
Abstract
Bombyx mori parvo-like virus (BmPLV) has two complementary single-stranded DNA genome (VD1 and VD2) and owns a self-encoding DNA polymerase motif, but its replication mechanism is unclear. In our previous research, a protein encoded by VD1-ORF1 was indentified in the midgut of BmPLV China Zhenjiang isolate-(BmPLV-Z) infected silkworm larvae via two-dimensional gel electrophoresis (2-DE). This protein was named as non-structural protein 2 (NS2), which showed no similarity to that of parvoviruses. To date, little is known about it. In this study, sequence alignment results showed that NS2 shared homology with some chromosomal replication initiator protein dnaA and DNA-binding response regulators. The ns2 was cloned and expressed in E. coli, and then a polyclonal antibody of the NS2 protein was prepared successfully. The data from real-time quantitative PCR displayed that the transcription of VD1-ORF1 from BmPLV-Z-infected midguts started from 28-h post inoculation (h p.i.) in low amounts, but in high amounts at late stages of infection. Immunofluorescence showed that NS2 ultimately concentrated on the nuclear membrane in BmN cells at late stages, indicating that NS2 might be associated with integral membrane protein.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bombyx mori parvo-like virus (BmPLV) termed as B. mori densovirus type II (BmDNV-II), which can infect the columnar cells of midgut epithelium and causes the fatal flacherie disease of silkworm [3]. The virus particles are small icosahedral, non-enveloped particles 19–24 nm in diameter. These features are very close to B. mori densovirus type I (BmDNV-1) which has a linear single-stranded complementary DNA genome [about 5000 nucleotides (nts) in length]. Unlike densovirus, BmPLV has two sets of complementary DNAs (VD1; about 6600 nts, VD2; about 6100 nts) [3]. VD1 and VD2 share a common terminal sequence of 53 nts. Both DNAs have imperfect inverted terminal repeats which were unable to form terminal hairpins like that of parvoviruses, and these complementary terminal repeats would allow these molecules to form panhandles [3, 31]. Furthermore, the viral DNA could encode a DNA polymerase [15, 38]. These unusual properties imply a replication mechanism different from other densoviruses or vertebrate parvoviruses [3, 4]. Previous reports predicted that this virus could replicate like adenoviruses [31]. In this model, DNA synthesis initiates with a protein covalently linked at the 5′-terminal base of a panhandle structure. Up to date, the replication mechanism of this virus is unclear.
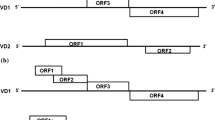
Bombyx mori parvo-like virus includes two isolates, Japan Yamanashi isolate (BmDNV-2) and China Zhenjiang isolate (BmPLV-Z). Their genome show high similarity about 98% in VD1 and 97% in VD2 [35]. Six ORFs (VD1; four ORFs, VD2; two ORFs) with the potential of encoding a polypeptide longer than 100 amino acids were identified in the virus genomes [4]. There are four non-structural proteins encoded by VD1-ORF1, VD1-ORF2, VD1-ORF4, and VD2-ORF2, respectively, in BmPLV-Z [35]. VD1-ORF2 (NS1) has a conservative structural domain as NS1s from parvoviruses, and recombinant BmPLV-Z NS1 protein possesses ATP-binding, ATPase, DNA-binding, and helicase activities in vitro [20]. VD1-ORF4 contains a conserved domain of protein-primed DNA polymerase belonging to DNA polmerase of B family [17]. VD2-ORF2 encodes a deduced 222 amino acid protein NS3 which shows low homology to that of Junonia coenia densovirus (JcDNV), Galleria mellonella densovirus (GmDNV), Mythimna loreyi densovirus (MlDNV), and Diatraea saccharalis densovirus (DsDNV) [37]. The study of JcDNV NS3 biochemical properties suggested that JcDNV NS3 was essential for viral DNA replication [1].
VD1-ORF1 of BmPLV-Z encodes a 126 amino acid peptide. A protein from BmPLV-Z-infected silkworm midguts was identified by comparative proteomics, and it was encoded by VD1-ORF1 (our unpublished data) and this protein was not detected in the proteins of virions [21]. In addition, analysis of viral transcripts showed that VD1-ORF1 and VD1-ORF2 were in the same transcript, from which two proteins could be generated by alternative translation [35]. BmPLV-Z-NS2 showed no homology to other NS2s from parvoviruses and little is known about it.
