
Abstract
A fluorinated FI Zr-based catalyst of bis[N-(3,5-dicumylsalicylidene)-2′,6′-flouroanilinato]zirconium(IV) dichloride was prepared and used for polymerization of ethylene. It was revealed that ortho-F-substituted phenyl ring on the N electronically plays a key role in the suppression of chain transfer reactions especially β-hydride transfer which resulted in an increase in the molecular weight of the obtained polymer and moderation of the catalyst activity as well. Methylaluminoxane (MAO) and triisobuthylaluminum (TIBA) were used as a cocatalyst and a scavenger, respectively. The catalyst showed the maximum activity at about [Al]:[Zr] = 32000:1 M ratio and further addition of MAO did not affect the activity of the catalyst. Ortho-F not only impressed the activity, but also reduced the [Al]:[Zr] molar ratio needed to reach the highest activity in comparison with the similar non-fluorinated FI catalysts. The highest activity of the prepared catalyst was obtained at 35 °C. At the monomer pressure of 3 bars polyethylene was obtained with the viscosity average molecular weight (M v) of 1.3 × 106 indicating the dramatic effect of ortho-F substitution on the polymerization mechanism. The polymerization was carried out using different amounts of hydrogen. Neither the activity of the catalyst nor the viscosity average molecular weight (M v) of the obtained polymer was sensitive to the hydrogen concentration. However, higher amount of hydrogen could slightly increase the activity of the catalyst.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Development of new catalysts with transition metals has played a substantial role in the fast-growing polyolefins industry. Improvement of new, better performing, less costly polyolefins has often been a result of catalyst development. However, polymerization processes of olefins, beside the requirement of higher activity catalyst to control the particle size, particle size distribution, and morphology of the resultant polyolefin are quite important. In the other words, success in these developments requires an appropriate integration of catalyst selections with reactor type and process parameters. Following the great success of the metallocene catalysts, significant efforts have been directed toward the discovery and application of new, highly active, single-site catalysts (post-metallocene catalysts) [1, 2]. These research efforts have led to the introduction of quite a few high-activity, single-site catalysts based on both early and late transition metal complexes with various ligand environments [3–9]. In association with appropriate cocatalysts, many of these catalysts show ethylene polymerization activities that are superior or comparable to those seen with early group 4 metallocene catalysts. These post-metallocene catalysts can produce a wide array of distinctive polymers (e.g., hyper-branched PEs, ethylene–methyl acrylate copolymers, monodisperse poly(1-hexene)s, and block copolymers based on α-olefins), many of which were inaccessible using metallocene catalysts [2, 10].
Although a large number of families of high-performance single-site catalysts have been developed thus far, improvements in some aspects of catalytic performance (e.g., temperature stability, precise control of chain transfer, comonomer sequence distribution control, precise control of polymer stereochemistry, and the ability to incorporate sterically encumbered monomers and polar monomers) are still required to achieve both greater control over polymer microstructures and extension of generic polyolefinic materials by introducing new monomer combinations.
In particular, bis(phenoxy-imine) group 4 metal catalysts, developed by Fujita [11–13] caused a new revolution in the field of catalytic olefin polymerization. These complex catalysts exhibit unique characteristics for production of new polymers that are not prepared by conventional Ziegler–Natta catalysts, as well as by ordinary metallocene-type catalysts [14]. The FI catalysts display a very high ethylene polymerization activity even at ambient temperature [12–15]. The ligand structure has a central role, correlations between the ligand structure and the polymerization activity have been interpreted. The introduction of an electron-donating methoxy group para to the phenoxy-O was found to increase the thermal stability of FI catalysts, making them available for polymerizations at industrially practical higher temperatures [15, 16].
Recently fluorinated Ti–FI catalysts have been discovered that can promote unprecedented living ethylene and propylene polymerization, resulting in the formation of functionalized polymers and block copolymers from ethylene, propylene, and higher α-olefins [17, 18]. It has been reported that fluorinated Ti–FI catalysts are capable of mediating the highly controlled, thermally robust living polymerization of ethylene and propylene [18, 19]. Electronic attractive interaction between a fluorinated phenoxy-imine ligand and a growing polymer chain has significant effects on the catalytic properties of Ti–FI catalysts [19]. Additionally, the presence of electron withdrawing fluoro substituents on aniline is considered to be highly beneficial for increasing the catalyst activity. It is known that introduction of electron-withdrawing F atoms on the ligand structure results in an increase in metal–carbon reactivity leading to reduced activation energy for ethylene insertion [20].
