
Abstract
In this study, we classified the five strains (ChDC F128T, ChDC F145, ChDC F174, ChDC F206, and ChDC F300) as a novel species of genus Fusobacterium by DNA–DNA hybridization and multi-locus phylogenetic analysis (MLPA), based on a single sequence (24,715 bp) of 22 concatenated housekeeping genes, with morphological and chemotaxonomic characteristics. DNA–DNA hybridization data showed that the values of genomic relatedness between ChDC F128T and each of the other novel strains were ranged from 79.0 to 82.6 %, while those of genomic relatedness between ChDC F128T and type strain of each of subspecies of F. nucleatum or Fusobacterium periodonticum were ranged from 40.9 to 54.4 %. MLPA revealed that the 5 strains were clustered as one group and clearly discriminated with F. nucleatum and F. periodonticum with 100 % bootstrap value. The DNA G+C content of the five novel strains were ranged from 26.9 to 27.0 mol%. The cellular fatty acid analysis of clinical isolates and type strains revealed C14:0, C16:0, and cis-9 C16:1 as the major fatty acids. The cell wall peptidoglycan of the 5 strains was comprised of meso-lanthionine. These results show that the 5 strains are novel species and belong to the genus Fusobacterium. Strain ChDC F128T (=KCOM 1249T = KCTC 5108T = JCM 30218T) is suggested to be the type strain of a novel species of genus Fusobacterium, for which the name Fusobacterium hwasookii sp. nov. is proposed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Fusobacterium nucleatum is a gram-negative, anaerobic, fusiform-shaped bacterium and is normally isolated from the oral cavity in human [13]. F. nucleatum may play an important role in dental plaque formation, acting as a bridge of early colonizers such as gram-positive, facultative anaerobes, and late colonizers such as gram-negative, strict anaerobes [1, 2]. F. nucleatum was classified into 5 subspecies (nucleatum, polymorphum, vincentii, animalis, and fusiforme) based on the polyacrylamide gel electrophoretic pattern of the whole-cell proteins and DNA homology [3] or electrophoretic patterns of glutamate dehydrogenase and 2-oxoglutarate reductase and DNA–DNA hybridization patterns [5, 6]. F. nucleatum subsp. fusiforme has been reclassified into the F. nucleatum subsp. vincentii based on phylogenetic analysis using a single sequence (24,715 bp) of concatenated 22 housekeeping genes of 8 F. nucleatum strains, including type strains of 5 F. nucleatum subspecies [12]. Recently, 5 strains (ChDC F128T, ChDC F145, ChDC F174, ChDC F206, and ChDC F300) of F. nucleatum were isolated from subgingival plaques of Korean patients and a novel F. nucleatum subspecies has been suggested, based on the comparison of nucleotide sequences of RNA polymerase beta subunit gene (rpoB) and zinc protease gene [10]. However, these results were not adequate to classify the 5 novel strains as a novel subspecies of F. nucleatum or a novel species of the genus Fusobacterium. Based on the phylogenetic, biochemical, and chemotaxonomic findings presented here, the 5 novel strains were found to represent a novel species of the genus Fusobacterium.
Materials and Methods
Five clinical strains, ChDC F128T (=KCOM 1249T = KCTC 5108T = JCM 30218T), ChDC F145 (=KCOM 1253 = KCTC 5166 = JCM 30219), ChDC F174 (=KCOM 1256 = KCTC 5111 = JCM 30220), ChDC F206 (=KCOM 1258 = KCTC 5112 = JCM 30221), and ChDC F300 (=KCOM 1268 = KCTC 5174 = JCM 30222) as well as comparative reference type strains, F. nucleatum subsp. polymorphum ATCC 10953T, F. nucleatum subsp. vincentii ATCC 49256T, Fusobacterium nucleatum subsp. nucleatum ATCC 25586T, F. nucleatum subsp. animalis ATCC 51191T, and F. periodonticum ATCC 33693T were obtained from Korean Collection for Oral Microbiology (KCOM, Korea) or American Type Culture Collection (ATCC, USA). All strains were cultured and maintained in brain heart infusion (BHI, Difco Laboratories, USA) broth or on BHI agar containing sheep blood and incubated at 37 °C in an anaerobic chamber (Model Bactron I, USA) with an atmosphere of 10 % H2, 10 % CO2, and 80 % N2.
