
Abstract
A bacterial strain was taxonomically characterised by means of a genomic approach comprising 16S rRNA gene sequence analysis, multilocus sequence analysis (MLSA), the DNA G+C content, whole genome analyses (ANI and GGDC) and phenotypic characterisation. The strain CAIM 1540T was isolated from a cultured oyster Crassostrea corteziensis in La Cruz, Sinaloa state, México. The isolate was found to be catalase and oxidase positive, cells were observed to be motile, O/129-sensitive and facultatively anaerobic. The almost-complete 16S rRNA gene sequence placed this strain within the genus Vibrio; the closest related species were found to be Vibrio aestivus, Vibrio marisflavi, Vibrio maritimus and Vibrio variabilis with similarity values of 99.02, 97.05, 96.70, and 96.59 % respectively. MLSA of four housekeeping genes (ftsZ, gapA, recA, and topA) was performed with the closely related species. A draft genome sequence of strain CAIM 1540T was obtained. The DNA G+C content of this strain was determined to be 43.7 mol%.The ANI values with V. aestivus were 89.6 % (ANIb), 90.6 % (ANIm) and a GGDC value of 39.5 ± 2.5 % was obtained; with V. marisflavi the genomic similarities were 71.5 % (ANIb), 85.5 % (ANIm) and 20.2 ± 2.3 % (GGDC); with V. maritimus 72.6 % (ANIb), 85.7 % (ANIm) and 22.0 ± 2.0 % (GGDC); and with V. variabilis 72.6 % (ANIb), 85.8 % (ANIm) and 21.6 ± 1.6 % (GGDC). These ANI and GGDC values are below the threshold for the delimitation of prokaryotic species, i.e. 95–96 and 70 %, respectively. Phenotypic characters also showed differences with the closest related species analysed. The results presented here support the description of a novel species, for which the name Vibrio mexicanus sp. nov. is proposed, with strain CAIM 1540T (= CECT 8828T, = DSM 100338T) as the type strain. In addition, we found that the recently described species Vibrio thalassae and Vibrio madracius might be a single species because the values of ANIb 95.8 %, ANIm 96.6 % and GGDC 70.2 ± 2.9 % are above the accepted species thresholds.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Members of the family Vibrionaceae are Gram-negative Gammaproteobacteria and are found in a wide variety of aquatic biotopes, including extremophile species that can live at great depths such as Photobacterium profundum or symbionts such as Aliivibrio fischeri (Thompson and Swings 2006).Most members of the family Vibrionaceae are free-living marine bacteria but many can also form biofilms on marine invertebrate exoskeletons or other surfaces (Thompson and Swings 2006). The members of this family have also an important impact on aquaculture; Aliivibrio salmonicida, Vibrio anguillarum and Vibrio vulnificus are major fish pathogens, while Vibrio harveyi and Vibrio parahaemolyticus are the most important pathogens of shrimp (Austin and Austin 1999; Gomez-Gil et al. 1998, 2003; Tran et al. 2013; Soto-Rodriguez et al. 2015).
Notably, the genus Vibrio consists of over 90 species (http://www.bacterio.net/vibrio/html). However, within this genus there are closely related species that are difficult to identify. The genus is divided in many clades according to their phylogenetic relationships established by Multilocus Sequence Analyses (MLSA, Sawabe et al. 2007, 2013). In particular, the so-called Marisflavi clade (Lucena et al. 2012) consists of three species, Vibrio aestivus, Vibrio marisflavi and Vibrio stylophorae based on the 16S rRNA gene sequences. These species were recently isolated from seawater and corals (Wang et al. 2011; Sheu et al. 2011, Lucena et al. 2012). However, the robustness of this clade has not yet been supported by MLSA. Furthermore, the closely related Mediterranei clade consists of five species, Vibrio maritimus, Vibrio variabilis, Vibrio mediterranei, Vibrio madracius and Vibrio thalassae (Sawabe et al. 2013; Moreira et al. 2014; Tarazona et al. 2014).
