
Abstract
Three novel isolates (A-354T, A-328, and A-384) were retrieved from apparently healthy scleractinian Madracis decactis in the remote St Peter & St Paul Archipelago, Mid-Atlantic Ridge, Brazil. The novel isolates formed a distinct lineage based on the phylogenetic reconstruction using the 16S rRNA and pyrH gene sequences. They fell into the Mediterranei clade and their closest phylogenetic neighbour was V. mediterranei species, sharing upto 98.1 % 16S rRNA gene sequence similarity. Genomic analysis including in silico DDH, MLSA, AAI and genomic signature distinguished A-354T from V. mediterranei LMG 19703 (=AK1) with values of 33.3, 94.2, 92 %, and 11.3, respectively. Phenotypically, the novel isolates can be differentiated from V. mediterranei based on the four following features. They do not grow at 8 % NaCl; use d-gluconic acid but not l-galactonic acid lactone as carbon source; and do not have the fatty acid C18:0. Differentiation from both the other Mediterranei clade species (V. maritimus and V. variabilis) is supported by fifteen features. The novel species show lysine decarboxylase and tryptophan deaminase, but not gelatinase and arginine dihydrolase activity; produce acetoin; use α-d-lactose, N-acetyl-d-galactosamine, myo-Inositol, d-gluconic acid, and β-hydroxy-d,l-butyric acid; and present the fatty acids C14:0 iso, C15:0 anteiso, C16:0 iso, C17:0 anteiso, and C17:1x8c . Whole-cell protein profiles, based on MALDI-TOF, showed that the isolates are not clonal and also distinguished them from the closes phylogenetic neighbors. The name Vibrio madracius sp. nov. is proposed to encompass these novel isolates. The G+C content of the type strain A-354T (=LMG 28124T=CBAS 482T) is 44.5 mol%.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Madracis decactis is a zooxanthellate scleractinian coral, affiliated to a genus widely distributed along the Brazilian and Caribbean coasts (from Parcel de Manuel Luiz Reefs, in the north, to the coast of Santa Catarina, in the south), in all Brazilian oceanic Islands (Fernando de Noronha, Roca Atol, St Peter & St Paul Archipelago (SPSPA) and Trindade/Martim Vaz), and West Africa [5, 15, 16, 18]. M. decactis accounts for relevant reef structures in the southeastern Brazilian coast (São Paulo) and these populations were seriously affected by a bleaching event during the 1993/4 summer [13]. The study of the culturable heterotrophic microbiota in the remote SPSPA analyzed 403 isolates from healthy and bleached M. decactis and the corallivore polychaete Hermodice carunculata (fireworm). Three Vibrio isolates (A-354T, A-328, and A-384) retrieved from healthy M. decactis could not be precisely assigned to any known Vibrio species [14]. The novel isolates shared at maximum 98.1 % 16S rRNA gene sequence similarity toward the species V. mediterranei. The independent position of the novel isolates was confirmed by pyrH gene based on phylogenetic reconstruction.
The aim of the present study was to perform a detailed taxonomic characterization of the three novel isolates originated from healthy M. decactis in SPSPA (Online Resource 2), based on the polyphasic and genomic approaches described previously [26, 28]. A-354T was isolated using thiosulfate-citrate-bile salt-sucrose (TCBS) medium, whereas A-328 and A-384 using Marine Agar (DIFCO), the three strains at ambient temperature (~27 °C), after 48-h incubation. Gene sequences of 16S rRNA and uridylate kinase (pyrH), were obtained as described previously [14]. Raw sequence data were assembled using ChromasPro V. 1.7.1 (Technelysium Pty. Ltd, Australia) to generate consensus sequences. The sequences were blasted against the GenBank world database to retain closest matches to include in the phylogenetic analysis. Pairwise similarities of these sequences with those of the closest phylogenetic neighbors were calculated using Jalview V.2 [30]. Pairwise and multiple alignments were performed using ClustalW [12]. Similarity matrices and phylogenetic analysis were performed using MEGA 5 (Molecular Evolutionary Genetics Analysis) [25]. Trees were drawn using the neighbour-joining statistical method [22]. The robustness of each topology was checked by 1,000 bootstrap replications [7]. Overall mean distances indicated no correction (uniform) for rates among sites. Gaps missing data treatment was complete deletion. The evolutionary distances were computed using Kimura 2-parameter, (Fig. 1); Jukes–Cantor (Fig. 2), and the p-distance (Online Resource 4) methods. All substitutions (transitions and transversions) were included.