No highly conserved domains were detected in NS2s from the densoviruses, and the functions of NS2s have been studied in far less detail than those of the vertebrate parvoviruses. In the studies of mosquito densonucleosis virus Aedes aegypti densovirus (AeDNV), mutations ranging from complete NS2 knock-out to a single missense amino acid substitution in NS2 can significantly reduce viral replication and production of viable progeny [2]. Probably the best studied to date is the NS2 protein of minute virus of mice (MVM). Roles have been ascribed to NS2 of MVM in viral DNA amplification, efficient translation of viral mRNA, capsid formation, and packaging of virion ssDNA [9, 24, 25]. The distribution of NS2 proteins within the cell is regulated according to their phosphorylation state [7], and the interaction of these polypeptides with members of the 14-3-3 family of cellular proteins is dependent on distinct phosphorylation events. Eichwald et al. [13] have provided evidence that the NS2 proteins of MVM are required for efficient nuclear egress of progeny virions in mouse cells.
In this study, we developed some studies to characterize the features and functions of BmPLV-Z NS2. First, the functional and structural domains were analyzed according to the sequence alignment and motif scanning. Second, VD1-ORF1 was cloned and expressed in E. coli. The expressed His-NS2 protein was purified and used to prepare polyclonal antibody. Moreover, RNA transcripts of VD1-ORF1 in infected silkworm midguts were detected by real-time quantitative PCR (RT-qPCR). Finally, subcellular localization of NS2 in BmN cells was investigated. Immunofluorescence showed that NS2 ultimately concentrated on the nuclear membrane in BmN cells, indicating that NS2 might be associated with integral membrane protein.
Materials and Methods
Virus, Insect, and Cell
Bombyx mori parvo-like virus China Zhenjiang isolate (BmPLV-Z), silkworm strain HuaBa35 (susceptible to BmPLV-Z), and BmN cells were maintained in our laboratory. The silkworm larvae were reared with fresh mulberry leaves at 25 ± 2°C under a 12-h light/dark photoperiod. The BmN cells were cultured at 27°C in TC-100 insect medium (Gibco, USA) supplemented with 10% (v/v) fetal bovine serum (Gibco, USA).
Bioinformatics Analysis
The protein sequence was analyzed using ExPASy Proteomics server (http://www.expasy.org/tools) for the prediction of motifs, domains (MotifScan tool), transmembrane regions, and signal peptides (Topology prediction). Homologs were explored using BLAST2 searching tool in normal SMART (http://smart.embl-heidelberg.de/) SWISS-PROT databases. Multiple sequence alignment of proteins was performed with the Clustal W software [30], and edited with Genedoc.
Cloning, Expression of NS2 in E. coli, and Preparation of an Antiserum
The ns2 coding region was amplified from the BmPLV-Z genomic DNA using the ns2-specific primers NS2-F: 5′-AAGGATCCATGGCATTCAACGCTCTC-3′ (BamH I site was underlined) and NS2-R: 5′-GGCTCGAGCTTAGAGCTCTTTGCACT-3′ (Xho I site was underlined), and cloned into vector pET30a(+) digested with BamH I and Xho I to produce plasmid pET30-ns2. The recombinant plasmid pET30-ns2 was transformed into competent cells of E. coli BL21 (DE3) LysS for expression of the NS2 protein. The 6× His-tagged protein, His-NS2 was induced by addition of 0.6–2 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Sigma-Aldrich, USA), analyzed by 15% SDS-PAGE, and stained with Coomassie brilliant blue. A matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometer was used to identify the fusion peptides. The fusion peptides were analyzed by Western blot using anti-6× his monoclonal antibody (Qiagen, Germany) and goat antirabbit IgG conjugated with horseradish peroxidase (Invitrogen, USA). The immunoreactive proteins were visualized by DAB staining.
6× His-NS2 was purified using Ni2+–NTA agarose beads (Novagen, USA) under native conditions following the manufacturer’s instructions, and the polyclonal antibodies were raised in rabbits according to the procedure as described previously [37]. NS2 antiserum was purified using protein-A-sepahrose CL-4B (Sigma, USA). The sensitivity of the purified NS2 antiserum was identified by Western blotting.