Although the catalytic behavior of ethylene and propylene polymerization, as well as ethylene–propylene copolymerization, using fluorinated Ti–FI complexes are extensively described in literature [21–23], but less attention has been paid to the fluorinated Zr–FI complexes that prompted us to study the catalytic properties of a fluorinated Zr–FI catalyst in ethylene polymerization.
In this study in order to investigate the influence of ligand on catalyst activity and catalytic behavior during the polymerization, a fluorinated Zr-based FI catalyst was prepared and used for polymerization of ethylene. The catalytic performance is sensitive to the conditions of polymerization which are influenced by factors such as temperature, monomer concentration, and catalyst:cocatalyst molar ratio. Therefore, mentioned factors in ethylene polymerization using the prepared catalyst were studied precisely. Furthermore, influence of hydrogen concentration on the polymerization activity was investigated. Also some specifications of resulting polymer were studied.
Experimental
Materials and ethylene polymerization
Zirconium tetrachloride, dichloromethane, methanol, 4-toluenesulfonic acid, phenol and amine derivatives were supplied by Merk Chemical Co. (Darmstadt, Germany) and were used as received. Toluene was obtained from the Merck Chemicals Co., n-hexane was supplied by Arak Petrochemical Co (Arak, Iran), the chemicals were prepared from distilling over sodium wire, stored over 13× and 4A activated molecular sieves and degassed by bubbling with dried nitrogen gas before use. Polymerization grade ethylene (purity 99.9%) was supplied by Iranian Petrochemical Co. (Tehran, Iran). Nitrogen gas (purity 99.99%) was supplied by Roham (Tehran Iran). Methylaluminoxane (MAO) (10% solution in toluene) and triisobutylaluminum (T1BA) (purity 93%) was supplied by Sigma–Aldrich Chemicals (Steinheim, Germany).
All the catalyst preparation and polymerization procedure were carried out under dried N2. Catalyst handling and polymerization procedures were carried out in 1-L stainless steel Buchi reactor (bcp 250) equipped with controllers systems as described before [24]. Toluene (250 mL) was introduced into the nitrogen-purged reactor and stirred (450 rpm) and the reactor was kept at the appropriate temperature and then the ethylene gas feed (100 L/h) was started. After 5 min, the ethylene gas feed was stopped and the toluene solution was kept under N2. To the resulting toluene solution were added toluene solutions of TIBA, MAO, and catalyst to start ethylene polymerization. Ethylene gas feed was started and the pressure of reactor was kept constant at the applied monomer pressure for each run. After 15 min, isobutyl alcohol (10 mL) was added and reaction was terminated by shutting off the feed stream followed by nitrogen purge and polymer precipitation using acidified (HCl) methanol. The obtained polymer was recovered by filtration, washed with methanol (100 mL × 3), and dried in a vacuum oven at 70 °C overnight.
Polyethylene and complex characterization
1H NMR spectrum was recorded on a Bruker BRX-100 AVANCE spectrometer. Elemental analysis was performed on a Thermo Finnigan Flash 1112EA microanalyzer. The viscosity average molecular weight (Mv) of some polymer samples was determined according to the literature [25, 26]. Intrinsic viscosity [η] was measured in decaline at 135 °C using an Ubbelohde viscometer. Mv values were calculated through equation [η] = 6.2 × 10−4 M 0.7v [25]. Differential scanning calorimetry (DSC) (Universal V4IDTA) with a rate of 10 °C/min instrument was used for polymer characterization. The degree of crystallinity of a polyethylene sample can be calculated from its heat of fusion which can be determined by differential scanning calorimetry. Comparison of the measured heat of fusion with that estimated for 100% crystalline polyethylene yields the fraction of the sample in the crystal lattice. In practice, the heat of fusion of pure crystalline polyethylene is taken to be in the range of 66–70 cal/g, with a commonly accepted value being 69 cal/g [26]. According to the literature [27], calculation of ΔHf/ΔHf* × 100 gives the values of crystallinity where ΔHf is the heat of fusion and ΔHf* = 69 cal/g is the heat of fusion of 100% crystalline polyethylene.