Phylogenetic Analysis
Multi-locus phylogenetic analysis (MLPA), based on 22 housekeeping genes (concatenated 24,715 bp), was performed, and the nucleotide sequence of each gene was obtained from GenBank (Table 1). Multiple sequences were aligned using the CLUSTAL W algorithm and sequence similarities were calculated using the MegAlign program (DNAStar LasergeneTM 8.0, DNAStar Inc., USA). The evolutionary distance was calculated according to the Kimura 2-parameter model and phylogenetic trees were constructed by maximum-likelihood and neighbor-joining methods in MEGA 6.06 software [17]. The stability of phylogenetic trees was assessed by a bootstrap analysis of 1,000 replicates.
Genomic DNA Isolation
The bacterial cells were harvested and suspended in STES buffer [0.5 M NaCl, 0.01 M EDTA, and 0.2 M Tris–HCl [pH 8.0] without 1 % sodium dodecyl sulfate (SDS)]. Lysozyme (Sigma, USA) was added at a final concentration of 2 mg/ml and incubated at 55 °C for 1 h. Once the cells were lysed, SDS (final concentration, 1 %) and then proteinase K (Bioneer, Korea) (final concentration 100 μg/ml) were added, and the solution was incubated at 55 °C for 1 h. After preliminary extraction with phenol:chloroform:isoamyl alcohol (25:24:1), RNase was added to the extract at a final concentration of 50 μg/ml and incubated at 37 °C for 2 h. Genomic DNA was purified using phenol:chloroform:isoamyl alcohol (25:24:1). DNA was precipitated with 3 volumes of cold ethanol and centrifuged at 15,000×g for 15 min at 4 °C. The precipitated DNA was air-dried and dissolved in TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 7.4). DNA concentration was determined by the Epoch™ Microplate Spectrophotometer (BioTek Instruments Inc., USA) at wavelengths of 260 and 280 nm.
DNA–DNA Hybridization
DNA–DNA hybridization was performed as described by Ezaki et al. with modification [4]. Briefly, heat-denatured genomic DNA solution (1 μg of DNA per ml) in phosphate-buffered saline (PBS) containing 0.1 M MgCl2 (PBSM) was incubated at 37 °C for 2 h in a microplate. After each solution was discarded, the microplate was washed once with PBS and then dried at 45 °C overnight. Photo-biotinylated genomic DNA was prepared as follows: 7 µl photobiotin (Sigma) and 1 μg denatured genomic DNA were mixed in a 1.5-ml tube and then irradiated with UV (100× μJ/cm2) for 20 min. After irradiation, free photobiotin was removed by 1-butanol. For quantitative detection of biotinylated genomic DNA in the microplate, 200 μl of prehybridization solution (2× SSC, 0.3 M NaCl, 0.03 M sodium citrate, 5× Denhardt solution, and 50 % formamide) containing 100 μg denatured salmon sperm DNA (Sigma) was added to microplate wells that were previously coated with genomic DNA. The microplate was incubated at 37 °C for 1 h. The prehybridization solution was removed from microplate and replaced with hybridization solution (prehybridization solution with 5 % dextran sulfate) containing biotinylated genomic DNA. The microplate was covered with sealing tape and incubated at 37 °C overnight. After hybridization, the microplate was washed three times with 200 μl of 2× SSC. Then, 100 μl streptavidin-β-d-galactosidase solution (250 μg/ml in PBS containing 0.5 % bovine serum albumin and 0.1 % Triton X-100) was added to each well and then incubated at 37 °C for 30 min. After incubation, the microplate was washed three times with PBS containing 0.1 % Triton X-100, and 100 μl of 3 × 10−4 μM 4-methylumbelliferyl-β-d-galactopyranoside in PBS containing 1 mM MgCl2 was added. The microplate was incubated at 37 °C for several periods of time. The fluorescence intensity was measured with a Fluoroskan Ascent Microplate Fluorometer (Thermo Scientific, USA) at an excitation wavelength of 360 nm and an emission wavelength of 450 nm.