Transcriptional Analysis
The total RNA was extracted from the midgut of silkworm strain HuaBa35 infected with BmPLV-Z at different time points (0, 2, 4, 6, 12, 24, 28, 36, 40, 48, 52, and 72 h p.i.) by adding Trizol reagent (Invitrogen, USA), according to the manufacturer’s protocol, and then treated with 0.2 U/μl of DNase I (TaKaRa) at 37°C for 2 h. The first-strand cDNA was synthesized with 2 μg of total RNA as templates using SYBR RT-PCR Kit A (TaKaRa).
Real-time quantitative PCR was conducted to examine viral replication on an MX3000P thermal cycler amplification and detection system (Stratagene). Amplification of the cDNA was performed by RT-qPCR using the SYBR RT-PCR Kit B (TaKaRa). Specific primers for virus gene VD1-ORF1 were: 5′-CATGGCATTCAACGCTCTC-3′ and 5′-CTTAGAGCTCTTTGCACT-3′, and the length of the PCR product is 373 bp. As an internal control for adjustment of template RNA quantity, a 284-bp fragment of B. mori cytoplasmic actin gene A3 was amplified in parallel with the same RNA samples using the following primers: Bm-actinA3-F: 5′-GCGCGGCTACTCGTTCACTACC-3′ and Bm-actinA3-R: 5′-GGATGTCCACGTCGCACTTCA-3′. Substitute the ct number of VD1-ORF1 and actin obtained from RT-qPCR separately into 2−Δct (Δct = ctactinA3 − ctVD1-ORF1) at different time points, and the result is the relative copy number of the mRNA belonging to VD1-ORF1 at different time points. The data were represented as arithmetic means ± standard deviations (±SDs).
Western blot analysis
Time courses of NS2 expression after BmPLV-Z infection were analyzed by Western blot, for which fifth instar larvae of silkworm strain HuaBa35 (2 days) were infected with 5 μl infected midguts lysis solution. Silkworm midguts were then excised at different time points (0, 2, 12, 24, 36, 48, 60, and 72 h p.i.), and the total proteins were extracted by RIPA lysis buffer (Beyotime, China) according to the manufacturer’s protocol. A total of 240 μg proteins were separated by 15% SDS-PAGE gel and subsequently transferred onto PVDF membrane for Western blotting. Anti-NS2 antibodies were diluted with pre-antiserum as 1:1000, secondary antibodies conjugated HRP were diluted as 1:5000, and the immunoreactive proteins were visualized by enhanced chemoluminescence (ECL) method (Thermo Scientific Pierce).
Subcellular Localization Analysis of NS2
The vector pFastBacHTb was introduced to construct a transient expression plasmid. The B. mori nucleopolyhedrovirus (BmNPV) ie-1 promoter region was amplified from the BmNPV (T3 isolate) genomic DNA using the ie-1-specific primers, ie-1-F: 5′-GTATACGATTTGCAGTTCGGGAC-3′ (Bst1107 I site was underlined) and ie-1-R: 5′-GGATCCAGTCGTTTGGTTGTTCA-3′ (BamH I site was underlined). The polyhedrin promoter of pFastBacHTb vector, between Bst1107 I site and BamH I site, was replaced with BmNPV ie-1 promoter to generate the vector pFastBac-ie1p. The ns2 coding region was amplified from the BmPLV-Z genomic DNA using the ns2-specific primers ns2-F: 5′-AAGGATCCATGGCATTCAACGCTCTC-3′ (BamH I site was underlined) and ns2-R2: 5′-CCAAGCTTCTTAGAGCTCTTTGCACT-3′ (Hind III site was underlined), and the amplified product was subcloned into vector pFastBac-ie1p digested with BamH I and Hind III sites to generate transient expression plasmid pFastBac-ie1-ns2. Purified plasmids were used to transfect BmN cells, which were grown on sterile coverslips in cell culture capsules using Cellfectin® Reagent (Invitrogen, USA) as previously described [20]. The untransfected BmN cells were used as negative control.