DNA–DNA hybridization (DDH) is still considered a gold standard for species delineation, despite that other methods have greater resolving power and are easier to perform. Such methods comprise of genomic taxonomy techniques such as genome-to-genome distance (GGD), average nucleotide identity (ANI), and genotype-to-phenotype on the basis of whole genome sequences (Amaral et al. 2014; Thompson et al. 2015). In this study, a genomic taxonomy study was performed to classify a novel Vibrio strain, CAIM 1540T.
Materials and methods
Bacterial strain and growth conditions
The strain CAIM 1540T was isolated on Marine Agar from a cultured oyster (C. corteziensis) in La Cruz, Sinaloa state, México (23°55′05.9″N 106°53′24.7″W) on February 13th, 2004. The type strains of V. aestivus CAIM 1861T and V. marisflavi CAIM 1886T were obtained from the Collection of Aquatic Important Microorganisms (CAIM). The strains were grown on Tryptone Soy Agar (TSA; Oxoid) + 2 % NaCl (w/v) and incubated at 30 °C for 24 h. Cultures were maintained frozen at −80 °C in Tryptone Soy Broth (TSB) supplemented with 2 % NaCl (w/v) and 15 % (v/v) glycerol as preservative.
Phenotypic analyses
The following phenotypic tests were performed with the strains (MacFaddin 1993; Noguerola and Blanch 2008): Gram staining, catalase and oxidase activities, cell morphology, motility, oxidation/fermentation test, methyl red test, Voges–Proskauer test, utilisation of citrate, arginine dihydrolase, lysine and ornithine decarboxylation, and nitrate reduction. Further characterisation was done using API 20E test strips (bioMérieux) and Biolog GN MicroPlate™. The strains were grown onTSB to determine salt tolerance (0–10 % NaCl), growth at different temperatures (8, 20, 30, 37, 40 °C) and pH (2–14). The sensitivity to the vibriostatic agent O/129 (2,4-diamino-6, 7- diisopropylpteridine, 150 µg per disc) and the use of 44 substrates as sole carbon and energy sources were determined as described previously (Macián et al. 2001).
Analysis of fatty acid methyl esters (FAMEs) was performed according to the Microbial Identifications Systems (MSI) (MIDI, Newark, DE, USA) protocol as described by Sasser (1990). The cells were grown on TSA supplemented with 2.0 % NaCl (w/v) and incubated at 25 °C for 24 h.
DNA isolation, amplification, sequencing, and sequence analysis
Genomic DNA was extracted from 24 h cultures on TSA supplemented with 2 % NaCl (w/v) using a Promega kit (Wizard® Genomic DNA Purification Kit). The amplification and sequencing of the 16S rRNA gene, and of the genes ftsZ (cell division protein), gapA (glyceraldehyde 3-phosphate dehydrogenase), recA (recombinase A gene) and topA (topoisomerase I) were performed as previously described (Pascual et al. 2010; Cano-Gomez et al. 2010; Yoshizawa et al. 2010). Sequence data analysis was carried out with the DNASTAR Lasergene SEQMAN program. Sequence similarity of the 16S rRNA was determined using the EzTaxon-e server (Kim et al. 2012). Phylogenetic trees were reconstructed using the neighbour-joining, maximum-likelihood, and maximum parsimony algorithms with MEGA ver. 5.05 (Tamura et al. 2011). Sequences of phylogenetically closely related species were obtained from GenBank/EMBL/DDBJ (Supplementary Table 1).
