
Abstract
The diversities of soil bacterial communities in the Phragmites australis, Phalaris arundinacea, and non-plantation constructed wetlands were compared and analyzed by through high-throughput Illumina sequencing. At the phylum level, the ten dominant bacterial phyla in the three types of constructed wetlands were the same. At the genus level, the three most dominant bacterial genera in the three types of constructed wetlands were the same. The non-plantation constructed wetland (Nop-cw) had the highest diversity of soil bacterial community. The introduction of P. australis or P. arundinacea did not increase diversity of the soil bacterial communities in the constructed wetlands but greatly changed the compositions and potential function of the soil bacterial communities, especially some bacterial genera involved in pollutant removal. So it was predicted that the P. australis constructed wetland (Pau-cw) had a larger capacity for the removal of heavy metals and sulfur than the P. arundinacea constructed wetland (Par-cw), and the nitrification capacity of the P. arundinacea constructed wetland was stronger than that of the P. australis constructed wetland. The above results not only clarified the differences among the soil bacterial communities of the constructed wetlands with different plants in terms of diversity but also revealed the decontamination mechanism of the constructed wetlands to some degree.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Constructed wetlands are wastewater treatment systems that simulate natural wetlands [1, 2]. It can achieve an extremely high wastewater purification effect through physical, chemical, and biological processes [3].
Microbes are the main promoters of material cycling in constructed wetlands. Constructed wetland microbes have important roles in wastewater purification and thus have been a focus of research [3,4,5]. However, there are relatively few studies on the diversity and function of bacterial communities in constructed wetlands from the perspective of the phylum and genus. Ibekwe et al. detected 31 different operational taxonomic units (OTUs) from soil bacterial communities in the constructed wetland by using denatured gradient gel electrophoresis and cloning library methods [6]. Li et al. detected 221 genera from 17 bacterial phyla in the constructed wetland soil by using the restriction fragment length polymorphism (T-RFLP) and cloning library methods [7]. However, given the limitations of the adopted technologies, these studies were only confined to the preliminary identification of microbial communities in constructed wetlands. Determining the overall picture of microbial communities to fully reveal the diversity of soil microbial communities in constructed wetlands remains difficult. This limitation is considered the main reason that the decontamination function of soil microbes in constructed wetlands is often underestimated. Some scholars have applied high-throughput sequencing technology to study microbes in natural or constructed wetland in recent years [8,9,10]. Compared with other research technologies, high-throughput sequencing technologies are more efficient and accurate in determining the relative abundance of all kinds of microbes in habitats through large-scale analysis of microbes in samples [11].
Phragmites australis and Phalaris arundinacea are the two most commonly used plant species in wetlands constructed for wastewater purification [12,13,14]. The constructed wetlands where P. australis and/or P. arundinacea are planted have large stronger decontamination capacities [15, 16]. However, the diversity and functions of microbial communities in the P. australis or P. arundinacea constructed wetlands have not been investigated through high-throughput sequencing technologies. In this present paper, we studied the diversity and functions of the soil bacterial communities in the P. australis, P. arundinacea, and non-plantation constructed wetlands by performing high-throughput Illumina sequencing on a Miseq sequencing platform. Our aim is to explore the mechanism of the highly efficient decontamination of constructed wetlands with P. australis or P. arundinacea or both from the perspective of microbes.
Materials and Methods
Constructed Wetland Simulation
On December 27, 2015, 12 plastic buckets (80 cm in diameter and 100 cm in height) were selected, and each plastic bucket was filled with about 95 kg of loamy soil (dry weight). The 12 plastic buckets were divided equally into three groups. The rhizomes of P. australis and P. arundinacea (collected from the Zhenjiang Waterfront Wetland of Yangtze River Lower Reach, China) were transplanted into four plastic buckets of the first and second group, respectively, whereas the four plastic buckets of the third group were not transplanted. Treatments with four replicates consisted of three different plant conditions in the constructed wetlands, namely treatments of the P. australis simulated constructed wetland (Pau-cw), P. arundinacea simulated constructed wetland (Par-cw), and Non-plantation simulated constructed wetland (Nop-cw).
