the bacterium Thermus thermophilus. J Biol Chem 284:15549–15556
Park H-S, Kilbane JJ 2nd (2004) Gene expression studies of Thermus thermophilus promoters PdnaK, Parg and Pscs-mdh. Lett Appl Microbiol 38:415–422
Taylor MP, van Zyl L, Tuffin IM, Leak DJ, Cowan DA (2011) Genetic tool development underpins recent advances in thermophilic whole-cell biocatalysts. Microb Biothechnol 4:438–448
Ulrich JT, McFeters GA, Temple KL (1972) Induction and characterization of β-galactosidase in extreme thermophile. J Bacteriol 110:691–698
Vian A, Carrascosa AV, Garcia JL, Cortes E (1998) Structure of the β-galactosidase gene from Thermus sp. strain T2: expression in Escherichia coli and purification in a single step of an active fusion protein. Appl Environ Microbiol 64:2187–2191
Vieira J, Messing J (1982) The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene 19:259–268
Acknowledgments
We are grateful to J. Hasegawa for technical assistance with illustrations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by L. Huang.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Fujita, A., Sato, T., Koyama, Y. et al. A reporter gene system for the precise measurement of promoter activity in Thermus thermophilus HB27. Extremophiles 19, 1193–1201 (2015). https://doi.org/10.1007/s00792-015-0789-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-015-0789-3