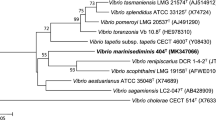
Neighbour-joining phylogenetic tree showing the phylogenetic position of V. madracius sp. nov. based on 16S rRNA gene sequences. The optimal tree with the sum of branch length = 0.22677584 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown next to the branches. The evolutionary distances were computed using the Kimura 2-parameter method and are in the units of the number of base substitutions per site. The analysis involved 22 nucleotide sequences. All ambiguous positions were removed for each sequence pair. There were a total of 1,386 positions in the final dataset. Photobacterium was used as outgroup. Bar, 1.0 % estimated sequence divergence

Neighbour-joining phylogenetic tree showing the phylogenetic position of V. madracius sp. nov. based on concatenated 16S rRNA, pyrH, rpoA, recA, mreB, and gyrB gene sequences. There were a total of 4,082 positions in the final dataset. The optimal tree with the sum of branch length = 0.58396460 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown next to the branches. The evolutionary distances were computed using the Jukes–Cantor method and are in the units of the number of base substitutions per site. The analysis involved 13 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + Noncoding. All ambiguous positions were removed for each sequence pair. Escherichia coli was used as outgroup. Bar, 2 % estimated sequence diverge
The whole genome of A-354T was sequenced by means of Ion Torrent technology as described by Quail et al. [21] with minor modifications, as follows. Library preparation was carried out using the Ion Plus Fragment Library Kit, with 1 μg DNA (in Low TE, 50 µL). DNA was fragmented using the BioRuptor® Sonication System as described in the Ion Plus Fragment Library Kit protocol. End repair, adapter ligation, nick repair, and amplification (8 cycles) were also performed as described in the Ion Plus Fragment Library protocol. 300- and 400-bp fragments were selected through agarose gel (2 % m/v) electrophoresis. Quality and concentration of the libraries were determined using Agilent 2100 Bioanalyzer with the associated High Sensitivity DNA kit (Agilent Technologies), as well as with Ion library Quantization kit using TaqMan® in a 7500 Real-Time PCR System (Applied Biosystems®). The amount of library required for template preparation was calculated using the Template Dilution Factor calculation described in the protocol. Emulsion PCR and enrichment steps were carried out in the Ion OneTouch™ 200 Template Kit v2. Ion Sphere Particle quality assessment was carried out as outlined in this protocol. Sequencing was undertaken using 314 chips without barcoding. The Ion PGM™ 200 Sequencing Kit was used for sequencing reactions following the recommended protocol and Torrent Suite 1.5 was used for analyses. The reads were assembled using De novo Assembly in the Ion Torrent Platform based on MIRA software. DNA G+C content was determined through Rapid Annotations Using Subsystems Technology (The RAST server version 4.0) [3].
MLSA was performed with partial gene sequences of the 16S rRNA, pyrH, recombination repair protein (recA), RNA polymerase alpha subunit gene (rpoA), actin-like cytoskeleton protein (mreB), and DNA gyrase subunit B (gyrB) (4,082 nts) as described previously [23]. recA (JN039146) and rpoA (JN039148) gene sequences described for LMG 19703 were employed as templates for ortholog search against A-354T genome using BLAST algorithm in RAST environment. The template for mreB search was retrieved from AK1 (=LMG 19703) genome (gi|149189046:34693-35736). Strain A-203 was included in pyrH- and MLSA-based phylogenies, as a representative of all AK1-related strains isolated from the fireworms in SPSPA [14]. Strains and accession numbers of the sequences employed for concatenated tree building are listed in Online Resource 3. Pairwise similarities, alignment, and phylogenetic reconstruction were performed as above mentioned. AAI was calculated as described previously [10]. The genomic signature was determined by the dinucleotide relative abundance value for each genome [8, 9]. The genome distance was calculated using GGDC [1, 2].