The draft genome of CAIM 1540T was sequenced by means of the Ion Torrent PGM platform as described earlier (Quail et al. 2012; Moreira et al. 2014) with minor modifications as follows. Library preparation was carried out using the Ion Plus Fragment Library Kit, with 1 μg DNA (in Low TE, 50 μL). DNA was fragmented using the BioRuptor®Sonication System as described in the Ion Plus Fragment Library Kit protocol. End repair, adapter ligation, nick repair, and amplification (10 cycles) were also performed as described in the Ion Plus Fragment Library protocol. 300 and 350 bp fragments were selected through agarose gel (2 % m/v) electrophoresis (E-Gel SizeSelect, Life technologies). Quality and concentration of the libraries were determined using an Agilent 2100 Bioanalyzer with the associated High Sensitivity DNA kit (Agilent Technologies), as well as with an Ion library Quantization kit using TaqMan® in a CFX96™ Real-Time PCR System (Bio-Rad). The amount of library required for template preparation was calculated using the Template Dilution Factor calculation described in the manufacturers protocol. Emulsion PCR and enrichment steps were carried out in the Ion OneTouchTM 200 Template Kit v2. Ion Sphere Particle quality assessment was carried out as outlined in this protocol. Sequencing was done using a 318 chip with barcoding. The Ion PGM™ 200 Sequencing Kit was used for sequencing following the recommended protocol and Torrent Suite 1.5 was used for analyses. The reads were assembled de novo with Newbler (RunAssembly ver. 2.3).
The DNA G+C mol% was estimated using a method described previously (Moreira et al. 2011) and from the draft genome sequence.
Accession numbers
The GenBank/EMBL/DDBJ accession numbers for the 16S rRNA, ftsZ, gapA, recA, and topA gene sequences of strain CAIM 1540T are JQ434105, KP698215, KP698212, KP698213, and KP698214 respectively; all other gene sequences used in this study are listed in the supplementary Table 1. The genome accession number of the type strain CAIM 1540T is JYJP00000000.
Result and discussion
Bacterial characterisation
Strain CAIM 1540T was isolated from a cultured oyster (C. corteziensis) in La Cruz, Sinaloa state, México. The strain showed phenotypic characteristics that place it clearly asa member of the genus Vibrio: the cells were observed to be motile small rods, Gram-negative, facultatively anaerobic, oxidase and catalase positive. The strain was found to require sodiumions for growth; the optimal salinity range obtained was between 3 and 5 % (Fig. S1). No growth was observed at NaCl concentrations of 7 % or higher. Optimal pH was established at a range of 6–8 (Fig. S2). The optimal temperature was found to be 30 °C (Fig. S3) and no growth was observed at 40 °C. The strain was found to grow on thiosulfate-citrate-bile-sucrose agar (TCBS) agar (Difco) as green colonies. Strain CAIM 1540T was also found to be able to reduce nitrates to nitrites, to be positive for methyl red test, urea hydrolysis, l-tryptophan deaminase, indole production, and bovine gelatin hydrolysis. Strain CAIM 1540T was found to be negative for citrate utilisation, the Voges–Proskauer test, arginine dihydrolase and lysine and ornithine decarboxylase. Several phenotypic tests could be selected to clearly differentiate CAIM 1540T from the closest related species Vibrio species (Table 1).
The fatty acid profile of strain CAIM 1540T showed the main features (Table 2) of the members of the genus Vibrio.
Phylogenetic analysis
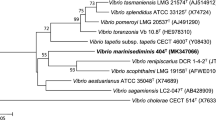
The closest related species identified by 16S rRNA gene sequence were found to be V. aestivus and V. marisflavi, with similarity values of 99.02–97.05 % respectively (Fig. 1); the third member of the proposed Marisflavi clade (Lucena et al. 2012), V. stylophorae, was found to show a more distant relationship (95.04 %) and this species was well separated in the phylogenetic tree (Fig. 1). Similarities with members of the Mediterranei clade were 96.7 % with V. maritimus and 96.6 % with V. variabilis. The 16S rRNA gene sequence analysis clearly separates CAIM 1540T from V. aestivus forming a separate branch, supported with a high bootstrap value(Fig. 1).

