
Abstract
α-Diimine nickel dibromide complexes of dibromo[N,N’-bis(2,6-diisopropylphenyl)-2,3-butanediimine]nickel(II) (A) and dibromo[N,N’-(phenanthrene-9,10-diylidene)bis(2,6-diisopropylaniline)]nickel(II) (B) were synthesized under controlled conditions. The catalysts A, B and mixed of 1:1 weight ratio of them (C) were activated by methylaluminoxane and were used for propylene polymerization in a semi-batch reactor. In study of the catalytic systems behavior, activities, glass transition temperature (T g) and viscosity average molecular weight (M v) values related to the catalyst C were between those related to the catalysts A and B under same conditions. The highest and the lowest molecular weights of the obtained polymers were produced by the catalysts B and A which were about 80,000 and 70,000 (g/mol) under same conditions, respectively. The T g of polypropylenes produced by the catalysts was about −34 °C. The resulting polymers had similar behavior with ethylene-propylene copolymer which it could be due to formation of ethylene-type units in the polymer chain.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In 1995, Brookhart and coworkers synthesized a new class of Ni(II) and Pd(II) polymerization catalysts stabilized by bulky α-diimine ligands. The α-diimine complexes based on nickel(II) activated by MAO are able to polymerize α-olefins to form high molecular weight polymers [1–6]. Compared to the early Ziegler–Natta and metallocene catalysts, the late transition metal complexes show better tolerance to the functional groups, which make them have an ability to form copolymers with a variety of polar-functionalized comonomers [1, 7, 8].
Marked chain isomerization mechanism (chain-walking) of nickel and palladium complexes allows them to produce polymers with a broad spectrum of branching topologies, ranging from relatively linear to hyperbranched or dendritic. Recently, a new family of polypropylenes (PPs) has been synthesized by bulky α-diimine/nickel catalysts, which those differ from PP produced by the early transition metal catalysts due to chain-walking events that often result in unique microstructures with ethylene-type runs (sequences of CH2) in the PP chain [3, 9–12]. The nickel α-diimine catalysts produce moderately syndiotactic PPs with an array generated by inversions and 3,1-enchainments as well as different types of short- and long-chain branches [9, 12, 13].
In this study, the catalysts A and B were synthesized as late transition metal α-diimine nickel complexes. The catalysts were bearing bulky substituent (isopropyl) in the ortho-position of the aryl group. Propylene polymerization was carried out using the catalysts A, B and mixed of 1:1 w/w of them (C) in the presence of MAO. The catalytic performance in propylene polymerization is influenced by factors such as reaction temperature, monomer pressure, and cocatalyst/catalyst molar ratio. Therefore, for all the catalysts the effect of mentioned factors on catalyst activity, M v and the apparent property and behavior of produced polymers were studied.
Experimental
Materials and propylene polymerization
Methanol and dichloromethane were supplied by Merck Chemical Co. (Darmstadt, Germany) and were used without any purification. Toluene was obtained from Arak Petrochemical Co. (Arak, Iran), it was dried by refluxing over sodium wire/benzophenone and then distilled under dry N2 and stored over activated 4 Å/13× type molecular sieves. Toluene was used as solvent of the catalyst slurry and polymerization medium. 4-Toluenesulfonic acid, diacetyl(2,3-butanedione), 9,10-phenanthrenequinone and aniline compounds were supplied by Merck Chemical Co. (Darmstadt, Germany) and were used in ligands synthesis. Methylaluminoxane (MAO) was purchased from Sigma Aldrich Chemicals (Steinheim, Germany) as 10 wt% (1.5 M) solution in toluene. 1,2-Dimethoxyethane nickel(II) bromide [(DME)NiBr2] were supplied by Sigma Aldrich Chemicals (Steinheim, Germany) with purity 97 % and was applied in the synthesis of the catalysts. Polymer-grade propylene was obtained from Arak Petrochemical Co. (Arak, Iran) and it was used after passing through a column of activated 4 Å/13× type molecular sieves. Nitrogen was obtained from Roham Gas (Tehran, Iran) dried by passage through KOH, activated silica gel and 4 Å/13× type molecular sieve columns. All manipulations were carried out under an atmosphere of dried and purified nitrogen using Schlenk or dry box techniques.