The gene sequence data obtained in this study are available through the open access website TAXVIBRIO (http://www.taxvibrio.lncc.br/). The GenBank accession numbers for the 16S rRNA, pyrH gene sequences for V. madracius strains, and for A-354T genome, are listed in Online Resource 1.
Phenotypic characterization was performed using commercially available miniaturized kits (API 20E and API ZYM; BioMerieux) as described previously [11, 29] and by BIOLOG GEN III metabolic fingerprinting, following the manufacturer’s instructions. These tests included determination of temperature, salinity growth ranges, several biochemical responses and 71 carbon source utilization assays. Unless indicated otherwise, isolates were grown onto MA for 24 h at 30 °C. The optimal growth temperature was determined using MA at pH 7.5, and the optimal salinity was determined in peptone water (1.5 % peptone, 30 °C, pH 7.5). Growth under anaerobic conditions was determined after incubation in an anaerobic atmosphere (Microanaerobac, PROBAC, Brasil) on MA at 30 °C. Fatty acid methyl ester analyses (FAME) were performed using the Sherlock Microbial Identification System according to the standard protocol. To this end, isolates were harvested from MA after 24 h of incubation at 30 °C. Protein analysis by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS was performed as described previously [31]. Briefly, isolates were subcultured twice on MA for 24 h at 30 °C. MALDI-TOF MS was conducted using a 4800 Plus MALDI-TOF/TOF Analyzer (Ab Sciex NV) in linear mode and the 4000 Series Explorer Software v3.5.3 (Applied Biosystems). Mass spectra were analyzed using BioNumerics v5.1 (Applied Maths) and clustered with unweighted pair group method with arithmetic averages (UPGMA) using Pearson’s correlation coefficient. [24]. Strains of the closest related neighbors were included for comparison.
The 16S rRNA gene sequence analysis revealed that the three novel isolates (A-354T, A-328, A-384) formed a tight monophyletic branch affiliated to the genus Vibrio (Fig. 1). The three novel isolates shared more than 99 % 16S rRNA gene sequence similarity. The species V. mediterranei (LMG 19710) was the closest, sharing 98.1 % similarity with the novel isolates. However, the pyrH gene sequence similarity between the novel isolates and LMG 19710 was at maximum 89.5 % (Online Resource 4). The MLSA including 16S rRNA, pyrH, recA, rpoA, mreB, and gyrB gene sequences confirmed that A-354T is closely related to V. mediterranei, but can be clearly distinguished from the closest neighbors, (Fig. 2) at lower similarity levels than the cut-offs determined to define a species of the family Vibrionaceae [27, 28]. Trees based on partial sequences of the housekeeping gene pyrH (531 bp) and on the six genes MLSA (4,082 bp) confirmed the phylogenetic position in the genus Vibrio, in a separate branch, clearly indicating that they belong to a new Vibrio species (Fig. 2 and Online Resource 4). In addition, the genomic taxonomy analysis distinguished A-354T from LMG 19710 (=AK1) with values of 33.3, 94.2, 92 %, and 11.3 for in silico DDH, MLSA, AAI and genomic signature, respectively, clearly supporting the proposal of a new species.
Four phenotypic features distinguish the novel isolates from V. mediterranei species. V. madracius sp. nov. can grow at 8 % NaCl; uses d-gluconic acid, but not l-galactonic acid lactone as carbon source; and does not present the fatty acid C18:0 (Online Resource 5). Differentiation from both the other Mediterranei clade species (V. maritimus and V. variabilis) is supported by the following 15 traits: lysine decarboxylase and tryptophan deaminase, but not gelatinase and arginine dihydrolase activity; acetoin production; use of α-d-lactose, N-acetyl-d-galactosamine, myo-inositol, d-gluconic acid, and β-hydroxy-d,l-butyric acid; and presence of the fatty acids C14:0 iso, C15:0 anteiso, C16:0 iso, C17:0 anteiso and C17:1x8c . MALDI-TOF MS protein profiles distinguished the novel isolates among each other, suggesting that they represent different populations in nature. The protein profiles also allowed their discrimination from the closest related species of the Mediterranei clade (Online Resource 6). MLSA was more discriminative than MALDI-TOF, and FAME for strains differentiation. Based on the phenotypic, genomic, and phylogenetic evidence provided in the present study, we propose to classify the three isolates as a new species, V. madracius sp. nov. (Table 1).