Phylogenetic tree based on partial 16S rRNA gene sequences obtained by the neighbour joining method based on the Jukes–Cantor model. GenBank sequence accession numbers are given in parentheses. Numbers at nodes denote the level of bootstrap based on 1000 replicates; only values greater than 50 % are shown. Vibrio cholerae was used as bacterial out-group. Bar 0.5 % estimated sequenced divergence. Scale bar, base substitutions per site
MLSA has been proposed as a valuable technique for the identification and classification of vibrios (Cano-Gomez et al. 2010). In this study, sequences of the following four housekeeping genes were obtained: cell division protein (ftsZ, 432 pb), glyceraldehyde-3-phosphate dehydrogenase A (gapA,602 bp), protein recombinase A ( recA, 494 bp), and DNA topoisomerase 1 (topA, 402 bp); these sequences were compared with those of related species. The phylogenetic trees based on the housekeeping genes ftsZ (Fig. S4), gapA (Fig. S5), recA (Figs. S6, S10), topA (Fig. S7) and the 16S rRNA gene (Fig. 1), and on the concatenated sequences of these five genes (Figs. 2, S8, S9), confirmed the clustering of the proposed novel Vibrio species as a independent branch with high bootstrap values, and its distinction from the closest phylogenetic neighbours. In all individual trees the isolate formed a monophyletic group with V. aestivus. Notably these analyses suggest that this lineage is well separated from V. marisflavi, suggesting that neither V. aestivus nor CAIM 1540T should be considered members of the Marisflavi clade.

Phylogenetic tree based on concatenated sequences (3131 bp) of the four housekeeping genes ftsZ (432 bp), gapA (602 bp), recA (494 bp) and topA (402 bp), and the 16S rRNA gene (1261 bp) sequences available from the GenBank (accession numbers are listed in Supplementary Table S1) Neighbour Joining. Numbers at nodes denote the level of bootstrap based on 1000 replicates; only values greater than 50 % are shown. V. cholerae was used as bacterial out-group. Scale bar, base substitutions per site
Each MLSA gene was analysed for recombination events with the program SplitsTree v4 (Huson and Bryant 2006) (Figs. S11–S18).Recombination analyses are useful to detect recombination events that may affect the topology of phylogenetic trees; a recombination event increases the distance between species. Several recombination events were observed, especially in recA as seen for other vibrios (González-Castillo et al. 2014). Nevertheless, the analysis of concatenated gene sequences provides more informative data and minimizes the weight of recombination events, making it a tool that increases the quality of phylogenetic analyses and provides a greater power of taxonomic resolution (Pascual et al. 2010). In this study recombination events were minimized by using concatenated gene sequences and, therefore, phylogenetic tree topology was unaffected.
Genomic analysis
DDH has been the gold standard for prokaryotic classification at the genomic level as it offers a numerical and relatively stable limit, but in the era of genomics this method seems to be out-dated and could be replaced by a comparison between sequenced genomes. ANI with at least 20 % of the genome of the query strains rather than its complete sequence, is enough to clearly differentiate species (Richter and Rosselló-Móra 2009).ANI is calculated with two algorithms, BLAST and MUMmer. The proposed limits for the definition of species are set at 95–96 % of ANI values (Lucena et al. 2012). Therefore, shotgun genome sequencing was performed on strain CAIM 1540T. The subsequent assembly produced 167 contigs (N 50 = 88,690 bp, G+C 43.7 %) for a 5.4 Mb genome.
Comparison of the draft genome of strain CAIM 1540T yielded ANI values (Table 3) of 89.6 % (ANIb) and 90.6 % (ANIm) with the closest related species V. aestivus, 71.5 % (ANIb) and 85.5 % (ANIm) with V. marisflavi, 72.6 % (ANIb) and 85.7 % (ANIm) with V. maritimus, and 72.6 % (ANIb) and 85.8 % (ANIm) with V. variabilis. These values are clearly below the species-delineating threshold of 96 %, indicating that strain CAIM 1540T does not belong to these previously described species.