Polymerization runs were carried out in a stainless steel Buchi AG reactor (CH-8610) using the catalysts A, B and mixed of 1:1 w/w of them (C). The reactor temperature was adjusted by a thermostatic bath. The dried toluene (100 ml) was used as solvent. MAO solution (10 wt% in toluene) was used as a cocatalyst. Monomer injection system was equipped with mass flow controller (MFC) that loading of reactor was accomplished with the flow control of propylene gas. Toluene was saturated with the monomer gas prior to the polymerization. The catalysts A (3.0 × 10−3 mmol), B (3.0 × 10−3 mmol) and mixed of 1:1 w/w of them (3.0 × 10−3 mmol) were suspended in toluene and was transferred into the reactor under flow of nitrogen. After the desired reaction time, polymerization ended with transfer of reaction mixture into methanol containing HCl (5 %). Reaction mixture was filtered and the polymer was washed with methanol and water several times and was dried for 12 h at 40 °C.
PP and complex characterization
Direct application of Mark–Houwink equation of the form [η] = 1.58 × 10−4 M 0.77v dl g−1, displayed the so-called viscosity average molecular weight, M v [14]. M v of polymer samples were measured in decaline (decahydronaphthalene) at 135 °C by means of Ubbelohde suspended level dilution viscometer [15]. Differential scanning calorimetry (DSC) measurements were performed on a Netzsch 200 F3 DSC instrument under an N2 atmosphere. The samples were heated from −100 to +200 °C with a rate of 10 °C/min. The T g of polymers were determined by DSC. Melting points (mp) were determined in open capillary tubes in an Electrothermal IA 9000 melting point apparatus. 1H NMR and 13C NMR analyses of ligands were performed on Bruker BRX-100 Avance and Bruker Avance 400 spectrometers, respectively. Mass spectra (MS) were recorded with a Varian MAT CH7 mass spectrometer. Elemental analysis for CHN was carried out by CHNS type Thermo Finnigan 1112EA microanalyzer.
Ligands and catalysts preparation
Synthesis of ligand 1, [N,N′-bis(2,6-diisopropylphenyl)-2,3-butanediimine]
The ligand was synthesized through the reaction of 2,3-butanedione (5.0 mmol, 0.43 ml in 15 ml methanol), 2,6-diisopropylaniline (10.0 mmol, 1.88 ml) and in the presence of trace amount of 4-toluenesulfonic acid as a catalyst. The solution was stirred for 24 h at 40 °C. A yellow solid was obtained by solvent removing method. The solid was washed and crystallized with cold methanol and dried. The yield of the reaction was about 85 %; mp: 100 °C. 1H NMR (CDCl3, 100 MHz): δ 7.05–7.30 (m, 6H, Ar–H), 2.72 (septet, 4H, CH–(Me)2), 2.24 (s, 6H, N=C–CH 3), 1.24, 1.17 (d, 24H, CH–(CH 3)2). 13C NMR (CDCl3, 100.6 MHz): δ 167.2 (C=N), 146.7, 135.1, 123.9, 123.2 (Ar–C), 28.5 (CH–(Me)2), 23.6, 23.1 (CH–(CH3)2), 16.8 (CH3–C=N). EI-MS: m/z 404 [M+]. Anal. Calc. for C28H40N2: C, 83.11; H, 9.96; N, 6.93. Found: C, 83.07; H, 10.02; N, 6.88.