Determination of DNA G+C Content
The G+C content of the genomic DNA was determined by the modified method described by Tamaoka and Komagata [16]. Briefly, genomic DNA was hydrolyzed into nucleosides using nuclease P1 and alkaline phosphatase. The resultant nucleosides were analyzed by high-performance liquid chromatography (HPLC; Shimadzu, Japan) equipped with a reversed-phase column (YMC pack ODS-A, 150 × 4.6 mm, Japan). Elution of the nucleosides was carried out with 0.55 M NH4H2PO4/acetonitrile (20:1, v/v), at a flow rate of 1 ml/min at 30 °C. Each nucleotide was detected by UV absorbance at 270 nm. The relative G+C contents were calculated by using DNA isolated from Escherichia coli KCTC 2441T.
Characterization of Morphology and Phenotype
Cell shape and size of the bacterial strains were determined by scanning electron microscopy. The cultured cells were fixed in 2.5 % glutaraldehyde (Sigma) in 0.1 M phosphate buffer (pH 7.4) for 1 h at 4 °C. After pre-fixation, the specimens were washed in the same buffer solution and then post-fixed in 1.5 % osmium tetroxide solution in 0.1 M phosphate buffer solution for 3 h at room temperature. Specimens were dehydrated in increasing concentrations of ethanol, and freeze-dried cells were mounted directly on stubs by using double-sided adhesive tape and coated with platinum by using a sputter coater. The cells were observed under a scanning electron microscope (S-4800, Hitachi, Japan) at 5 kV.
API 20A and API ZYM test strips (bioMerieux, France) were used to analyze the biochemical and physiological traits, enzyme activities, and sugar fermentation patterns of the strains, as per the manufacturer’s instructions. All tests were performed using fresh cultures of the strains under the same experimental conditions.
Analysis of Fatty Acids, Peptidoglycan, and Polar Lipids
Cellular fatty acids were saponified, methylated, and extracted using the MIDI/Hewlett Packard Microbial Identification System (MIDI Microbial ID, USA), according to the manufacturer’s instructions. The fatty acids were analyzed using gas chromatography (model 6890N and Auto-sampler 7683; Agilent) and identified using the Microbial Identification Sherlock software package (classical method, MIDI Sherlock system 4.1, TSBA library version 4.0, USA). The results were validated by NMR spectra, recorded on a UNITY Inova 400 (Varian Inc., USA) with methanol-d4. For peptidoglycan analysis, 5 mg of freeze-dried cells were hydrolyzed with 1 ml of 6 N HCl in a screw-capped tube at 100 °C for 18 h. The hydrolysate was filtered and concentrated by drying in a rotary evaporator at 65 °C. The residue was dissolved in distilled water. Each sample was analyzed by two-dimensional thin-layer chromatography on cellulose plates. Polar lipid analysis was carried out by the Identification Service of the DSMZ (Braunschweig, Germany).