Synthesis of 3,5-dicumylsalicylaldehyde
To a stirred ethylmagnesium bromide (3.0 M in Et2O, 30 mmol) a solution of 2,4-dicumylphenol (in THF, 28.0 mmol) was added dropwise over a 15 min at 0 °C. The mixture was stirred for 2 h at room temperature. Dried toluene (50 mL) and a mixture of triethylamine (41.6 mmol) and paraformaldehyde (purity 94%, 93.9 mmol) were added. The mixture was stirred for further 2 h at 80 °C. HCl (6 N, 20 mL) was added at 0 °C. The organic phase was separated, dried over MgSO4, and its solvent was removed. The yield of the reaction was about 65%. 1H NMR (CDCl3, 100 MHz): δ 2.5 (s, 12H, Me), 7.08–7.41 (m, 12H, aromatic-H), 10.14 (s, 1H, CH=O), 12.97 (s, 1H, OH).
Synthesis of N-(3,5-dicumylsalicylidene)-2′,6′-difluoroanilin
To a stirred mixture of 3,5-dicumylsalicylaldehyde (10 mmol) in methanol (50 mL), 2,6-difluoroaniline (10.5 mmol) was added over a 5 min at room temperature in the presence of trace amount of 4-toluenesulfonic acid as a catalyst. The solution was refluxed for 5 h, concentrated then recrystallized to a yellow solid using methanol. The yield of the reaction was about 73%. 1H NMR (CDCl3, 100 MHz): δ 2.32 (s, 12H, Me), 7.05–7.38 (m, 15H, aromatic-H), 8.59 (s, 1H, CH=N), 13.50 (s, 1H, OH). Anal. found: C, 79.32; H, 6.30; N, 2.95; O, 3.38;. Calcd.: C31H29F2NO; C, 79.29; H, 6.22; N, 2.98; O, 3.41%.
Synthesis of bis[N-(3,5-dicumylsalicylidene)-2′,6′-flouroanilinato]zirconium(IV) dichloride
To a stirred solution of N -(3,5-dicumylsalicylidene)-2′,6′-difluoroanilin (5 mmol) in dried THf (20 mL) at −78 °C, n-BuLi (1.6 M in n-hexane, 5.2 mmol) was added dropwise in 10 min. The solution was allowed to warm to room temperature. Solution of ZrCl4 (2.5 mmol in THF) was added at 0 °C dropwise. The solution was stirred for 16 h at room temperature. Dried CH2Cl2 (50 mL) was added to the solution and mixed for 15 min, the byproduct of LiCl was filtered out. The solid was washed with n-hexane and the organic filtrates was added to the solution and concentrated under vacuum to afford a green solid of the catalyst in 48% yield. 1H NMR (CDCl3, 100 MHz): δ 2.23 (s, 24H, Me), 7.18–7.54 (m, 30H, aromatic-H), 8.54 (s, 2H, CH=N). Anal. found: C, 67.66; H, 5.17; N, 2.51; O, 2.94;. Calcd.: C62H56Cl2F4N2O2Zr; C, 67.74; H, 5.13; N, 2.55; O, 2.91. The adopted procedure in the synthesis of the catalyst is outlined in Scheme 1.

Synthetic route to prepare bis[N-(3,5-dicumylsalicylidene)-2′,6′-flouroanilinato]zirconium(IV) dichloride
Result and discussion
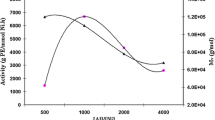
Polymerization of ethylene was carried out using the prepared catalyst at different conditions in toluene. The catalyst activity, R p (average), is expressed as g PE/mmol Zr h was determined after polymerization for 15 min for each run. TIBA (2 mmol), the scavenger, was added prior to the addition of MAO, the cocatalyst while the amount of TIBA was not included in the [Al]:[Zr] ratios. The polymerization activity was continuously increased with increasing the [Al]:[Zr] molar ratio in the range studied (Fig. 1).