Soil Sample Collection
The soil samples were collected in September 2017. Three points from each bucket were selected according to the equilateral triangle. The soil samples (sampling depth of 0–15 cm) were collected, and then the 12 soil samples from every treatment were fully mixed into a mixed sample. The three mixed samples from the three different treatments were placed in an ice box for DNA extraction.
DNA Extraction, PCR Amplification and High-Throughput Sequencing
The microbial genomic DNA was extracted in triplicate from 0.3 g of well-mixed soil samples by using Isoil beads beating kit (WAKO Japan) according to the manufacturer’s protocol. The DNA concentrations of the Pau-cw, Par-cw, and Nop-cw were 12.54, 11.6, and 8.98 ng/uL, respectively.
515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) were used as the primer set to amplify the bacterial 16S rRNA V4 region. The PCR products were subjected to high-throughput DNA sequencing on Illumina MiSeq platform at Novo-source Technology Co. Ltd. (Beijing, China).
Raw data for high-throughput sequencing, OTUs and Chao 1, Shannon, and Simpson Indexes of the samples were analyzed and processed by using the ribosomal database project (RDP) database, Mothur and QIIME software to obtain accurate and high-quality DNA sequence information [17,18,19]. The raw sequences obtained in this study have been deposited in the short-read archive database of GenBank with accession number PRJNA503666.
Results and Discussion
Species Diversity
The high-throughput DNA sequencing library of soil bacterial communities in the three treatments is shown in Table 1. A total 69,290, 65,745, 73,587 raw sequences and 4766, 4876, and 4995 of bacterial OTUs were obtained from the soil samples of the PAu-cw, Par-cw, and Nop-cw, respectively. The Chao1, Simpson, and Shannon index of the soil bacterial community in the Nop-cw were consistently higher than those in the other two treatments. Statistical analysis shows a significant difference in raw sequence, OTU number, Chao1 and Shannon index between the Pau-cw and Nop-cw, and a significant difference in raw sequence and OTU number between the Par-cw and Nop-cw. Meanwhile, no significant difference in any of the above parameters was observed between the Pau-cw and Par-cw.
Figure 1 shows the dilution curves of bacterial 16S rDNA sequencing in the three treatments. Although a small amount of new OTUs were still found even when the number of sequencing exceeded 4000, the whole curve was extremely smooth, indicating that the sequencing library should be saturated. Additionally, as the same sequence number was used, the Nop-cw always had the highest bacterial OTU number, whereas the Pau-cw always had the least.

Dilution curves of bacterial OTUs of high-throughput DNA sequencing library from the three types of constructed wetlands
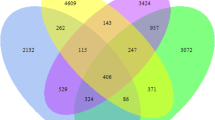
The Venn diagram of soil bacterial OTUs in the three treatments (Fig. 2) shows that the number of common bacterial OTUs in the three treatments was 2764. A total 3777 common bacterial OTUs existed in the Pau-cw and Par-cw. The other two treatments had a lower common OTUs. The number of the specific bacterial OTUs in the Nop-cw was 1388, which was 2.2 and 2.3 times of specific bacterial OTUs in the Pau-cw and Par-cw, respectively.

Venn diagram of bacterial OTUs of high-throughput DNA sequencing library in the three types of constructed wetlands
All the above diversity parameters of soil bacterial community in the Nop-cw were higher than those in the plantation constructed wetlands to varying degrees, indicating that the diversity of soil bacterial community would decrease after some plants, such as P. australis or P. arundinacea, are introduced into the constructed wetlands. However, the number of OTUs shared by the two plantation constructed wetlands was higher than that shared by the Nop-cw and either of the two plantation constructed wetlands. It indicates that an obvious change in the structure of soil bacterial community occurred after P. australis or P. arundinacea had been transplanted into the constructed wetlands, and a large number of new shared OTUs with specific functions appeared in the constructed wetlands and potentially improved of decontamination function for the plantation constructed wetlands.