Synthesis of ligand 2, [N,N′-(phenanthrene-9,10-diylidene)bis(2,6-diisopropylaniline)]
To prepare the ligand, suspension of 9,10-phenanthrenequinone (4.8 mmol, 1.0 g) in methanol (10 ml) was added to a stirred solution of 2,6-diisopropylaniline (14.4 mmol, 2.70 ml) in methanol (10 ml) containing a trace amount of 4-toluenesulfonic acid. The reaction mixture was heated to reflux for 48 h. After removal of the solvent, the precipitated green solid was washed with cold methanol several times and dried. The yield of the reaction was about 50 %; mp: 159 °C. 1H NMR (CDCl3, 100 MHz): δ 6.97–8.24 (m, 14H, Ar–H), 3.21 (septet, 4H, CH–(Me)2), 1.42, 1.27 (d, 24H, CH–(CH 3)2). 13C NMR (CDCl3, 100.6 MHz): δ 161.5, 159.7 (C=N), 150.2, 147.3, 141.6, 139.3, 136.5, 135.1, 134.2, 132.1, 131.4, 129.7, 129.5, 125.3, 124.2, 118.6 (Ar–C), 29.5, 27.7 (CH–(Me)2), 24.4, 23.5 (CH–(CH3)2). EI-MS: m/z 526 [M+]. Anal. Calc. for C38H42N2: C, 86.64; H, 8.04; N, 5.32. Found: C, 86.57; H, 8.09; N, 5.30.
Synthesis of catalyst A, dibromo[N,N′-bis(2,6-diisopropylphenyl)-2,3-butanediimine]nickel(II)
(DME)NiBr2 (1.2 mmol, 0.37 g) and ligand 1 (1.2 mmol, 0.48 g) were combined in a Schlenk flask under a nitrogen atmosphere to prepare catalyst A. Methylene chloride (25 ml) was added to the solid mixture. The produced suspension was stirred for 24 h at room temperature. Solvent removal of the suspension resulted brown solid. The solid was washed with Et2O several times and dried in vacuum. The yield of the reaction was about 71 %; decomposed at temperature above 300 °C. EI-MS: m/z 543 [M+−Br], 463 [M+−2Br], 404 [M+−NiBr2]. Anal. Calc. for C28H40N2NiBr2: C, 53.97; H, 6.47; N, 4.49. Found: C, 53.76; H, 6.53; N, 4.45.
Synthesis of catalyst B, dibromo[N,N′-(phenanthrene-9,10-diylidene)bis(2,6-diisopropylaniline)]nickel(II)
The synthesis of complex B was carried out according to the complex A, using the ligand 2 (1.2 mmol, 0.63 g) and (DME)NiBr2 (1.2 mmol, 0.37 g). The complex B was isolated as a brown powder. The yield of the reaction was about 80 %; decomposed at temperature above 300 °C. EI-MS: m/z 665 [M+−Br], 585 [M+−2Br], 526 [M+−NiBr2]. Anal. Calc. for C38H42N2NiBr2: C, 61.24; H, 5.68; N, 3.76. Found: C, 60.97; H, 5.75; N, 3.71.
Structures of the catalysts A and B are shown in Fig. 1.

Possible structure of the catalysts used for polymerization of propylene
Result and discussion
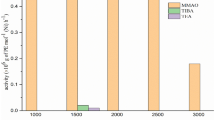
Propylene polymerization reactions using the prepared catalysts A, B and mixed of 1:1 w/w of them (C) were carried out at the different polymerization conditions. The activity of the catalyst, R p (average) is expressed as g PP/mmol Ni.h. Figure 2 shows the effect of Al/Ni molar ratio on the polymerization behavior of propylene using of the catalysts A, B, and C. MAO was used as a cocatalyst. In general, the role of MAO in polymerization can be included alkylation, generation of cationic active species and stabilization of these species by coordinative contact with its counterion. However, excess MAO inhibits the coordination of monomer with the active center and results in a slight decrease in the catalytic activity [1, 16].