Results and Discussion
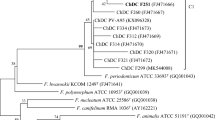
The phylogenetic tree constructed with concatenated 22 housekeeping genes showed that all the F. nucleatum strains were clearly separated into 5 distinct lineages with 100 % bootstrap value (Fig. 1 and supplementary Fig. S1). The group distance between 5 novel strains and F. nucleatum subsp. nucleatum, F. nucleatum subsp. polymorphum, F. nucleatum subsp. vincentii, F. nucleatum subsp. animalis, or F. periodonticum was 6.5, 4.8, 7.2, 8.0, and 10.2 %, respectively. Similarity of the concatenated 22 housekeeping genes between ChDC F128T and F. nucleatum subsp. nucleatum ATCC25586T, F. nucleatum subsp. polymorphum ATCC10953T, F. nucleatum subsp. vincentii ATCC51190T, F. nucleatum subsp. animalis ATCC51191T, or F. periodonticum ATCC33693T was 93.6, 94.5, 92.5, 91.8, and 90.6 %, respectively. In a previous study, the phylogenetic tree complied from the 16S rDNA sequences could not delineate the F. nucleatum subspecies from F. periodonticum, but the novel F. nucleatum subspecies strains (ChDC F128T, ChDC F145, ChDC F174, ChDC F206, and ChDC F300) were clustered as one group and were clearly separated from the other F. nucleatum subspecies and F. periodonticum [10]. The values of genomic relatedness between ChDC F128T and ChDC F206, ChDC F145, ChDC F174, or ChDC F300 were 82.9, 79.0, 82.6, and 86.1 %, respectively, which indicated that these 5 strains represented the same species. The values of genomic relatedness between ChDC F128T and F. nucleatum subsp. nucleatum ATCC25586T, F. nucleatum subsp. polymorphum ATCC10953T, F. nucleatum subsp. vincentii ATCC51190T, F. nucleatum subsp. animalis ATCC51191T, or F. periodonticum ATCC33693T were 45.6, 53.7, 54.4, 49.6, and 40.9 %, respectively. It was recommended that values of 70 % and DNA–DNA relatedness with ΔTm of 5 °C or less are reasonable borders for species circumscription [19]. The 5 clinical strains of F. nucleatum used in this study were suggested to be a novel subspecies of F. nucleatum by comparing nucleotide sequences of rpoB and zinc protease gene [10]. However, DNA–DNA hybridization values were lower than the suggested threshold for species delineation [19]. These results reveal that the 5 strains should be classified as a novel Fusobacterium species.

Maximum-likelihood phylogenetic tree based on a single sequence (24,715 bp) by concatenating 22 housekeeping genes. The stability of phylogenetic trees was assessed by a bootstrap analysis of 1,000 replicates by using MEGA version 6.06 [17]. Bar indicates 0.01 changes per nucleotide position
The DNA G+C content of strains ChDC F128T, ChDC F206, ChDC F145, ChDC F174, and ChDC F300 were 26.9, 27.0, 27.0, 26.9, and 27.0 mol%, respectively. These results are similar to the values reported previously for F. nucleatum subspecies [6, 8, 15].
The cell lengths of ChDC F128T, ChDC F145, ChDC F174, ChDC F206, and ChDC F300 were 28.9 (7.3–65.5) × 4.9 (4.5–5.0), 10.3 (5.5–40.7) × 4.4 (4.3–4.6), 65.7 (11.2–165.5) × 3.7 (3.4–4.4), 12.7 (5.2–50.7) × 4.6 (4.3–5.0), and 14.9 (4.9–35.8) × 4.6 (4.3–5.0) μm, respectively (Supplementary Fig. S2).
The cellular fatty acid analysis of the clinical isolates and type strains revealed C14:0, C16:0, and cis-9 C16:1 as being the major fatty acids. Among the analyzed strains, the major fatty acids of clinical isolates were similar to those of F. nucleatum subsp. nucleatum ATCC25586T, F. nucleatum subsp. polymorphum ATCC10953T, and F. periodonticum ATCC33693T, whereas cis-9 C16:1 was found to be relatively low in F. nucleatum subsp. vincentii ATCC49256T and F. nucleatum subsp. animalis ATCC51191T. The major fatty acids detected in the strains, C14:0, C16:0, and cis-9 C16:1, were consistent with those found in F. nucleatum [7, 11, 18]. A comparison between the major fatty acids of the clinical isolates and those of the 5 Fusobacterium type strains is shown in Table 2.