Digital DDH calculations were also performed for these genome sequences. The genomic distance was calculated using the Genome-To-Genome Distance Calculator (GGDC) (Auch et al. 2010; Meier-Kolthoff et al. 2013). In silico DDH is calculated by the Genome Blast Distance Phylogeny (GBDP), which was devised as an approach for the inference of phylogenetic trees or networks from a given set of wholly (or even incompletely) sequenced genomes (Henz et al. 2005) and was subsequently revisited and enhanced (Auch et al. 2010). Strains from the same prokaryotic species share >70 % in silico GGDC (Thompson et al. 2013). Comparisons with the draft genome of strain CAIM 1540T yielded GGDC values as low as 39.5 ± 2.5 % with V. aestivus and 20–23 % with other closely related species (Table 4), confirming that the strain does not belong to any of these species.
Another tool with a high resolving power, which uses genome sequences is vibrio phenotyping, this program is based on the search for those enzymes related to the phenotype of interest. It uses BLAST to assign a positive match if the identity is greater than 40 % with a sequence query length greater than 70 %. It is an alternative for the phenotypic identification of vibrios (Amaral et al et al. 2014; Thompson et al 2015). CAIM 1540T showed similar results to those obtained with commercial systems (Table 5).
In addition to our genomic studies of strain CAIM 1540T, we found that the recently described species V. thalassae, which was assigned to the Mediterranei clade (Tarazona et al. 2014; Validation List 160, Oren and Garrity 2014) and V. madracius (Moreira et al. 2014; Validation List 161, Oren and Garrity 2015) comprise a single species because the values of ANIb 95.8 %, ANIm 96.6 % and GGDC 70.2 ± 2.9 % (Tables 3, 4) are above the accepted thresholds for delineating prokaryotic species. Thus, V. madraciuscan likely be considered a later heterotypic synonym of V. thalassae.
The DNA G+C mol% range reported for members of the genus Vibrio is between 38 and 51 % (Dieguez et al. 2011).The precise DNA G+C mol% for strain CAIM 1540T was obtained from the draft genome sequence to be 43.7 %; this value was also estimated by a real time-PCR method (Moreira et al. 2011) to be 44.3 mol%.
The polyphasic taxonomic study which involved phenotypic, genotypic, genomic and phylogenetic analyses support the proposal of a novel species, for which the name Vibrio mexicanus sp. nov. is proposed, with CAIM 1540T as the type strain.
Description of Vibrio mexicanus sp. nov
Vibrio mexicanus (me.xi.ca´nus N.L. masc. adj. mexicanus from México).
Gram-negative, curved bacilli that grow as green colonies on TCBS agar, motile and facultatively anaerobic, not luminescent and do not swarm on marine agar or on TSA with 2.0 % NaCl. Growth occurs at 1–6 % NaCl (optimally in 4 % NaCl), no growth without NaCl or with more than 7 % NaCl. Grows at 20, 30, 37 °C (optimum 30 °C). Grows at 4–11 pH (optimally at pH 6–8). Sensitive to the vibriostatic agent O/129 (150 µl per disc). Oxidase and catalase positive. Negative for arginine dihydrolase, lysine and ornithine decarboxylases, positive for nitrate reduction, methyl red test, urea, l-tryptophan, indole, and