Description of Vibrio madracius sp. nov
Vibrio madracius (ma.dra’ci.us. L. masc. adj. madracius, after Madracis, the coral host genus)
Colonies are pale beige, regular shaped, with smooth and translucent edges, 1 mm in diameter after 24 h at 30 °C on MA under aerobic conditions. On TCBS colonies are yellow, round, with a smooth border and 2 mm in diameter. Cells are Gram-negative, non-spore-forming rods (0.9 × 2.3 µm), motile, facultative anaerobic, oxidase, and catalase-positive. Grows well between 16 and 40 °C, shows poor growth at 15 °C, and does not grow at 4 and 42 °C. No growth occurs in the absence of NaCl, but grows well under NaCl concentrations of 1–8 % (w/v). Grows at pH 6–11. Positive for alkaline phosphatase, esterase (C4), esterase lipase (C8), lipase (C14), leucine arylamidase, naphthol-AS-BI-phosphohydrolase, β-galactosidase, α-glucosidase, N-acetyl-β-glucosaminidase, lysine decarboxylase, tryptophane deaminase, and indole production; but negative for cystine arylamidase, trypsin, α-chemotrypsin, β-glucosidase, β-glucuronidase, α-mannosidase, α-fucosidase, H2S production, arginine dihydrolase, ornithine decarboxylase, urease, acetoin production (Voges–Proskauer), and gelatinase activity. Variable reactions were obtained for valine arylamidase (+), acid phosphatase (−), and α-galactosidase (−) (whenever variable within species, result for the type strain is in parentheses). Reduces nitrate to nitrite, but not nitrite to N2. Produces acid from glucose, sucrose, mannitol, sorbitol, melibiose, and amygdalin, but not from rhamnose and arabinose. Acid from inositol was variable (+). Dextrin, d-maltose, d-trehalose, d-cellobiose, gentiobiose, sucrose, d-turanose, stachyose, d-raffinose, α-d-lactose, d-melibiose, β-methyl-d-glucoside, N-acetyl-d-glucosamine, d-fructose, d-salicin, α-d-glucose, d-mannose, d-galactose, inosine, d-mannitol, d-glucose-6-PO4, d-fructose-6-PO4, l-alanine, l-arginine, l-aspartic acid, l-glutamic acid, l-histidine, l-serine, l-pyroglutamic acid, d-gluconic acid, d-glucuronic acid, methyl pyruvate, l-lactic acid, citric acid, bromo-succinic acid, tween-40, α-hydroxy-butyric, β-hydroxy-d,l-butyric and α-keto-butyric acids, N-acetyl-β-mannosamine, N-acetyl-d-galactosamine, N-acetyl neuraminic acid, 3-methyl glucose, l-fucose, d-sorbitol, d-arabitol, myo-inositol, glycerol, glycyl-l-proline, pectin, d-galacturonic acid, quinic acid, d-saccharic acid, d-lactic acid methyl ester, α-keto-glutaric acid, l-malic acid, acetoacetic acid, and acetic acid are used as sole energy sources. Does not utilize d-serine, gelatin, citrate, l-galactonic acid lactone, d-galacturonic acid, mucic acid, p-hydroxy-phenylacetic acid, d-malic acid and formic acid. The following reactions are variable within the species: d-fucose (−), l-rhamnose (w), d-aspartic acid (+), glucuronamide (−), γ-amino-butyric acid (+), and propionic acid (+). The most abundant cellular fatty acids are summed in feature 3 (27.84 %; comprising C16:1 ω7c and/or iso-C15 2-OH), C18:1ω7c (14.10 %), C16:0 (12.97 %), C12:0 (6.54 %), C16:0 iso (6.20 %), C14:0 (6.10 %), summed feature 2 (4.31 %; comprising C14:0 3-OH and/or iso-C16:1 I), C12:0 3-OH (2.98 %), Iso-C14:0 (2.38 %), iso-C15:0 (2.09 %), Anteiso-C15:0 (1.87 %), Iso-C13:0 (1.85 %), iso-C17:0 (1.60 %), Anteiso-C17:0 (1.32 %), C14:0 3-OH (1.08 %); and in minor amounts Iso-C18:0, iso-C15 3-OH, and C17:1 x8c (<1 %). The G+C content of the type strain (A-354T) is 44.5 mol%. The type strain is A-354T (=LMG 28124T = CBAS 482T). It was isolated from the tissues of healthy M. decactis (Scleractinia) in St Peter & St Paul Archipelago, Mid-Atlantic Ridge, Brazil.