Effect of MAO concentration on the activity of the catalyst. Polymerization conditions: temperature = 35 °C, polymerization time = 15 min, monomer pressure = 3 bar, [Zr] = 7 × 10−5 mmol, toluene = 250 mL
The value of [Al]:[Zr] = 32000:1 obtained for the catalyst with reasonable activity in comparison with the non-fluorinated FI catalysts such as bis[N-(3-tert-butylsalicilidene)anilinato]zirconium(IV) dichloride [28] which needs the value more than [Al]:[Zr] = 120000:1 to show the highest activity could be an advantage of the prepared catalyst. It is important to point out that using the [Al]:[Zr] molar ratio more than [Al]:[Zr] = 32000:1 did not cause any obvious further enhancement in the activity of the catalyst.
It has been reported that the steric bulky substitution on ortho position to the phenoxy oxygen of the ligand provides steric protection toward the anionic phenoxy oxygen from coordination with lewis acidic compounds [29, 30]. Introduction of bulky cumyl group on ortho position to the phenoxy oxygen in the prepared catalyst prevents the catalyst from coordination to the cocatalyst even at higher concentrations of cocatalyst and also induces effective ion-separation between the active cationic species and the anionic cocatalyst which provides more space for polymerization and increases the electrophilicity of the active species [30, 31].
Influence of polymerization temperature on activity was studied at the temperature between 20 and 55 °C, while the [Al]:[Zr] molar ratio was kept constant at [Al]:[Zr] = 32000:1. As it can be seen in the Fig. 2, the highest activity of the catalyst was obtained at 35 °C. The reduction of catalyst activity in the polymerization performed at the lower and upper temperature than the optimum value could be attributed to a low propagation rate and catalyst irreversible deactivation, respectively [31]. However, higher temperature promotes easy transfer of the monomer to the catalytic active centers [32] as well as reducing the solubility of the monomer gas in the polymerization medium. Similar trends have been already mentioned in the literature using homogeneous ansa-metallocene catalysts [33].

Relationship between the polymerization temperature and the activity. Polymerization conditions: polymerization time = 15 min, monomer pressure = 3 bar, [Al]:[Zr] = 32000:1, [Zr] = 7 × 10−5 mmol, toluene = 250 mL
At the monomer pressure of 3 bars the catalyst could produce polyethylene with the viscosity average molecular weight (M v) of 1.3×106 which is surprisingly a high value in comparison with the similar non-fluorinated FI catalysts [34, 35] indicating the dramatic electronic effect of ortho-F substitution on the polymerization mechanism [35] (Fig. 3). The M v values of 36 × 104, 10.4 × 104, and 1.84 × 104 have been reported already for the polyethylene obtained using the non-fluorinated bis[N-(3-t-butyl-5-methoxysalicylidene)anilinato]zirconium(IV) dichloride, bis[N-(3-cumyl-5-methoxysalicylidene)cyclohexylaminato]zirconium(IV) dichloride and bis[(3-cumyl-5-methylsalicylidene)anilinato]zirconium(IV) Dichloride, respectively [28, 36]. It has been proposed that attractive interaction between the ortho-F and the β-H on a growing polymer chain, which is expected to effectively curtail β-H transfer to the central metal and incoming monomer, is responsible for the unprecedented behavior [22, 34]. Although this interaction suppresses the β-H elimination, but the transition state of interaction between ortho-F and the β-H on a growing polymer chain is probably disfavored for coordination of ethylene to the active centers. This fact was proved with the moderation of activity of the catalyst.

The electronically interaction between H-β and ortho-F-atom-substituted phenyl ring on the N
The highest activity of the catalyst was 3.2 × 105 g PE/mmol Zr h at the temperature of 35 °C and monomer pressure of 3 bars. It has been accepted that the bulky substitutions effectively increase the activity of the catalyst through ion separation that results in increasing the unsaturation degree of the active species that leads to facilitate the coordination of ethylene to the active centers [12–14, 37]. The bulky cumyl groups on the structure of the catalyst may effectively play role to increase the activity. Both of steric effect causing by cumyl bulky groups and electronic effect causing by F groups influenced the activity of the catalyst profoundly.