Effect of Al/Ni molar ratio on activity of the catalytic systems. Polymerization conditions: [Ni] = 3 × 10−5 mol/l, temperature = 30 °C, monomer pressure = 4 bar, reaction time = 30 min, toluene = 100 ml
As displayed in Fig. 2, activity of the catalytic systems increased significantly with increasing Al/Ni molar ratio up to 2,000:1 while decreased in the molar ratios higher than 2,000. However, activity of the catalyst C in the molar ratio of 2,000 was about 138 (g PP/mmol Ni.h) between activity of the catalysts A and B which were about 117 and 164 (g PP/mmol Ni.h), respectively. The influence of Al/Ni molar ratio on the M v of the polymers obtained using the catalytic systems were investigated (Fig. 3). Increasing the Al/Ni molar ratio up to 2,000 increased M v values, while further increase in the ratio decreased M v of the obtained polymers. The competition between chain transfer to aluminum and propagation reactions increases in the ratio higher than 2,000 as reported [17, 18]. As shown in Fig. 3, the M v values related to catalyst C are between the M v values related to the catalysts A and B, in the Al/Ni molar ratio range from 1,000 to 3,000 which studied.

Influence of Al/Ni molar ratio on M v of the obtained polymers. Polymerization conditions: [Ni] = 3 × 10−5 mol/l, temperature = 30 °C, monomer pressure = 4 bar, reaction time = 30 min, toluene = 100 ml
The influence of polymerization temperature on activity of the catalytic systems and M v of the resulting polymers was investigated in temperature range from 30 to 60 °C (for the catalysts A and B) and from 20 to 60 °C (for the catalyst C), while the [Al]/[Ni] ratio was kept constant at 2,000:1. As displayed in Figs. 4 and 5, increasing temperature in the studied range decreased activity of the catalytic systems and M v values of the obtained polymers. The decrease of the catalyst activity with increasing temperature in the polymerization medium could be attributed to the catalyst irreversible deactivation and reducing the solubility of the monomer gas in the polymerization medium. Also higher rate of chain transfer and termination reactions than propagation reaction could be result of increasing polymerization temperature that leads to form shorter chains [16, 19, 20]. Figures 4 and 5 show that the M v values and activities related to the catalyst C are between those related to the catalysts A and B, in the temperature range from 30 to 60 °C. The M v values of polymers formed by the catalysts A, B, and C were about 58,000, 75,000 and 70,000 (g/mol), also activities of the catalysts A, B, and C were about 70, 94, and 81 (g PP/mmol Ni.h) in the polymerization temperature of 40 °C, respectively.

Effect of reaction temperature on activity of the catalytic systems. Polymerization conditions: [Ni] = 3 × 10−5 mol/l, [Al]/[Ni] = 2,000:1, monomer pressure = 4 bar, reaction time = 30 min, toluene = 100 ml

Influence of reaction temperature on M v of the obtained polymers. Polymerization conditions: [Ni] = 3 × 10−5 mol/l, [Al]/[Ni] = 2,000:1, monomer pressure = 4 bar, reaction time = 30 min, toluene = 100 ml
The influence of monomer pressure in the range of 2.5–8 bars on behavior of the catalysts A and B was studied, while the [Al]/[Ni] ratio and polymerization temperature were kept constant at 2,000:1 and 30 °C, respectively. According to runs 1–8 (Table 1), the highest productivity was obtained at the monomer pressure of +4 bars. The higher of the pressure the higher of the monomer concentration close to the catalyst active centers which could increase temperature in the polymerization environment. Increasing activity in the higher pressure may encapsulate the active centers because of the formed rubber-like polymer. Therefore, the higher activity and the higher increase of the reaction temperature could decay the active centers quickly [21, 22]. In order to study increasing temperature with increasing pressure from 2.5 to 8 bars, the propylene polymerization was carried out using the catalyst C at low temperature (20 °C). The highest activity of the catalyst C was about 222 (g PP/mmol Ni.h) at a monomer pressure of +4 bars and reaction temperature of 20 °C, it was higher than activity of the catalysts A and B which were about 117 and 164 (g PP/mmol Ni.h), respectively, at a monomer pressure of +4 bars and reaction temperature of 30 °C (Table 1). As expected, the M v values of the polymers obtained using the catalytic systems increased with increasing pressure from 2.5 to 8 bars (runs 1–13, Table 1).