Functional Diversity
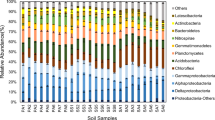
At the phylum level, the ten most dominant bacterial phyla in the soils from the three treatments are shown in Fig. 3. Although the ten most abundant bacterial phyla in the three treatments were identical, a distinguished difference in the proportion of every phylum in the bacterial community was found among the three treatments. Protectorate (31.7%) predominated in the Nop-cw, followed by Actinobacteria (26.6%), Acidobacteria (10.7%), Firmicutes (7.4%), Gemmatimonadetes (5.2%), Thaumarchaeota (4.1%), Chloroflexi (4.0%), Nitrospirae (2.2%), Bacteroidetes (1.8%), and Verrucomicrobia (1.3%). Proteobacteria was also the absolute dominant phylum in the Pau-cw and Par-cw, with the proportions of 45.6% and 44.0%, respectively. The dominance order of the bacterial phyla in the Pau-cw was Acidobacteria (14.5%), Firmicutes (8.3%), Actinobacteria (7.0%), Gemmatimonadetes (3.6%), Bacteroidestes (3.4%), Chloroflexi (3.2%), Thaumarchaeota (3.0%), Nitrospirae (3.0%), and Verrucomicrobia (2.7%), whereas that in the Par-cw was Acidobacteria (12.4%), Firmicutes (11.4%), Actinobacteria (9.1%), Bacteroidestes (3.9%), Chloroflexi (3.5%), Nitrospirae (3.2%), Gemmatimonadetes (3.1%), Verrucomicrobia (2.0%), and Thaumarchaeota (1.6%).

The clustering tree of bacterial community composition at phylum level in the three types of constructed wetlands
Of the 10 most common bacterial phyla, Proteobacteria, Actinobacteria, Acidobacteria, and Firmicutes were simultaneously the main dominant bacterial phyla in the three treatments. It indicates that these phyla have a wide range of ecological adaptability, which is consistent with other related research results [10, 20,21,22]. After P. australis and P. arundinacea were transplanted into the constructed wetland, the proportions of Proteobacteria in the soil bacterial communities were increased to 45.6% and 44.0%, respectively, from 31.7% and for Acidobacteria to 14.5% and 12.4% from 10.7%, that of Actinobacteria to 3.4% and 3.9% from 1.8%, Verrucomicrobia to 2.7% and 2.0% from 1.3%, and Nitrospirae to 3.0% and 3.2% from 2.2%. As the most dominant bacterial phylum, Proteobacteria is considered to be highly active in promoting nitrogen cycling in nature and is one of the major nitrifying microbes [23]. It was found its’ abundance has a significant linear positive relationship with nitrification rate during long-term natural dissolved organic matter decomposition [24]. Acidobacteria, Bacteroidetes, Verrucomicrobia, and Nitrospirae were reported as important nitrogen-translating bacterial phyla [25], and were found to enhance the nitrification strength of the sludge systems [26]. Acidobacteria was further pointed out to use nitrite as the main source of N to meet its own growth and metabolism requirements [27]. Nitrospira was found to be a class of Gram-negative bacteria, and can oxidize nitrite into nitrates [28]. All these results show that a considerable change in the structure of soil bacterial community occurred despite the decrease in diversity in soil bacterial community to a certain extent after some plants were transplanted into the constructed wetlands, and the abundance of bacteria associated with nitrogen transformation increased significantly. This finding explains why the constructed wetlands with some plants have higher nitrogen removal efficiency from the perspective of bacterial phylum.