In the API ZYM panel, tests for naphthol-AS-BI-phosphohydrolase were positive in only strain ChDC F206 and weakly positive in the other strains (ChDC F128T, ChDC F145, ChDC F174, and ChDC F300), while most reactions were negative. API 20A test for indole production was positive, while the remaining 18 tests, including the test for catalase, were negative in the 5 strains (Table 3). The biochemical test results for the 5 strains were almost the same as those of F. nucleatum and F. periodonticum, except for the results of the test for naphthol-AS-BI-phosphohydrolase.
Lanthionine has been reported as an essential component of F. nucleatum peptidoglycan [9]. Lanthionine has also been found to exist in the cell wall peptidoglycan of clinical isolates, F. nucleatum subspecies, and F. periodonticum ATCC33693T (Supplementary Fig. S3). The data indicate that the clinical isolates belong to the genus Fusobacterium.
Polar lipids consisted of phosphatidylethanolamine, diphosphatidylglycerol, phosphatidylglycerol, 3 unknown phospholipids, and an unknown aminolipid, with one unknown aminophospholipid in ChDC F128T. The major polar lipids of F. nucleatum using fast-atom-bombardment mass spectrometry (FAB-MS) were phosphatidylethanolamine and phosphatidylglycerol [14]. However, the major polar lipid in ChDC F128T was diphosphatidylglycerol (Supplementary Fig. S4).
On the basis of the molecular, chemical, and phenotypic evidence presented in this study, we propose that the strains ChDC F128T, ChDC F145, ChDC F174, ChDC F206, and ChDC F300 should be assigned to a novel species of Fusobacterium, which we propose should be named Fusobacterium hwasookii sp. nov.
Description of Fusobacterium hwasookii sp. nov.
Fusobacterium hwasookii [hwa.so.ok’i.i. N.L. gen. masc. n. hwasookii named after Hwa-sook Kim, a Korean microbiologist who isolated these strains and has contributed to the description of this species]
Fusobacterium hwasookii is a gram-negative, anaerobic, and fusiform-shaped bacterium with variable size. The cell length of ChDC F128T was 28.9 (7.3–65.5) × 4.9 (4.5–5.0). The API ZYM system showed negative reaction for alkaline phosphatase, esterase (C4), esterase lipase (C8), lipase (C14), leucine arylamidase, cystine arylamidase, trypsin, α-chymotrypsin, acid phosphatase, α-galactosidase, β-galactosidase, β-glucuronidase, β-glucosidase, α-lucosidase, N-acetyl-β-glucosaminidase, α-mannosidase, and α-fucosidase, whereas naphthol-AS-BI-phosphohydrolase was weakly positive for strain ChDC F128T. In the API 20A system, strain ChDC F128T showed indole production but were negative for acid production from d-glucose, d-mannitol, d-lactose, d-saccharose, d-maltose, salicin, d-xylose, l-arabinose, glycerol, d-cellobiose, d-mannose, d-melezitose, d-raffinose, d-sorbitol, d-rhamnose, and d-trehalose. For strain ChDC F128T, esculin was not hydrolyzed, gelatin was not digested, catalase reaction was negative, and the cellular fatty acids were mainly composed of C14:0, C16:0, and cis-9 C16:1. The G+C content of ChDC F128T was 26.9 mol%. DNA–DNA relatedness between ChDC F128T and ChDC F145, ChDC F174, ChDC F206, or ChDC F300 was approximately 79.0, 82.6, 82.9, and 86.1 %, respectively. DNA–DNA relatedness between ChDC F128T and F. nucleatum subsp. nucleatum ATCC25586T, F. nucleatum subsp. polymorphum ATCC10953T, F. nucleatum subsp. vincentii ATCC51190T, F. nucleatum subsp. animalis ATCC51191T, or F. periodonticum ATCC33693T was 45.6, 53.7, 54.4, 49.6, and 40.9 %, respectively. The cell-wall diamino acid of strain ChDC F128T was found to be meso-lanthionine. The major polar lipids of ChDC F128T were phosphatidylethanolamine, diphosphatidylglycerol, and phosphatidylglycerol.