bovine gelatin; negative reaction for utilisation of citrate and the Voges–Proskauer test. Ferments d-glucose and amygdalin but not d-mannitol, l- rhamnose, inositol, d-sorbitol, d-melibiose, l-arabinose and d-sucrose (in API 20E tests). Utilises the following substrates as sole sources of carbon: 2-ketoglutarate, l-arabinose, l-aspartate, d-cellobiose, d-glucosamine, d-mannitol, d-fructose, glycerol, d-glucose, l-alanine, d-galactose, l-glutamate, lactose, d,l-lactate, malate, maltose, d-mannose, l-leucine, d-melibiose, N-acetyl-d-glucosamine, l-ornithine, l-rhamnose, d-ribose, succinate, sucrose, d-xylose and d-trehalose. Negative for utilisation ofacetate, citrate, d-galacturonate, ϒ-aminobutyrate, d-gluconate, pyruvate, d-glucuronate, salicin, glycine, p-hydroxybenzoate, l-lysine, l-histidine, m-inositol, propionate, putrescine, l-threonine and tyrosine. Using Biolog GN2 MicroPlates, oxidizes the following substrates: d-melibiose, glycerol, d,l, α glycerolphosphate, d-fructose, d-cellobiose, glucose-1-phosphate, glucose-6-phosphate, p-hydroxyphenylacetic acid, bromosuccinic acid, β-methyl-d-glucoside, itaconic acid, succinamic acid, hydroxy-l-proline, l-fucose, glucuronamide, l-leucine, l-rhamnose, glycogen, d-galactose, formic acid, α-ketoglutaric acid, l-alaninamide, l-ornithine, Tween 40, Tween 80, α-d-glucose, d-sorbitol, d-galacturonic acid, d,l-lactic acid, l-alanine, sucrose, d-gluconic acid, malonic acid, N-acetyl-d-glucosamine, α-d-lactose, d-trehalose, propionic acid, adonitol, lactulose, turanose, d-glucuronic acid, quinic acid, l-aspartic acid, l-arabinose, maltose, d-saccharic acid, l-glutamic acid, d-mannitol, sebacic acid and d-mannose; weak positive reactions for inosine, thymidine and phenylethylamine. Negative for I-erythritol, d-alanine, m-inositol, putrescine, 2-aminoethanol, acetic acid, l-histidine, α-cyclodextrin, cis-aconitic acid, dextrin, d-psicose, citric acid, α-ketobutyric acid, d-raffinose, gentiobiose, d-galactonic acid lactone, α-ketovaleric acid, l-phenylalanine, l-proline, N-acetyl-d-galactosamine, l-alanyl-glycine, l-pyroglutamic acid, d-glucosaminic acid, l-asparagine, d-serine, l-serine, 2,3-butanediol, urocanic acid, uridine, xylitol, α-hydroxybutyric acid, l-threonine, d-arabitol, methyl pyruvate, β-hydroxybutyric acid, glycyl-l-aspartic acid, d,l-carnitine, mono-methyl-succinate, γ-hydroxybutyric acid, succinic acid, glycyl-l-glutamic acid, and γ-aminobutyric acid. The major fatty acids are summed feature 3 (comprising C16:1 w7c and/or C16:1 w6c and/or C15:0 iso 2–OH), C16:0, summed feature 8 (C18:1 w6c and/or C18:1 w7c) and C14:0. The following fatty acids are present in small amounts: C15:0 iso, C17:0 iso, C12:0, summed feature 2 (C14:0 3OH and/orC16:1iso) and C12:0 3OH.
The type strain is CAIM 1540T. The type strain was isolated from a cultured oyster, C. corteziensis, in La Cruz, Sinaloa state, México and deposited as CAIM 1540T and as CECT 8828T. The GenBank/EMBL/DDBJ accession numbers for the 16S rRNA, ftsZ, gapA, recA, and topA gene sequences of strain CAIM 1540T are JQ434105, KP698215, KP698212, KP698213, and KP698214 respectively. The genome accession number of the type strain CAIM 1540T is JYJP00000000.