The phylogenetic analysis of the top 100 genera in the soils from the three treatments is shown in Fig. 4. At the genus level, a distinguished difference in the proportion of the bacterial community was found among the three treatments. The dominance order in the Nop-cw was Arthrobacter (3.3%), Pseudomonas (2.7%), Bacillus (2.6%), Gaiella (1.8%), Sphingomonas (1.4%), Lactobacillus (1.2%), Microvirga (1.0%), Candidatus-Alysiosphaera (0.9%), Solirubrobacter (0.9%), and Blastococcus (0.8%), and that in the Pau-cw was Geobacter (3.4%), Bacillus (2.5%), Desulfurivibrio (1.4%), Desulfocapsa (1.3%), Ramlibacter (1.0%), Thermomonas (1.0%), Anaeromyxobacter (1.0%), an unidentified-Nitrospiraceae (0.9%), Sphingomonas (0.8%), Azoarcus (0.8%), Geothrix (0.8%), Steroidobacter (0.7%), and Gaiella (0.6%), while that in the Par-cw was Thiobacillus (3.3%), Bacteria (2.2%), Ramlibacter (1.6%), an unidentified-Nitrospiraceae (1.3%), Sulfuricurvum (1.1%), Geobacter (1.1%), Gaiella (1.0%), Blautia (0.9%), Anaeromyxobacter (0.8%), Steroidobacter(0.8%), and Streptococcus (0.8%).

Phylogenetic analysis of the top 100 bacterial genera in the three types of constructed wetlands
A ternary plot analysis was performed among the top ten most common genera in the soils from the three treatments (Fig. 5). Of the above ten most common genera, Bacillus was most abundant and is distributed symmetrically in the every treatments; Arthrobacter, Pseudomonas, and Lactobacillus were most abundant in the Nop-cw; Geobacter and Desulfocapsa were most abundant in the Pau-cw; and Ramlibacter, Thiobacillus, Thermomonas, and Sulfuricurvum were most abundant in the Par-cw.

Specific bacterial genera in the three types of constructed wetlands
Among the three treatments, only the two plantation constructed wetlands shared more common bacterial genera. A T-test of the relative abundance of all common genera in the two plantation constructed wetlands was performed, and nine of all common genera, such as Nitrosospira, Nitrosomonas, Candidatus Solibacter, Candidatus Entotheonella, Desulfobulbus, Ferritrophicum, Pirellula, Chthoniobacter, and Anaerobacterium, were found to have significant difference in the relative abundance between the two plantation constructed wetlands (Fig. 6). Other studies have shown that Bacillus is one of the dominant bacterial genera in the Paus-cw [29]. In the present paper, Bacillus was the only common dominant genus in the three treatments. It indicates that Bacillus has a strong adaptability. It is often regarded as dominant native bacterial genera in different types of constructed wetlands and plays an important role in maintaining the stability of the microbial communities in constructed wetlands. Anaeromyxobacter, Steroidobacter, Geobacter, and Ramlibacter, as common dominant bacterial genera in the two plantation constructed wetlands, are considered to be important nitrogen-transforming bacteria and are indispensable participants in the process of nitrogen removal in constructed wetland systems [30]. Anaeromyxobacter couples nitrate, nitrite, humus, or O2 reduction for anaerobic respiration and is thus a potential functional bacterium for water restoration [30]. Geobacter was found to be a sulfur reducer, and play an important role in sulfur removal [31]. As a specific dominant bacterial genus in the Pau-cw, Desulfocapsa is a sulfide bacterium that obtains the energy needed for its growth by oxidizing reduced sulfur compounds, which results in a decrease in pH value and a change in the redox of the surrounding environment. As a result, Desulfocapsa transforms heavy metals and sulfur in a system from their respective original organic states to free states before their removal [32]. Thus, the Pau-cw had larger removal capacity for heavy metals and sulfur than the other two types of constructed wetlands and this was partially confirmed by other studies [12]. Nitrosospira and Nitrosomonas, two common bacterial genera in the two types of plantation constructed wetlands, are the main ammonia-oxidizing bacteria (AOB), which oxidize ammonium (NH4+) to nitrite (NO2−), which is a key stage of nitrification [33]. However, the relative abundances of Nitrosospira and Nitrosomonas in the Par-cw were significantly higher than those in the Pau-cw, so the former may have stronger nitrification capacity than the latter. This observation explains why the constructed wetlands with different plant conditions have different decontamination effects in terms of bacterial genus.