The cell lengths of ChDC F145, ChDC F174, ChDC F206, and ChDC F300 were 10.3 (5.5–40.7) × 4.4 (4.3–4.6), 65.7 (11.2–165.5) × 3.7 (3.4–4.4), 12.7 (5.2–50.7) × 4.6 (4.3–5.0), and 14.9 (4.9–35.8) × 4.6 (4.3–5.0) μm, respectively. In API ZYM system, naphthol-AS-BI-phosphohydrolase was positive for only strain ChDC F206 and weakly positive for ChDC F145, ChDC F174, and ChDC F300 strains. The G+C content of ChDC F145, ChDC F174, ChDC F206, and ChDC F300 ranged between 26.9 and 27.0 mol%.
These results show that the 5 strains are novel species that belong to genus Fusobacterium. The type strain, ChDC F128T (=KCOM 1249T = KCTC 5108T = JCM 30218T), was isolated from subgingival plaques of Korean patient.
References
Albandar JM, Brown LJ, Löe H (1997) Putative periodontal pathogens in subgingival plaque of young adults with and without early-onset periodontitis. J Periodontol 68:973–981
Bolstad AI, Jensen HB, Bakken V (1996) Taxonomy, biology, and periodontal aspects of Fusobacterium nucleatum. Clin Microbiol Rev 9:55–71
Dzink JL, Sheenan MT, Socransky SS (1990) Proposal of three subspecies of Fusobacterium nucleatum Knorr 1922: Fusobacterium nucleatum subsp. nucleatum subsp. nov., comb. nov.; Fusobacterium nucleatum subsp. polymorphum subsp. nov., nom. rev., comb. nov.; and Fusobacterium nucleatum subsp. vincentii subsp. nov., nom. rev., comb. nov. Int J Syst Bacteriol 40:74–78
Ezaki T, Hashimoto Y, Yabuuchi E (1989) Fluorometric deoxyribonucleic acid-deoxyribonucleic acid hybridization in microdilution wells as an alternative to membrane filter hybridization in which radioisotopes are used to determine genetic relatedness among bacterial strains. Int J Syst Bacteriol 39:224–229. doi:10.1099/00207713-39-3-224
Gharbia SE, Shah HN (1990) Heterogeneity within Fusobacterium nucleatum, proposal of four subspecies. Lett Appl Microbiol 10:105–108. doi:10.1111/j.1472-765X.1990.tb00276.x
Gharbia SE, Shah HN (1992) Fusobacterium nucleatum subsp. fusiforme subsp. nov. and Fusobacterium nucleatum subsp. animalis subsp. nov. as additional subspecies within Fusobacterium nucleatum. Int J Syst Bacteriol 42:296–298. doi:10.1099/00207713-42-2-296
Jantzen E, Hofstad T (1981) Fatty acids of Fusobacterium species: taxonomic implications. J Gen Microbiol 123:163–171
Karpathy SE, Qin X, Gioia J, Jiang H, Liu Y, Petrosino JF, Yerrapragada S, Fox GE, Haake SK, Weinstock GM, Highlander SK (2007) Genome sequence of Fusobacterium nucleatum subspecies polymorphum: a genetically tractable fusobacterium. PLoS One 2:e659
Kato K, Umemoto T, Sagawa H, Kotani S (1979) Lanthionine as an essential constituent of cell wall peptidoglycan of Fusobacterium nucleatum. Curr Microbiol 3:147–151
Kim HS, Lee DS, Chang YH, Kim MJ, Koh S, Kim J, Seong JH, Song SK, Shin HS, Son JB, Jung MY, Park SN, Yoo SY, Cho KW, Kim DK, Moon S, Kim D, Choi Y, Kim BO, Jang HS, Kim CS, Kim C, Choe SJ, Kook JK (2010) Application of rpoB and zinc protease gene for use in molecular discrimination of Fusobacterium nucleatum subspecies. J Clin Microbiol 48:545–553. doi:10.1128/JCM.01631-09
Könönen E, Kanervo A, Salminen K, Jousimies-Somer H (1999) Beta-lactamase production and antimicrobial susceptibility of oral heterogeneous Fusobacterium nucleatum populations in young children. Antimicrob Agents Chemother 43:1270–1273