To investigate the subcellular localization of NS2, BmN cells grown on sterile coverslips were used for immunofluorescence staining. In brief, the cells were washed with PBS at 72 h and 96-h post transfection (p.t.), and fixed with 2 ml of 4% paraformaldehyde in PBS for 20 min. Then, cells were washed three times with PBS, permeabilized with 0.2% Triton X-100 in PBS for 15 min, and further incubated in blocking buffer (1% skimmed milk powder in PBS) for 1 h. After washing four times with cold PBS, cells were incubated with anti-NS2 polyclonal antiserum (1:1000) for 2 h at 37°C. Cells were washed three times with PBS, and then incubated with the secondary antibody, fluorescein isothiocyanate-(FITC) conjugated goat anti-rabbit IgG (1:3000) (Qualex, Inc), for 1 h at 37°C. Cells were then washed three times with PBS. After nuclear DNA stained with DAPI (Invitrogen, USA) for 15 min, cells were examined with a confocal laser scanning microscope (Zeiss lsm 5 live).
Results
Sequence Analyses and Homology Searching
MotifScan searching results of one Asn-N-glycosylation site (87–90), three Casein kinase II phosphorylation sites (24–27, 36–39, and 91–94), three Protein kinase C phosphorylation sites (64–66, 119–121, and 122–124), and one 1,4,5-trisphosphate receptor (4–23) were detected (Fig. 1a). No signal peptide, transmembrane regions, mitochondrial targeting sequences, nuclear localization signals, or membrane retention signals were found by any of motif searching engines.

Amino acid sequences alignment and putative motifs of NS2. a Alignment between NS2 and chromosomal replication initiator protein sequences. The putative 1,4,5-trisphosphate/ryanodine receptor (a), Casein kinase II phosphorylation site (b), Protein kinase C phosphorylation site (c), and Asn-N-glycosylation site (d) are framed. b Alignment between NS2 and DNA-binding response regulator sequences
The results of BLAST2 searching showed that there were weak homology between NS2 and chromosomal replication dnaA initiators and DNA-binding response regulators from several bacteriums. The sequences of chromosomal replication initiators are from DNAA VIBCH (Vibrio cholerae, Q9KVX6) (identity = 27%), DNAA BUCAP (Buchnera aphidicola (Schizaphis graminum), P29434) (identity = 26%), DNAA PROMI (Proteus mirabilis, P22837) (identity = 25%), and DNAA SHEON (Shewanella oneidensis, Q8EKT2) (identity = 25%) (Fig. 1a). And DNA-binding response regulator sequences are from BACCZ (Bacillus cereus E33L, Q632K5) (identity = 37%), BACC1 (B. cereus ATCC 10987, Q72YN2) (identity = 37%), BACHK (Bacillus thuringiensis serovar konkukian, Q6HC47) (identity = 37%), and BACCR (B. cereus ATCC 14579, Q816J5) (identity = 37%). The results of sequence alignment are as following (Fig. 1b), and all identity scores are no less than 25%.
Expression of NS2 Protein in E. coli and its Polyclonal Antibody Preparation
The recombinant NS2 protein was expressed in E. coli BL21 harboring the expressing plasmid pET-30a-ns2 and separated in 15% SDS-PAGE. After induced by 1 mM IPTG at 28°C for approximate 4 h, the highest amount of 6× His-NS2 proteins about 21 kDa were produced (Fig. 2a). Induced protein bands were confirmed by Western blot using anti-6× His antibody (Fig. 2a). The fusion proteins excised were used for mass spectrometer analysis, which confirmed it target protein (Fig. 2b). The purified fusion protein (Fig. 2a) was used to prepare the specific antiserum against NS2. The specificity of the purified polyclonal antibodies was examined by Western blot (Fig. 2a), and the result showed that the obtained antibody could serve as a good tool to detect NS2 protein.