Significant analysis of difference in the relative abundance between the two plantation constructed wetlands
References
Li B, Chen H, Li N, Wu Z, Wen Z, Xie S, Liu Y (2017) Spatio-temporal shifts in the archaeal community of a constructed wetland treating river water. Sci Total Environ 605–606:269–275
Wang M, Zhang D, Dong J (2017) Application of constructed wetlands for treating agricultural runoff and agro-industrial wastewater: a review. Hydrobiologia 1:1–31
Weber K (2016) Microbial community assessment in wetlands for water pollution control: past, present, and future outlook. Water 8(11):503–522
Chang JJ, Wu SQ, Liang K, Wu Z, Liang W (2015) Comparative study of microbial community structure in integrated vertical-flow constructed wetlands for treatment of domestic and nitrified wastewaters. Environ Sci Pollut Res 22(5):3518–3527
Zhou XH, Zhang JP, Wen CZ (2017) Community composition and abundance of anammox bacteria in cattail rhizosphere sediments at three phenological stages. Curr Microbiol 74:1349–1357
Ibekwe AM, Lyon SR, Leddy M, Jacobson-Meyers M (2007) Impact of plant density and microbial composition on water quality from a free water surface constructed wetland. J Appl Microbiol 102(4):921–936
Li X, Zhang M, Liu F, Li Y, Li Y (2017) Bacterial community dynamics in a Myriophyllum elatinoides purification system for swine wastewater in sediments. Appl Soil Ecol 119:56–63
Bai SY, Tao L, Ding YL, Li ZL, Wang DQ, You SH, Xie QL (2017) Campus sewage treatment in multilayer horizontal subsurface flow constructed wetlands: nitrogen removal and microbial community distribution. Clean - Soil Air Water 45(11):254–263
Ligi T, Oopkaup K, Truu M, Preem JK, Nõlvak H (2014) Characterization of bacterial communities in soil and sediment of a created riverine wetland complex using high-throughput 16S r RNA amplicon sequencing. Ecol Eng 72:56–66
Wang F, Zhao HW, Xiang HY, Wu LJ, Men X, Qi C, Chen GQ, Zhang HB, Wang Y, Xian M (2018) Species diversity and functional prediction of surface bacterial communities on aging flue-cured tobaccos. Curr Microbiol 75:1306–1315
Peralta RM, Ahn C, Gillevet PM (2013) Characterization of soil bacterial community structure and physicochemical properties in created and natural wetlands. Sci Total Environ 443(3):725
Vymazal J, Svehla J, Kröpfelová L, Chrastný V (2007) Trace metals in Phragmites australis and Phalaris arundinacea growing in constructed and natural wetlands. Sci Total Environ 380(1):154–162
Fu WG, Li PP, Wu YY (2011) Mechanism of the plant community succession process in the Zhenjiang Waterfront Wetland. Plant Ecol 212:1339–1347
Brzezinska MS, Lalke-Porczyk E, Kalwasin A (2012) Extracellular enzyme activity in a willow sewage treatment system. Curr Microbiol 65:776–783
Chen Y (2015) Experimental study on the optimal operating conditions of vertical subsurface flow constructed wetland technology in cold regions of Northwest China. Dissertation, Lanzhou Jiaotong University
Vymazal J, Kröpfelová L (2015) Multistage hybrid constructed wetland for enhanced removal of nitrogen. Ecol Eng 84:202–208
Rodrigues NF, Kästle J, Coutinho TJ, Amorim AT, Campos GB, Santos VM, Marques LM, Timenetsky J, Farias ST (2015) Qualitative analysis of the vaginal microbiota of healthy cattle and cattle with genital-tract disease. Genet Mol Res 14(2):6518–6528
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267