Abbreviations
- ANI:
-
Average nucleotide identity
- GGDC:
-
Genome-to-genome distance calculator
- MLSA:
-
Multilocus sequence analysis
References
Amaral GRS, Dias GM, Wellington-Oguri M, Chimetto L, Campeão ME, Thompson FL, Thompson CC (2014) Genotype to phenotype: identification of diagnostic vibrio phenotypes using whole genome sequences. Int J Syst Evol Microbiol 64:357–365
Auch AF, von Jan M, Klenk H-P, Göker M (2010) Digital DNA–DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genom Sci 2:117–134
Austin B, Austin DA (1999) Bacterial fish pathogens: disease of farmed and wild fish, 3rd edn. Springer, Berlin
Cano-Gomez A, Goulden EF, Owens L, Høj L (2010) Vibrio owensii sp. nov., isolated from cultured crustaceans in Australia. FEMS Microbiol Lett 302:175–181
Chimetto LA, Cleenwerck I, Moreira APB, Brocchi M, Willems A, De Vos P, Thompson FL (2011) Vibrio variabilis sp. nov. and Vibrio maritimus sp. nov., isolated from Palythoa caribaeorum. Int J Syst Evol Microbiol 61:3009–3015
Dieguez AL, Beaz-Hidalgo R, Cleenwerck I, Balboa S, de Vos P, Romalde JL (2011) Vibrio atlanticus sp. nov. and Vibrio artabrorum sp. nov., isolated from the clams Ruditapes philippinarum and Ruditapes decussates. Int J Syst Evol Microbiol 61:2406–2411
Gomez-Gil B, RoqueA TurnbullJF, Tron-Mayen L (1998) Species of Vibrio isolated from hepatopancreas, hemolymph and digestive tract of a population of healthy juvenile Penaeus vannamei. Aquaculture 163:1–9
Gomez-Gil B, Thompson FL, Thompson CC, Swings J (2003) Vibrio rotiferianus sp. nov., isolated from cultures of the rotifer Brachionus plicatilis. Int J Syst Evol Microbiol 53:239–243
González-Castillo A, Balboa S, Romalde JL, Gomez-Gil B (2014) Vibrio crosai sp. nov., isolated from a cultured oyster Crassostrea gigas. Antonie Van Leeuwenhoek 106:457–463
Henz S, Huson D, Auch AF, Nieselt-Struwe K, Schuster S (2005) Whole-genomeprokaryotic phylogeny. Bioinformatics 21(10):2329–2335
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23(2):254–267
Kim OS, Cho YJ, Lee K, Yoon SH, Kim M, Na H, Park SC, Jeon YS, Lee JH, Yi H, Won S, Chun J (2012) Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol 62:716–721
Lucena T, Ruvira MA, Arahal DR, Macián MC, Pujalte MJ (2012) Vibrio aestivus sp. nov. and Vibrio quintilis sp. nov., related to Marisflavi and Gazogenes clades, respectively. Syst Appl Microbiol 35:427–431
MacFaddin JF (1993) Pruebas bioquímicas para la identificación de bacterias de importancia clínica (translation by Médica Panamericana SA). Williams & Wilkins, Baltimore
Macián MC, Ludwig W, Aznar R, Grimont P, Schleifer KH, Garay E, Pujalte MJ (2001) Vibrio lentus sp. nov., isolated from Mediterranean oysters. Int J Syst Evol Microbiol 51:1449–1456
Meier-Kolthoff JP, Alexander AF, Hans-Peter K, Markus K (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform 14:60
Moreira AP, Pereira N Jr, Thompson FL (2011) Usefulness of a real-time PCR platform for G+C content and DNA–DNA hybridization estimations in vibrios. Int J Syst Evol Microbiol 61:2379–2383
Moreira AP, Duytschaever G, Tonon LAC, Dias GM, Mesquita M, Cnockaert M, Francini-Filho RB, DeVos P, Thompson CC, Thompson FL (2014) Vibrio madracius sp. nov. isolated from Madracisdecactis (Scleractinia) in St. Peter & St. Paul Archipelago, Mid-Atlantic Ridge, Brazil. Curr Microbiol 69(4):405–411
Noguerola I, Blanch AR (2008) Identification of Vibrio spp. with a set of dichotomous keys. J Appl Microbiol 105:175–185
Oren A, Garrity GM (2014) List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 64:3603–3606