In order to study the catalysts lifetime, propylene polymerization was carried out with the selected reaction times of 5, 15, 30, 45, and 60 min at reaction temperature of 30 °C for the catalysts A and B, and 20 °C for the catalyst C. The maximum activity was obtained after 15 min of the polymerization for the catalysts A and B at 30 °C and for the catalyst C it was obtained after 30 min at 20 °C (Table 1). The catalyst activity decreased after the maximum time of polymerization because of possible encapsulation of active centers of the catalyst in rubbery polymer and decay of some active centers due to the sudden increase of the reaction temperature [23, 24]. Increasing M v values during the polymerization time represented chain growth during the reaction time [23, 25]. The maximum values of M v for the polymers obtained by the catalysts A and B were about 122,000 and 140,000 (g/mol), respectively, at reaction time of 60 min and temperature of 30 °C. Also the maximum value of M v for the polymer formed by the catalyst C was about 124,000 (g/mol) at reaction time of 60 min and temperature of 20 °C (runs 17, 21, and 25, Table 1).
The activity of the catalyst B was higher than the catalyst A. For example, runs 1 and 3 (Table 2) show that activities of the catalysts A and B in the polymerization of propylene were 117 and 164 (g PP/mmol Ni.h) under same conditions, respectively. According to the structures of the catalysts A and B, presence of an electron-withdrawing phenanthrene group in the catalyst B instead of electron-donating methyl groups caused increase in positive charge of nickel and increase the catalyst activity [26, 27]. The M v value of the PP formed by the catalyst B was about 80,000 (g/mol) higher than 70,000 (g/mol) for M v value of the polymer obtained by the catalyst A under same conditions (runs 1 and 3, Table 2). The behavior suggests that the α-diimine nickel complexes based on 9,10-phenanthrenequinone (B) can introduce more steric demands on the axial faces than those based on 2,3-butanedione (A) [1]. The T g of PPs obtained by the catalysts A, B, and C were about −33.3, −34.4, and −33.6, respectively (runs 1, 3, and 5, Table 2). Tables and Figures show that the activities, T g and M v values related to the catalyst C were between those related to the catalysts A and B under same conditions.
PPs formed by the α-diimine nickel catalysts (A, B, and C) had approximately the rubber-like behavior and were completely transparent. Also, the T g of resulting PPs was near to T g of ethylene-propylene copolymer with ethylene content of about 20–30 % (Table 2) [21, 28]. It can be realized to the probability of formation of ethylene-type units (sequences of CH2) in polymer chain. The resemblance of behavior of these propylene homopolymers to ethylene–propylene copolymer suggests that the monomer was incorporated via 2,1-insertion of propylene and chain-walking based on 3,1-enchainment to give sequences of CH2 in the polymer chain. As displayed in Scheme 1, presence of ethylene-type runs is a result of 2,1-insertion of propylene and also chain-walking, in which the metal center migrates along the growing polymer chain through a series of β-hydride elimination and reinsertion events [9, 13, 29].

Mechanism of ethylene-type unit formation via 2,1-insertion of propylene and chain-walking based on 3,1-enchainment
Conclusion
Propylene polymerization was catalyzed with the α-diimine Ni(II) catalysts (A, B, and C) in the presence of MAO as cocatalyst. To obtain optimum conditions in the polymerization, effects of the Al/Ni molar ratio, temperature, pressure of monomer and reaction time were studied. The activities of the catalysts A, B, and C were about 117, 164, and 138 (g PP/mmol Ni.h) and M v values of the polymers formed by the catalysts A, B, and C were about 70,000, 80,000, and 73,000 (g/mol), respectively, at [Al]/[Ni] ratio of 2,000:1, temperature of 30 °C and monomer pressure of +4 bars. The activity of the catalyst B was higher than the catalyst A which was due to presence of an electron-withdrawing phenanthrene group in the catalyst B structure. The activities of the catalytic systems and M v of the obtained polymers decreased with increasing temperature, while those increased with increasing Al/Ni molar ratio up to 2,000:1. Increasing pressure of the monomer to 8 bars increased the M v of the resulting polymers. In addition, performance of the catalyst C was between performance of the catalysts A and B in the propylene polymerization under same conditions. PP formed by the catalysts (A, B, and C) had approximately the rubber-like behavior. The T g of resulting PPs was closed to the T g of reported ethylene-propylene copolymer. This behavior was due to formation of ethylene-type units in a result of 2,1-insertion of propylene and 3,1-enchainment (chain-walking) using α-diimine nickel-based catalyst. So it is possible with these kinds of catalyst structures to obtain propylene–ethylene copolymer structure like with the same T g behavior. The behavior was not observed with previous catalysts used such as metallocene and Ziegler–Natta catalysts.