Expressing and identification of His-NS2. a Prokaryotic expression of NS2. Lane M is the protein molecular weight marker; lanes 1, 2, and 3 are the crude extracts of induced E. coli BL21 with pET-30a, without pET-30a, and with pET-30a-NS2; lane 4 is the purified His-NS2 recombinant protein; lane 5 is Western blotting result of purification incubated with the mouse anti-6× His monoclonal antibody; lane 6 is the Western blotting result of purification incubated with prepared polyclonal antiserum against NS2. b Amino acid sequences of NS2. Matched peptide sequences are shown as bold characters
Transcriptional Analysis of VD1-ORF1 (ns2) in Infected Silkworm Midguts
The mRNA expression of VD1-ORF1 was examined by RT-qPCR. The results displayed that the synthesis of VD1-ORF1 could not be detected until 28-h p.i. in low amounts, but in high amounts at late stages of infection (Fig. 3). The relative expression level of VD1-ORF1 mRNA at 40 h p.i. reached a peak during early stages of infection from 28 to 52 h p.i., and then increased rapidly from 52 to 72 h p.i.

Transcriptional analysis of VD1-ORF1(ns2) in infected silkworm midgut. Each RT-qPCR analysis was repeated at least three times for each set of RNA samples. Each point represents the mean value ± SD. The relative amounts of VD1-ORF1 were normalized using the Bm-actinA3 as a standard. The numerical data on y-axis obtained were the relative copy number (relative level) obtained at different time points
Examination of NS2 in Midgut of BmPLV-Z-infected Larvae
To investigate the expression of NS2 in midgut of larvae infected with BmPLV-Z, silkworm midguts were excised from infected larvae at different time points, and the total proteins extracted were used for Western blot analysis using anti-NS2 antibody. Time courses analysis of NS2 expression showed that NS2 could not be detected by anti-NS2 antibody in the midgut of BmPLV-Z-infected larvae at any time point. However, the viral structural protein VP could be recognized by anti-VP antibody in the same samples (data not shown).
Subcellular Localization of NS2 in BmN Cells by Immunofluorescence
To investigate the subcelluar localization of NS2, a transient expression plasmid pFastBac-ie1-ns2 was constructed, and the ns2 was under the control of the BmNPV ie-1 promoter (Fig. 4a). BmN cells were transfected with the plasmid, and untransfected BmN cells were used as control. The fluorescence was observed in the nucleus near to the nuclear membrane at 72 h p.t. in some transfected cells, and ultimately concentrated on the inner nuclear membrane at 96 h p.t. (Fig. 4b).

Subcellular localization of NS2 in BmN cells transfected with pFastBac-ie1-ns2. a The construction of transient expression plasmid pFastBac-ie1-ns2; b Confocal images of BmN cells transfected with pFastBac-ie1-ns2. BmN cells were treated with anti-NS2 antibody at 72 and 96 h p.t., followed by treatment with FITC-conjugated goat anti-rabbit IgG and DAPI (blue) and examined under confocal laser scanning microscope. From left to right: green fluorescence for FITC-treated NS2, DAPI-treated nucleus and the overlay images, cells under norm light. As a control, untransfected cells were also treated as transfected cells and observed with confocal microscope. Scale bar is 10.0 μm
Discussion
The genome of BmPLV contains two linear single-stranded DNAs (VD1 and VD2). Although the host range and tissue tropism of BmPLV, even the composition of virion are similar to the BmDNVs, their replication mechanisms are completely different [3]. Structural analysis of the replicative intermediates of the viral DNAs strongly suggested that BmDNV-2 genomic DNAs did not replicate by self-priming and hairpin transfer mechanisms, which are thought to be used by all parvoviruses [14]. However, the details of the replication mechanism of the BmPLV have not been defined. It is presumed that its replication mechanism is similar to that of adenovirus, which employs a protein-priming initiation mechanism [31].
The virus non-structural proteins are essential for viral replication in parvovirus [6, 8]. Previous studies indicated that NS1 and NS3 of BmPLV-Z showed some homology to that from densovirus and could be involved in the replication of virus [35, 37]. However, BmPLV-Z-NS2 showed no homology to other NS2s in the group of densoviruses and parvoviruses. In this article, we presented the sequence analysis, prokaryotic expression, transcriptional analysis, and subcellular localization to characterize BmPLV-Z-NS2.