Oren A, Garrity GM (2015) List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 65:1–4
Pascual J, Macián MC, Arahal DR, Garay E, Pujalte MJ (2010) Multilocus sequence analysis of the central clade of the genus Vibrio by using the 16S rRNA, recA, pyrH, rpoD, gyrB, rctB and toxR genes. Int J Syst Evol Microbiol 60:154–165
Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y (2012) A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and IlluminaMiSeq sequencers. BMC Genom 13(1):341
Richter M, Rosselló-Móra R (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106:19126–19131
Sasser M (1990) Identification of bacteria by gas chromatography of cellular fatty acids. Microbial ID Inc, Newark
Sawabe T, Kita-Tsukamoto K, Thompson FL (2007) Inferring the evolutionary history of vibrios by means of multilocus sequence analysis. J Bacteriol 189:7932–7936
Sawabe T, Ogura Y, Matsumura Y, Feng G, Rohul Amin AKM, Mino S, Nakagawa S, Sawabe T, Kumar R, Fukui Y, Satomi M, Matsushima R, Thompson FL, Gomez-Gil B, Christen R, Maruyama F, Kurokawa K, Hayashi T (2013) Updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front Microbiol 4:(414)1–14
Sheu S-Y, Jiang S-R, Chen CA, Wang JT, Chen WM (2011) Vibrio stylophorae sp. nov., isolated from the reef-building coral Stylophora pistillata. Int J Syst Evol Microbiol 61:2180–2185
Soto-Rodriguez SA, Gomez-Gil B, Lozano-Olvera R, Betancourt-Lozano M, Morales-Covarrubias MS (2015) Field and experimental evidence of Vibrio parahaemolyticus as the causative agent of acute hepatopancreatic necrosis disease of cultured shrimp (Litopenaeus vannamei) in Northwestern Mexico. Appl Environ Microbiol 81:1689–1699
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tarazona E, LucenaT Arahal DR, Macián MC, Ruvira MA, Pujalte MJ (2014) Multilocus sequence analysis of putative Vibrio mediterranei strains and description of Vibrio thalassae sp. nov. Syst Appl Microbiol 37:320–328
Thompson FL, Swings J (2006) Taxonomy of the vibrios. In: Thompson FL, Austin B, Swings J (eds) The biology of vibrios. American Society for Microbiology, Washington, pp 29–43
Thompson CC, Amaral GR, Campeão M, Edwards RA, Polz MF, Dutilh BE, Ussery DW, Sawabe T, Swings J, Thompson FL (2015) Microbial taxonomy in the post-genomic era: rebuilding from scratch? Arch Microbiol 197:359–370
Thompson CC, Chimetto L, Edwards RA, Swings J, Stackebrandt E, Thompson FL (2013) Microbial genomic taxonomy. BMC Genom 14:913
Tran L, Nunan L, Redman RM, Mohney LL, Pantoja CR, Fitzsimmons K, Lightner DV (2013) Determination of the infectious nature of the agent of acute hepatopancreatic necrosis syndrome affecting penaeid shrimp. Dis Aquat Organ 105:45–55
Wang H, Liu J, Wang Y, Zhang X-H (2011) Vibrio marisflavi sp. nov., isolated from sea water. Int J Syst Evol Microbiol 61:568–573
Yoshizawa S, Wada M, Yokota A, Kogure K (2010) Vibrio sagamiensis sp. nov., luminous marine bacteria isolated from sea water. J Gen Appl Microbiol 56:499–507
Acknowledgments
This work was partially funded by CONACYT project CB-2009-01 132328 and Grant AGL–2010–18438 from the Ministerio de Ciencia e Innovación (Spain).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
González-Castillo, A., Enciso-Ibarrra, J., Bolán-Mejia, M.C. et al. Vibrio mexicanus sp. nov., isolated from a cultured oyster Crassostrea corteziensis . Antonie van Leeuwenhoek 108, 355–364 (2015). https://doi.org/10.1007/s10482-015-0488-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-015-0488-1