References
Li L, Jeon M, Kim SY (2009) Synthesis, characterization and ethylene polymerisation of 9,10-phenanthrenequinone-based nickel(II)-α-diimine complexes. J Mol Catal A 303:110–116
Johnson LK, Killian CM, Brookhart M (1995) New Pd(II)- and Ni(II)-based catalysts for polymerization of ethylene and α-olefins. J Am Chem Soc 117:6414–6415
Ittel SD, Johnson LK, Brookhart M (2000) Late-metal catalysts for ethylene homo- and copolymerization. Chem Rev 100:1169–1204
Yang H, Wang Q, Fan Z, Lou W, Feng L (2003) Ethylene polymerization catalyzed by nickel-based complex combined with various modified EBAOs. Eur Polym J 39:275–279
Junges F, De Souza RF, Dos Santos JHZ, Casagrande OL Jr (2005) Ethylene polymerization using combined Ni and Ti catalysts supported in situ on MAO-modified silica. Macromol Mater Eng 290:72–77
Cherkasov VK, Druzhkov NO, Kocherova TN, Shavyrin AS, Fukin GK (2012) N,N′-Disubstituted phenanthrene-9,10-diimines: synthesis and NMR spectroscopic study. Tetrahedron 68:1422–1426
Li XF, Li YG, Li YS, Chen YX, Hu NH (2005) Copolymerization of ethylene with methyl methacrylate with neutral nickel(II) complexes bearing β-ketoiminato chelate ligands. Organometallics 24:2502–2510
Warwel S, Wiege B, Fehling E, Kunz M (2001) Copolymerization of ethylene with ω-unsaturated fatty acid methyl esters using cationic palladium complex. Macromol Chem Phys 202:849–855
Galland GB, Silva LPD, Dias ML, Crossetti GL, Ziglio CM, Filgueiras CAL (2004) 13C NMR determination of the microstructure of polypropylene obtained with the DADNi(NCS)2/methylaluminoxane catalyst system. J Polym Sci A 42:2171–2178
Guan ZJ (2003) Control of polymer topology through late-transition-metal catalysis. J Polym Sci A 41:3680–3692
Rose JM, Cherian AE, Coates GW (2006) Living polymerization of α-olefins with an α-diimine Ni(II) catalyst: formation of well-defined ethylene-propylene copolymers through controlled chain-walking. J Am Chem Soc 128:4186–4187
McCord EF, McLain SJ, Nelson LTJ, Arthur SD, Coughlin EB, Ittel SD, Johnson LK, Tempel D, Killian CM, Brookhart M (2001) 13C and 2D NMR analysis of propylene polymers made with α-diimine late metal catalysts. Macromolecules 34:362–371
Ruiz-Orta C, Fernandez-Blazquez JP, Anderson-Wile AM, Coates GW, Alamo RG (2011) Isotactic polypropylene with (3,1) chain-walking defects: characterization, crystallization, and melting behaviors. Macromolecules 44:3436–3451
Brandrup J, Immergut EH, Grulke EA, Abe A, Bloch DR (1999) Polymer handbook. Wiley, New York
Sobhanmanesh K, Hajizadeh A (2005) Ethylene/propylene copolymerization using Cp2ZrCl2/MAO catalyst: effects of copolymerization conditions on viscosity average molecular weight. Iran Polym J 14:15–21
Dos Santos JHZ, Da Rosa MB, Krug C, Stedile FC, Haag MC, Dupont J, Forte MDC (1999) Effects of ethylene polymerization conditions on the activity of SiO2-supported zirconocene and on polymer properties. J Polym Sci A 37:1987–1996