Amino acid sequence alignment showed that there were weak homology between NS2 and chromosomal replication initiator protein dnaA and DNA-binding response regulators from several bacteriums. The functions of chromosomal replication dnaA were identified to play an important role in the initiation and regulation of chromosomal replication through binding to the origin of replication [22, 28]. To examine whether sequence-specific binding exists between terminal sequences of viral genome and BmPLV-Z-NS2, a electrophoretic mobility shift assay (EMSA) was used to test the binding ability of purified His-NS2 fusion protein with P1-289 (amplified 289 bp (1–289 bp) of BmPLV-Z-VD1), and no interaction was observed (data not shown). Potential functions of NS2 are still to be identified. In many bacterial genera, DNA-binding response regulator plays roles in transcriptional regulation [26, 29].
In BmPLV-Z genome, there are 294 nts overlap between the 3′-end of the ns2 and the 5′-end of the ns1. Besides, the transcript investigation of BmPLV-Z showed that ns2 and ns1 were in the same transcript, which means NS2 and NS1 could be generated by alternative initiation of ribosome [35]. NS1 proteins from parvoviruses contained the initiator (replicator), protein motifs (motif 2 (I) and motif 3 (II)) which were predicted to be involved in the initiation and termination of rolling circle replication [16], and contains the untwisting enzyme superfamily III motif in C-terminal which also serves as an initiator of viral DNA replication [5, 18]. Results of the previous study showed that purified recombinant 6× His-NS1 protein of BmPLV-Z possesses ATP-binding, ATPase, DNA-binding, and helicase activities, indicating that NS1 appears to participate in the initiation of virus DNA replication [20]. However, the ns1 of BmPLV-Z lacks the N-terminal replication initiator motif (I and II) comparing to that of parvovirus [35]. The BmPLV-Z-NS2 showed weak homology to the chromosomal replication initiator protein dnaA. It was possible that BmPLV-Z-NS2 and NS1 may act synergistically in initiation and regulation of viral DNA replication, which could be explained by the different replication mechanisms of BmPLV.
BmPLV-Z-NS2 contains a putative Asn-glycosylation site. Transcriptional analysis indicated that the ns2 gene was expressed in relative low level at early stages but in very high amounts at late stages of infection. Subcellular localization analysis showed the BmPLV-Z-NS2 ultimately localized to the nuclear membrane in BmN cells, suggesting that it might be associated with integral membrane protein. These aspects are similar to adenovirus death protein (ADP). Previous studies indicated that ADP was a Asn-glycosylated integral membrane protein which ultimately localized to nuclear membrane, and it expressed early but is greatly amplified at late stages of infection [27, 32]. ADP is required for efficient cell lysis and virus release [33, 34]. Overexpression of ADP protein induces cell killing and increases cell lysis and spread of virus [10–12]. In addition, the virus particles of BmPLV-Z are small icosahedral, non-enveloped particles 19–24 nm in diameter, and the package and maturation of virus are also occurred in the nucleus. Therefore, the release of the BmPLV-Z virions is the same dilemma to be faced with, and the death of host cell is essential to the release of virus particles. It is possible that NS2 protein can change the biological properties of nuclear membrane by its localization on the nuclear membrane. Many viruses encode integral membrane proteins which locate in membrane structure with various functions, for instance, NS4A protein of dengus virus and NS4A of bovine viral diarrhea virus are integral membrane protein inducing membrane rearrangement [23]. In general, these proteins contain hydrophobic transmembrane domain as the membrane sorting signal [36], but there is no hydrophobic transmembrane domain in BmPLV-Z NS2 protein. It is possible that BmPLV-Z NS2 localizes to nuclear membrane by different sorting signals or by interactions with other membrane proteins.
The expression of ns2 gene was readily detected by transcriptional analysis, and the NS2 protein was detected by immunofluorescence in cells transiently transfected with plasmids designed to express this protein. However, we did not succeed in our efforts to detect NS2 in the midgut of larvae infected with BmPLV-Z at different time points (data not shown). Most likely, the NS2 protein is synthesized in too small amounts to be detected in the midgut of virus-infected larvae. It is also possible that NS2 protein could be a glycosylated protein and glycosylation of the protein in BmPLV-Z-infected cells might hinder the interaction between the protein and the antiserum. In previous report, the Ad4E3-30K protein was glycosylated, and detection of this protein in cells infected with Adenovirus type 4 was possible only in the presence of tunicamycin which prevented glycosylation [19]. It is possible also that the protein turns over rapidly in BmPLV-Z-infected cells.