As expected, the activity of the catalyst increased with increasing the monomer pressure (Fig. 4). The behavior is mainly due to the high concentration of the monomer close to the catalyst active centers.

Effect of monomer pressure on the polymerization behavior. Polymerization conditions: polymerization time= 15 min, [Al]:[Zr] = 32000:1, [Zr] = 7 × 10−5 mmol, temperature = 35 °C, toluene = 250 mL
The effect of polymerization temperature on the viscosity average molecular weight (M v) was illustrated in Table 1. As activation energy for chain transfer reaction is greater than that for propagation, high temperature leads to a decrease in polymer molecular weight. It could be as a result of β-hydride elimination which is facilitated with increasing the temperature [37, 38].
Crystallinity and melting point of the obtained polymer were between 55–65% and 125–135 °C, respectively. Higher pressure increased both the crystallinity and the M v values of the resulting polymer, while higher temperature decreased the M v values (Table 1).
Polymerization was carried out using different amount of hydrogen as a chain transfer agent while the polymerization was carried out at the optimum conditions established before. As it can be seen in Fig. 5, higher amounts of hydrogen only could slightly increase the activity of the catalyst. It is presumable that interaction between the ortho-fluorine atoms on the aniline ring and the β-hydrogen of the growing polymer chain retards the chain transfer reaction to the hydrogen favoring the chain propagation reaction. The M v of the obtained polymer was not sensitive to hydrogen concentration. Similar results have been reported already [39].

Influence of the hydrogen concentration on the polymerization activity and M v. Polymerization conditions: polymerization time = 15 min, [Al]:[Zr] = 32000:1, [Zr] = 7 × 10−5 mmol, temperature = 35 °C, toluene = 250 mL
Conclusions
A FI catalyst consists of fluorinated bis(phenoxy-imine)Zr complex was prepared and used for ethylene polymerization. The highest activity of the catalyst and the M v of the obtained polymer were 3.2 × 105 g PE/mmol Zr h and 1.32 × 106 at the monomer pressure of 3 bars, respectively. The highest activity of the catalyst were obtained at about 35 °C of temperature and [Al]:[Zr]=32000:1 M ratio. However, due to the restriction of ethylene coordination caused by transition state of the interaction between β-H of the growing polymer chain and F atoms, activity of the catalyst was decreased in comparison with the non-fluorinated FI catalysts reported already. Higher pressure increased the activity, crystallinity, and M v values of the resulting polymer. Ortho-F-substituted phenyl ring on the N electronically plays a key role in the suppression of chain transfer reactions resulted in increasing the molecular weight and moderation of the catalyst activity. This interaction probably prohibited chain transfer to the hydrogen which caused low sensitivity of the catalyst activity and molecular weight to the hydrogen concentration.
References
Kaminsky W (2004) The discovery of metallocene catalysts and their present state of the art. J Polym Sci Part A 42:3911–3921
Gibson VC, Spitzmesser SK (2003) Advances in non-metallocene olefin polymerization catalysis. Chem Rev 103:283–315
Britovsek GJP, Gibson VC, Wass DF (1999) The search for new-generation olefin polymerization catalysts: life beyond metallocenes. Angew Chem Int Ed 38:428–447
Baumann R, Davis WM, Schrock RR (1997) Synthesis of titanium and zirconium complexes that contain the tridentate diamido ligand, [((t-Bu-d 6)N-o-C6H4)2O]2− ([NON]2-) and the living polymerization of 1-Hexene by activated [NON]ZrMe2. J Am Chem Soc 119:3830–3831