Dias ML, Silva LPD, Crossetti GL, Galland GB, Filgueiras CAL, Ziglio CM (2006) Propylene polymerization with nickel-diimine complexes containing pseudohalides. J Polym Sci A 44:458–466
Mortazavi MM, Arabi H, Zohuri GH, Ahmadjo S, Nekoomanesh M, Ahmadi M (2009) Ethylene homo- and copolymerization using a bis-IndZrCl2 metallocene catalyst: structural composition distribution of the copolymer. Macromol React Eng 3:263–270
Sandaroos R, Damavandi S, Farhadipour A (2010) A new family of high-performance Ti catalysts for olefin polymerization. Macromol Chem Phys 211:2339–2346
Jiang H, Lu J, Wang F (2010) Polymerization of ethylene using a nickel α-diimine complex covalently supported on SiO2–MgCl2 bisupport. Polym Bull 65:767–777
Zohuri GH, Mortazavi MM, Jamjah R, Ahmadjo S (2004) Copolymerization of ethylene-propylene using high-activity bi-supported Ziegler–Natta TiCl4 catalyst. J Appl Polym Sci 93:2597–2605
Zohuri GH, Damavandi S, Sandaroos R, Ahmadjo S (2011) Ethylene polymerization using fluorinated FI Zr-based catalyst. Polym Bull 66:1051–1062
Mortazavi SMM, Arabi H, Zohuri GH, Ahmadjo S, Nekoomanesh M, Ahmadia M (2010) Copolymerization of ethylene/α-olefins using bis(2-phenylindenyl)zirconium dichloride metallocene catalyst: structural study of comonomer distribution. Polym Int 59:1258–1265
Damavandi S, Zohuri GH, Sandaroos R, Ahmadjo S (2012) Novel functionalized bis(imino)pyridine cobalt(II) catalysts for ethylene polymerization. J Polym Res 19:9796–9801
Chadwick JC, Miedema A, Sudmeijer O (1994) Hydrogen activation in propene polymerization with MgCl2-supported Ziegler–Natta catalysts: the effect of the external donor. Macromol Chem Phys 195:167–172
Zhang T, Guo D, Jie S, Sun WH, Li T, Yang X (2004) Influence of electronic effect on catalytic activity of salicylaldiminato nickel(II) complexes. J Polym Sci A 42:4765–4774
Gao H, Pei L, Li Y, Zhang J, Wu Q (2008) Vinyl polymerization of norbornene with nickel catalysts bearing [N,N] six-membered chelate ring: important influence of ligand structure on activity. J Mol Catal A 280:81–86
Zohuri GH, Sadegvandi F, Jamjah R, Ahmadjo S, Nekoomanesh M, Bigdelli E (2002) Copolymerization of ethylene/propylene elastomer using high-activity Ziegler–Natta catalyst system of MgCl2 (ethoxide type)/EB/PDMS/TiCl4/PMT. J Appl Polym Sci 84:785–790
Jeon M, Han CJ, Kim SY (2006) Polymerizations of propylene with unsymmetrical (α-diimine)nickel(II) catalysts. Macromol Res 14:306–311
Acknowledgments
The authors express their appreciation to the Ferdowsi University of Mashhad for supporting the research project. Also, we thank the NMR laboratory of Tabriz University and DSC laboratory of Polymer and Petrochemical Institute of Iran.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pourtaghi-Zahed, H., Zohuri, G.H. Polymerization of propylene catalyzed by α-diimine nickel complexes/methylaluminoxane: catalytic behavior and polymer properties. Polym. Bull. 70, 1769–1780 (2013). https://doi.org/10.1007/s00289-013-0920-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-013-0920-5