References
Abd-Alla A, Jousset FX, Li Y, Fediere G, Cousserans F, Bergoin M (2004) NS-3 protein of the Junonia coenia densovirus is essential for viral DNA replication in an Ld 652 cell line and Spodoptera littoralis larvae. J Virol 78:790–797
Azarkh E, Robinson E, Hirunkanokpun S, Afanasiev B, Kittayapong P, Carlson J, Corsini J (2008) Mosquito densonucleosis virus non-structural protein NS2 is necessary for a productive infection. Virology 374:128–137
Bando H, Choi H, Ito Y, Nakagaki M, Kawase S (1992) Structural analysis on the single-stranded genomic DNAs of the virus newly isolated from silkworm: the DNA molecules share a common terminal sequence. Arch Virol 124:187–193
Bando H, Hayakawa T, Asano S, Sahara K, Nakagaki M, Iizuka T (1995) Analysis of the genetic information of a DNA segment of a new virus from silkworm. Arch Virol 140:1147–1155
Caruthers MJ, McKay DB (2002) Helicase structure and mechanism. Curr Opin Struct Biol 12:123–133
Cotmore SF, Tattersall P (1988) The NS-1 polypeptide of minute virus of mice is covalently attached to the 5′ termini of duplex replicative-form DNA and progeny single strands. J Virol 62:851–860
Cotmore SF, Tattersall P (1990) Alternate splicing in a parvoviral nonstructural gene links a common amino-terminal sequence to downstream domains which confer radically different localization and turnover characteristics. Virology 177:477–487
Cotmore SF, Christensen J, Nuesch JP, Tattersall P (1995) The NS1 polypeptide of the murine parvovirus minute virus of mice binds to DNA sequences containing the motif [ACCA]2–3. J Virol 69:1652–1660
Cotmore SF, D’Abramo AM Jr, Carbonell LF, Bratton J, Tattersall P (1997) The NS2 polypeptide of parvovirus MVM is required for capsid assembly in murine cells. Virology 231:267–280
Doronin K, Toth K, Kuppuswamy M, Ward P, Tollefson AE, Wold WS (2000) Tumor-specific, replication-competent adenovirus vectors overexpressing the adenovirus death protein. J Virol 74:6147–6155
Doronin K, Kuppuswamy M, Toth K, Tollefson AE, Krajcsi P, Krougliak V, Wold WS (2001) Tissue-specific, tumor-selective, replication-competent adenovirus vector for cancer gene therapy. J Virol 75:3314–3324
Doronin K, Toth K, Kuppuswamy M, Krajcsi P, Tollefson AE, Wold WS (2003) Overexpression of the ADP (E3–11.6K) protein increases cell lysis and spread of adenovirus. Virology 305:378–387
Eichwald V, Daeffler L, Klein M, Rommelaere J, Salome N (2002) The NS2 proteins of parvovirus minute virus of mice are required for efficient nuclear egress of progeny virions in mouse cells. J Virol 76:10307–10319
Hayakawa T, Asano S, Sahara K, Iizuka T, Bando H (1997) Detection of replicative intermediate with closed terminus of Bombyx densonucleosis virus. Arch Virol 142:1–7
Hayakawa T, Kojima K, Nonaka K, Nakagaki M, Sahara K, Asano S, Iizuka T, Bando H (2000) Analysis of proteins encoded in the bipartite genome of a new type of parvo-like virus isolated from silkworm—structural protein with DNA polymerase motif. Virus Res 66:101–108
Ilyina TV, Koonin EV (1992) Conserved sequence motifs in the initiator proteins for rolling circle DNA replication encoded by diverse replicons from eubacteria, eucaryotes and archaebacteria. Nucleic Acids Res 20:3279–3285
Kapitonov VV, Jurka J (2006) Self-synthesizing DNA transposons in eukaryotes. Proc Natl Acad Sci U S A 103:4540–4545
Koonin EV (1993) A common set of conserved motifs in a vast variety of putative nucleic acid-dependent ATPases including MCM proteins involved in the initiation of eukaryotic DNA replication. Nucleic Acids Res 21:2541–2547
Li Y, Wold WS (2000) Identification and characterization of a 30K protein (Ad4E3-30K) encoded by the E3 region of human adenovirus type 4. Virology 273:127–138
Li G, Sun C, Zhang J, He Y, Chen H, Kong J, Huang G, Chen K, Yao Q (2009) Characterization of Bombyx mori parvo-like virus non-structural protein NS1. Virus Genes 39:396–402