Nomura K, Sagara A, Imanishi Y (2002) Olefin polymerization and ring-opening metathesis polymerization of norbornene by (arylimido)(aryloxo)vanadium(V) complexes of the type VX2(NAr)(OAr¢). Remarkable effect of aluminum cocatalyst for the coordination and insertion and ring-opening metathesis polymerization. Macromolecules 35:1583–1590
Johnson LK, Killian CM, Brookhart M (1995) New Pd(I1)- and Ni(I1)-based catalysts for polymerization of ethylene and a-olefins. J Am Chem Soc 117:6414
Quijada R, Rojas R, Bazan G, Komon ZJA, Mauler RS, Galland GB (2001) Synthesis of branched polyethylene from ethylene by tandem action of iron and zirconium single site catalysts. Macromolecules 34:2411–2417
Azoulay JD, Bazan GC, Galland GB (2010) Microstructural characterization of poly(1-hexene) obtained using a nickel R-keto-β-diimine initiator. Macromolecules 43:2794–2800
Terao H, Ishii S, Mitani M, Tanaka H, Fujita T (2008) Ethylene/polar monomer copolymerization behavior of bis(phenoxy–imine)Ti complexes: formation of polar monomer copolymers. J Am Chem Soc 130:17636–17637
Mitani M, Saito J, Ishii SI, Nakayama Y, Makio H, Matsukawa N, Matsui S, Mohri JI, Furamaya R, Teroa H, Bando H, Tanaka H, Fujita T (2004) FI Catalysts: new olefin polymerization catalysts for the creation of value-added polymers. Chemical Rec 4:137–158
Busico V, Cipullo R, Friederichs N, Ronca S, Togrou M (2003) The first molecularly characterized isotactic polypropylene-block-polyethylene obtained via “quasi-living” insertion polymerization. Macromolecules 36:3806–3808
Suzuki Y, Terao H, Fujita T (2003) Recent advances in phenoxy-based catalysts for olefin polymerization. Bull Chem Soc Jpn 76:1493–1517
Furuyama R, Saito J, Ishii SI, Mitani M, Tohi Y, Makio H, Matsukawa N, Tanaka H, Fujita T (2003) Ethylene and propylene polymerization behavior of a series of bis(phenoxy–imine)titanium complexes. J Mol Cat A 200:31–42
Tian J, Hustad PD, Coates GW (2001) A new catalyst for highly syndiospecific living olefin polymerization: homopolymers and block copolymers from ethylene and propylene. J Am Chem Soc 123:5134–5135
Fujita M, Coates GW (2002) Synthesis and characterization of alternating and multiblock copolymers from ethylene and cyclopentene. Macromolecules 35:9640–9647
Makio H, Fujita T (2009) Development and application of FI catalysts for olefin polymerization: unique catalysis and distinctive polymer formation. Acc Chem Res 42(10):1532–1544
Saito J, Mitani M, Mohri J, Ishii S, Yoshida Y, Matsugi T, Kojoh S, Kashiwa N, Fujita T (2001) Highly syndiospecific living polymerization of propylene using a titanium complex having two phenoxy-imine chelate ligands. Chem Lett 576–582
Coates GW, Tian J, Hustad PD (2003) Bis(salicylaldiminato)titanium complex catalysts, highly syndiotactic polypropylene by a chain-end control mechanism, block copolymers containing this. US Patent 6562930
Sakuma A, Weiser MS, Fujita T (2007) Living olefin polymerization and block copolymer formation with FI catalyst. Polym J 39(3):193–207
Ishii SI, Saito J, Mitani M, Mohri JI, Matsukawa N, Tohi Y, Matsui S, Kashiwa N, Fujita T (2002) Highly active ethylene polymerization catalysts based on titanium complexes having two phenoxy-imine chelate ligand. J Macromol Catal 179:11–16
Nakayama Y, Saito J, Bando H, Fujita T (2005) Propylene polymerization behavior of fluorinated bis(phenoxy-imine) Ti complexes with an MgCl2-based compound (MgCl2-supported Ti-based catalysts). Macromol Chem Phys 206:1847–1852
Ishii SI, Furuyama R, Matsukawa N, Saito J, Mitani M, Tanaka H, Fujita T (2003) Ethylene and ethylene/propylene polymerization behavior of bis(phenoxy-imine) Zr and Hf complexes with perfluorophenyl substituents. Macromol Rapid Commun 24:452–456