Broszat M, Nacke H, Blasi R, Siebe C, Huebner J, Daniel R, Grohmann E (2014) Wastewater irrigation increases abundance of potentially harmful Gammaproteo bacteria in soils from Mezquital Valley, Mexico. Appl Environ Microbiol 80(17):5282–5291
Liu CL, Zuo WY, Zhao ZY, Qiu LH (2012) Bacterial diversity of different successional stage forest soils in Dinghushan. Acta Microbiol Sin 52(12):1489–1496
Yang J, Zhou GY, Tian YY, Liu QL, Liu CF, Yang Q, Zhou JC (2015) Differential analysis of soil bacteria diversity in different mixed forests of Dalbergia odorifera. Acta Ecol Sin 35(24):8117–8127
Du YX, Xie BM, Cai HS, Tang L, Guo CH (2016) Structural and functional diversity of rhizosphere microbial community of nine plant species in the Daqing Saline-alkali soil region. Acta Ecol Sin 36(3):740–747
Liu FP (2015) Archaea community and function of the nitrogen cycle in surface sediment from Poyang Lake, China, Dissertation, University of Nanchang
Teira E, Martinez-Garcia S, Lonborg C, Alvarez-Salgado XA (2011) Betaproteobacteria growth and nitrification rates during long-term natural dissolved organic matter decomposition experiments. Aquat Microb Ecol 63(1):19–27
Du T (2016) Change of Microbial Communities and Algal Facies in Large Water Pond for Sea Cucumber Apostichopus Japonicus Farming at All Seasons and Probiotics Screening. Dissertation, Shanghai Ocean University
Kielak AM, Barreto CC, Kowalchuk GA, Veen JAV, Kuramae EE (2016) The ecology of acidobacteria: moving beyond genes and genomes. Front Microbiol 7(16):744
Lei X, Li B, Li X, Wang L, Zhu J (2015) Rhizosphere microbial communities of three plants in vertical-flow constructed wetland. Chin J Ecol 34(5):1373–1381
Ducey TF, Vanotti MB, Shriner AD, Szogi A, Ellison A (2010) Characterization of a microbial community capable of nitrification at cold temperature. Bioresource Technol 101(2):491–500
Zhang HX (2017) Effects of water conditions on soil microbial diversity in coastal reed wetland. Ocean Sci 41(5):144–152
Wang H (2017) Study on the Nitrogen Removal Effects and Mechanism by Using Elodea Nuttallii-immobilized Nitrogen Cycling Bacteria Assemblage Technology in Qinshui River,Gonghu Bay. Dissertation. Nangjing University
Zhang L, Lin XJ, Zhang Z, Chen GH, Jiang F (2017) Elemental sulfur as an electron acceptor for organic matter removal in a new high-rate anaerobic biological wastewater treatment process. Chem Eng J 331:442–453
Carroza C, Hurtado C, Gutierrez F (2012) Nitrogenated compounds’ biofiltration under alternative bacterium fixation substrates. Lat Am J Aquat Res 40(SpecIssue):772–785
Vanotti MB, Szogi AA, Ducey TF (2013) High performance nitrifying sludge for high ammonium concentration and low temperature wastewater treatment. US Patent 8445253
Acknowledgements
This study is supported by the National Natural Science Foundation of China [31370448], Natural Science Foundation of Jiangsu Province of China [BK20150496], Major Projects of Natural Science Research in Colleges and Universities, Jiangsu Province [15KJA2100001], and the Priority Academic Program Development of Jiangsu Higher Education Institutions [PAPD].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fu, W., Wang, Y., Wei, W. et al. Species Diversity and Functional Prediction of Soil Bacterial Communities in Constructed Wetlands with Different Plant Conditions. Curr Microbiol 76, 338–345 (2019). https://doi.org/10.1007/s00284-019-01634-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-019-01634-7