Kook JK, Park SN, Lim YK, Choi MH, Cho E, Kong SW, Shin Y, Paek J, Chang YH (2013) Fusobacterium nucleatum subsp. fusiforme Gharbia and Shah 1992 is a later synonym of Fusobacterium nucleatum subsp. vincentii Dzink et al. 1990. Curr Microbiol 66:414–417. doi:10.1007/s00284-012-0289-y
Moore WEC (1987) Microbiology of periodontal disease. J Perio Res 22:335–341
Sadek F, Drucker DB, Boote V, Bennett KW, Eley A (1998) Phospholipids of Fusobacterium spp. J Appl Microbiol 85:302–308
Slots J, Potts TV, Mashimo PA (1983) Fusobacterium periodonticum, a new species from the human oral cavity. J Dent Res 62:960–963
Tamaoka J, Komagata K (1984) Determination of DNA base composition by reversed-phase high-performance liquid chromatography. FEMS Microbiol Lett 25:125–128. doi:10.1111/j.1574-6968.1984.tb01388.x
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi:10.1093/molbev/mst197
Tunér K, Baron EJ, Summanen P, Finegold SM (1992) Cellular fatty acids in Fusobacterium species as a tool for identification. J Clin Microbiol 30:3225–3229
Wayne LG, Brenner DJ, Colwell RR, Grimont PAD, Kandler O, Krichevsky MI, Moore LH, Moore WEC, Murray RGE, Stackebrandt E, Starr MP, Truper HG (1987) Report of the Ad Hoc Committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol 37:463–464. doi:10.1099/00207713-37-4-463
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and future Planning (NRF-2012R1A1A2003324) and in part by a Grant TGM0721311 funded by the Ministry of Trade, Industry and Energy.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Eugene Cho and Soon-Nang Park contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
284_2014_692_MOESM1_ESM.pptx
Supplementary Fig. S1. Neighbor-joining phylogenetic trees based on a single sequence (24,715 bp) by concatenating 22 housekeeping genes. The stability of phylogenetic trees was assessed by a bootstrap analysis of 1,000 replicates by using MEGA version 6.06 [17]. Bar indicates 0.01 changes per nucleotide position
284_2014_692_MOESM2_ESM.pptx
Supplementary Fig. S2. Scanning electron micrograph of strains (a) ChDC F128T, (b) ChDC F145, (c) ChDC F174, (d) ChDC F206, and (e) ChDC F300. The scale bars represent 30 μm (a), 10 μm (b), 100 μm (c), 20 μm (d), and 20 μm (e)
284_2014_692_MOESM3_ESM.pptx
Supplementary Fig. S3. Thin-layer chromatography of acid hydrolysates of peptidoglycan. Strains: 1, strain ChDC F128T; 2, F. nucleatum subsp. nucleatum ATCC25586T; 3, F. nucleatum subsp. polymorphum ATCC10953T; 4, F. nucleatum subsp. vincentii ATCC49256T; 5, F. nucleatum subsp. animalis ATCC51191T; 6, strain ChDC F128T; 7, F. periodonticum ATCC33693T. Arrow indicates meso-lanthionine (Lan)
284_2014_692_MOESM4_ESM.pptx
Supplementary Fig. S4. Polar lipid profile of ChDC F128T by two-dimensional thin-layer chromatography. Abbreviations: PE, Phosphatidylethanolamine; DPG, Diphosphatidylglycerol; PG, Phosphatidylglycerol; PL, unknown phospholipid; AL, unknown aminolipid; PN, unknown aminophospholipid; L, unknown polar lipid
Rights and permissions
About this article

Cite this article
Cho, E., Park, SN., Lim, Y.K. et al. Fusobacterium hwasookii sp. nov., Isolated from a Human Periodontitis Lesion. Curr Microbiol 70, 169–175 (2015). https://doi.org/10.1007/s00284-014-0692-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-014-0692-7