Yasunori Y, Shigekazu M, Terunori F (2005) Bis(pyrrolide-imine) Ti complexes with MAO: a new family of high performance catalysts for olefin polymerization. J Organometal Chem 690:4382–4397
Zohuri GH, Ahmadi Bonakdar M, Damavandi S, Eftekhar M, Askari M, Ahmadjo S (2009) Preparation of ultra high molecular weight polyethylene using bi-supported SiO2/MgCl2 (spherical)/TiCl4 catalyst: a morphological study. Iran Polym J 18:593–600
Brandrup J, Immergut EH (1989) Polymer handbook, vol VII, 3rd edn. Wiley, New York, pp 1–7
Peacock A (2000) Handbook of polyethylene: structures: properties, and applications, vol 57. Marcel Dekker Inc., New York
Justino J, Dias AR, Ascenso J, Marcues MM, Tait PJT (1997) Polymerization of ethylene using metallocene and aluminoxane systems. Polym Int 44:407–412
Matsui S, Mitani M, Saito J, Ishii SI, Mitani M, Matsui S, Tohi Y, Makio H, Matsukawa N, Tsuru K, Masatoshi N, Nakano T, Tanaka H, Kashiwa N, Fujita T (2001) A family of zirconium complexes having two phenoxy-imines chelate ligands for olefin polymerization. J Am Chem Soc 123:6847–6856
Nakayama Y, Bando H, Sonobe Y, Fujita T (2004) Olefin polymerization behavior of bis(phenoxy-imine) Zr, Ti, and V complexes with MgCl2-based cocatalysts. J Mol Catal A 213:141–150
Santos JHZD, Rosa MBD, Krug C, Stedile FCS, Haag MC, Dupont J, Forte MDC (1996) Effects of ethylene polymerization conditions on the activity of SiO2-supported zirconocene and on polymer properties. J Polym Sci A 37(13):1987–1996
Mazzeoa M, Lamberti M, Pappalardob D, Annunziataa L, Pellecchiaa C (2009) Polymerization of α-olefins promoted by zirconium complexes bearing bis(phenoxy-imine) ligands with ortho-phenoxy halogen substituents. J Mol Catal A 297:9–17
Chen Y, Rausch MD, Chien JCW (1995) Heptane-soluble homogeneous zirconocene catalyst: synthesis of a single diastereomer, polymerization catalysis, and effect of silica supports. J Polym Sci A 33:2093–2108
Matsui S, Fujita T (2001) FI catalysts: super active new ethylene polymerization catalysts. Catal Today 66:63–73
Matsukawa N, Mitani S, Mitani M, Saito J, Tsuru K, Kashiwa N, Fujita T (2001) Ethylene polymerization activity under practical conditions displayed by zirconium complexes having two phenoxy-imine chelate ligands. J Mol Catal A 169:99–104
Mitani M, Nakano T, Fujita T (2003) Unprecedented living olefin polymerization derived from an attractive interaction between a ligand and a growing polymer chain. Chem Euj J 9(11):2396–2403
Jochem TMP, Wickert G, Wim PMS (2002) Polymerization of liquid propylene with a 4th generation Ziegler–Natta catalyst: influence of temperature, hydrogen and monomer concentration and prepolymerization method on polymerization kinetics. Chem Eng Sci 57:3461–3477
Busico V, Talarico G, Cipullo R (2005) Living Ziegler–Natta polymerizations: true or false. Macromol Symp 226:1–16
Deck PA, Bewick CL, Marks TJ (1998) Highly electrophilic olefin polymerization catalysts. Quantitative reaction coordinates for fluoroarylborane/alumoxane methide abstraction and ion-Pair reorganization in group 4 metallocene and “constrained geometry” catalysts. J Am Chem Soc 120:1772–1784
Weiser MS, Wesolek M, Mulhaupt R (2006) The synthesis and X-ray structure of a phenoxyimine catalyst tailored for living olefin polymerisation and the synthesis of ultra-high molecular weight polyethylene and atactic polypropylene. J Organometall Chem 691:2945–2952
Acknowledgments
The authors thank Mr. Asghari of the DSC laboratory and Mrs. Sadeghian of the Central laboratory for their kind help with this study.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Zohuri, G.H., Damavandi, S., Sandaroos, R. et al. Ethylene polymerization using fluorinated FI Zr-based catalyst. Polym. Bull. 66, 1051–1062 (2011). https://doi.org/10.1007/s00289-010-0338-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-010-0338-2