Abbreviations
- SPSPA:
-
St Peter & St Paul Archipelago
- MLSA:
-
Multilocus sequence analysis
- ANI:
-
Average nucleotide sequence identity
- AAI:
-
Average amino acid identity
- DDH:
-
DNA–DNA hybridization
- GGDC:
-
Genome-to-genome distance calculator
- DSMZ:
-
German collection of microorganisms and cell cultures
- FAME:
-
Fatty acid methyl ester analyses
- MALDI-TOF:
-
Matrix-assisted laser desorption/ionization time-of-flight
References
Auch AF, von Jan M, Klenk H-P, Göker M (2010) Digital DNA–DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2(1):117
Auch AF, Klenk H-P, Göker M (2010) Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci 2(1):142–148
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O (2008) The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom 9(1):75
Balcazar JL, Pintado J, Planas M (2010) Vibrio hippocampi sp. nov., a new species isolated from wild seahorses (Hippocampus guttulatus). FEMS Microbiol Lett 307(1):30–34
Castro B, Pires D (2006) Reproductive biology of Madracis decactis (Lyman, 1859) (Cnidaria, Scleractinia) from southern Bahia reefs, Brazil. Arqs MN 64:19–27
Chimetto LA, Cleenwerck I, Moreira APB, Brocchi M, Willems A, De Vos P, Thompson FL (2011) Vibrio variabilis sp. nov. and Vibrio maritimus sp. nov., isolated from Palythoa caribaeorum. Int J Syst Evol Microbiol 61(12):3009–3015
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39(4):783–791
Karlin S, Mrázek J, Campbell AM (1998) Codon usages in different gene classes of the Escherichia coli genome. Mol Microbiol 29(6):1341–1355
Karlin S, Mrazek J, Campbell AM (1997) Compositional biases of bacterial genomes and evolutionary implications. J Bacteriol 179(12):3899–3913
Konstantinidis KT, Tiedje JM (2005) Towards a genome-based taxonomy for prokaryotes. J Bacteriol 187(18):6258–6264
Kushmaro A, Banin E, Loya Y, Stackebrandt E, Rosenberg E (2001) Vibrio shiloi sp. nov., the causative agent of bleaching of the coral Oculina patagonica. ISME J 51(4):1383–1388
Larkin M, Blackshields G, Brown N, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948
Migotto A. (1997) Anthozoan bleaching on the southeastern coast of Brazil in the summer of 1994. In: 6th International Conference of Coelenterate Biology, Leeuwenhorst, 1995 1997. Leeuwenhorst, pp 329–335
Moreira APB, Tonon LAC, Do Valle Cecilia PP, Alves N Jr, Amado-Filho GM, Francini-Filho RB, Paranhos R, Thompson FL (2014) Culturable heterotrophic bacteria associated with healthy and bleached scleractinian Madracis decactis and the fireworm Hermodice carunculata from the Remote St. Peter and St. Paul Archipelago, Brazil. Curr Microbiol 68(1):38–46
Neves E, Johnsson R (2009) Taxonomic revision of the southwestern Atlantic Madracis and the description of Madracis fragilis n. sp. (Scleractinia: Pocilloporidae), a new coral species from Brazil. Sci Mar 73(4):739–746
Nunes FLD, Norris RD, Knowlton N (2011) Long distance dispersal and connectivity in amphi-atlantic corals at regional and basin scales. PLoS One 6(7):e22298
Ortigosa M, Garay E, Pujalte Ma-Js (1994) Numerical taxonomy of Vibrionaceae isolated from oysters and seawater along an annual cycle. Syst Appl Microbiol 17(2):216–225