Lv M, Yao Q, Wang Y, Liu X, Liu H, Huang G, Chen K, Zhang J, Li X (2010) Identification of structural proteins of Bombyx mori parvo-like virus (China Zhenjiang isolate). Intervirology 54:37–43
Marczynski GT, Shapiro L (1992) Cell-cycle control of a cloned chromosomal origin of replication from Caulobacter crescentus. J Mol Biol 226:959–977
Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R (2007) The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J Biol Chem 282:8873–8882
Naeger LK, Cater J, Pintel DJ (1990) The small nonstructural protein (NS2) of the parvovirus minute virus of mice is required for efficient DNA replication and infectious virus production in a cell-type-specific manner. J Virol 64:6166–6175
Naeger LK, Salome′ N, Pintel DJ (1993) NS2 is required for efficient translation of viral mRNA in minute virus of mice-infected murine cells. J Virol 67:1034–1043
Rozhdestvenskaya AS, Totolian AA, Dmitriev AV (2010) Inactivation of DNA-binding response regulator Sak189 abrogates beta-antigen expression and affects virulence of Streptococcus agalactiae. PLoS One 5:e10212
Scaria A, Tollefson AE, Saha SK, Wold WS (1992) The E3-11.6K protein of adenovirus is an Asn-glycosylated integral membrane protein that localizes to the nuclear membrane. Virology 191:743–753
Skarstad K, Boye E (1994) The initiator protein DnaA: evolution, properties and function. Biochim Biophys Acta 1217:111–130
Stock AM, Robinson VL, Goudreau PN (2000) Two-component signal transduction. Annu Rev Biochem 69:183–215
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tijssen P, Bergoin M (1995) Densonucleosis viruses constitute an increasingly diversified subfamily among the parvoviruses. Virology 6:347–355
Tollefson AE, Scaria A, Saha SK, Wold WS (1992) The 11,600-MW protein encoded by region E3 of adenovirus is expressed early but is greatly amplified at late stages of infection. J Virol 66:3633–3642
Tollefson AE, Ryerse JS, Scaria A, Hermiston TW, Wold WS (1996) The E3-11.6-kDa adenovirus death protein (ADP) is required for efficient cell death: characterization of cells infected with adp mutants. Virology 220:152–162
Tollefson AE, Scaria A, Hermiston TW, Ryerse JS, Wold LJ, Wold WS (1996) The adenovirus death protein (E3-11.6K) is required at very-late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J Virol 70:2296–2306
Wang YJ, Yao Q, Chen KP, Wang Y, Lu J, Han X (2007) Characterization of the genome structure of Bombyx mori densovirus (China isolate). Virus Genes 35:103–108
Weiskircher E, Aligo J, Ning G, Konan KV (2009) Bovine viral diarrhea virus NS4B protein is an integral membrane protein associated with Golgi markers and rearranged host membranes. Virol J 6:185
Yin H, Yao Q, Guo Z, Bao F, Yu W, Li J, Chen K (2008) Expression of non-structural protei NS3 gene of Bombyx mori densovirus (China isolate). J Genet Genomics 35:239–244
Zhang J, Li G, Chen H, Li X, Lv M, Chen K, Yao Q (2010) Molecular cloning and expression of key gene encoding hypothetical DNA polymerase from B. mori parvo-like virus. Genet Mol Biol. doi:10.1590/S1415-47572010005000083
Acknowledgments
This study was supported by the National Nature Science Foundation of China (30871826) and the National Nature Science Foundation of China (31000080).
Author information
Authors and Affiliations
Corresponding author
Additional information
Fenghua Wang and Zhaoyang Hu contributed equally to this study.
Rights and permissions
About this article
Cite this article
Wang, F., Hu, Z., He, Y. et al. The Non-Structural Protein NS-2 of Bombyx mori Parvo-like Virus is Localized to the Nuclear Membrane. Curr Microbiol 63, 8–15 (2011). https://doi.org/10.1007/s00284-011-9933-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-011-9933-1