Pereira-Filho GH, Amado-Filho GM, de Moura RL, Bastos AC, Guimarães SM, Salgado LT, Francini-Filho RB, Bahia RG, Abrantes DP, Guth AZ (2011) Extensive Rhodolith beds cover the summits of southwestern atlantic ocean seamounts. J Coast Res 28(1):261–269
Pujalte M-J, Garay E (1986) Proposal of Vibrio mediterranei sp. nov.: a new marine member of the genus Vibrio. Int J Syst Evol Microbiol 36(2):278–281
Pujalte MJ, Ortiz-Conde BA, Steven SE, Esteve C, Garay E, Colwell RR (1992) Numerical taxonomy and nucleic acid studies of Vibrio mediterranei. Syst Appl Microbiol 15(1):82–91
Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y (2012) A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom 13(1):341
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Sawabe T, Ogura Y, Matsumura Y, Feng G, Amin AR, Mino S, Nakagawa S, Sawabe T, Kumar R, Fukui Y (2013) Updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front Microbiol 4:414
Strohalm M, Kavan D, Novák P, Volný M, Havlíček V (2010) mMass 3: a cross-platform software environment for precise analysis of mass spectrometric data. Anal Chem 82(11):4648–4651
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739
Thompson CC, Chimetto L, Edwards RA, Swings J, Stackebrandt E, Thompson FL (2013) Microbial genomic taxonomy. BMC Genom 14(1):913
Thompson CC, Vicente AC, Souza RC, Vasconcelos AT, Vesth T, Alves N, Ussery DW, Iida T, Thompson FL (2009) Genomic taxonomy of vibrios. BMC Evol Biol 9(1):258
Thompson F, Gevers D, Thompson C, Dawyndt P, Naser S, Hoste B, Munn C, Swings J (2005) Phylogeny and molecular identification of vibrios on the basis of multilocus sequence analysis. Appl Environ Microbiol 71(9):5107–5115
Thompson FL, Hoste B, Thompson CC, Huys G, Swings J (2001) The Coral Bleaching Vibrio shiloi Kushmaro et al. 2001 is a Later Synonym of Vibrio mediterranei Pujalte and Garay 1986. Syst Appl Microbiol 24(4):516–519
Waterhouse AM, Procter JB, Martin DM, Clamp Ml, Barton GJ (2009) Jalview Version 2, A multiple sequence alignment editor and analysis workbench. Bioinformatics 25(9):1189–1191
Wieme A, Cleenwerck I, Van Landschoot A, Vandamme P (2012) Pediococcus lolii DSM 19927T and JCM 15055T are strains of Pediococcus acidilactici. Int J Syst Evol Microbiol 62(Pt 12):3105–3108
Acknowledgments
This work was supported by CNPq, CAPES, and FAPERJ. The BCCM/LMG Bacteria Collection is supported by the Federal Public Service for Science Policy, Belgium. We are grateful to Leilei Li for her assistance with the MALDI-TOF MS data analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Nucleotide sequence data for Vibrio madracius sp. nov. are available in the DDBJ/EMBL/GenBank databases under the following accession number(s): KC751057, KC751062, KC751087 (16S rRNA); and KC751154, KC751309, KJ154030 (pyrH). ASHK00000000 is the accession number (Version ASHK00000000) for the genome of the type strain A-354T = LMG 28124T = CBAS 482 T (List in Online Resource 1). ASHJ00000000 is the accession number (Version ASHJ00000000) for the genome of V. mediterranei A-203.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Moreira, A.P.B., Duytschaever, G., Tonon, L.A.C. et al. Vibrio madracius sp. nov. Isolated from Madracis decactis (Scleractinia) in St Peter & St Paul Archipelago, Mid-Atlantic Ridge, Brazil. Curr Microbiol 69, 405–411 (2014). https://doi.org/10.1007/s00284-014-0600-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-014-0600-1