
Abstract
The genome of Bombyx mori densovirus (China isolate), termed as BmDNV-3, is composed of two kinds of different single-stranded linear DNA molecules (VD1 and VD2). In this study, the viral DNA molecules were purified and cloned into pUC119 vector, and the complete nucleotide sequence was determined. Sequence analysis showed that VD1 genome consisted of 6,543 nts including inverted terminal repeats (ITRs) of 224 nts, and VD2 genome consisted of 6,022 nts including ITRs of 524 nts. Comparison of the complete genome sequence between BmDNV-3 and BmDNV-2 (Yamanashi isolate) showed an identity of 98.4% in VD1 and 97.7% in VD2, with a total number of 228 bp substitutions, 11 bp deletions and 3 bp insertions found in BmDNV-3. A single nucleotide “A” deletion at nt 1589 in BmDNV-3 caused a frame shift mutation and brought about a premature stop codon, thus dividing VD2 of BmDNV-3 into two ORFs (named VD2 ORF1a and VD2 ORF1b) within that region, while there was only one ORF (named VD2 ORF1) in the corresponding region of BmDNV-2 (Yamanashi isolate). Comparative polymorphisms of ORFs and ITR regions of the two viral genomes showed that highly variable regions were mainly located in VD1 ORF3, VD1 ORF4, VD2 ORF2, and ITRs of BmDNV-3. Northern blots analysis revealed that VD1 had 1.1 kb and 1.5 kb transcripts from the left half of its plus strand, and one transcript about 3.3 kb from the right half of its minus strand. Sequencing of 3′ and 5′ RACE products showed that the 1.1 kb transcript started at nt 290 and ended at nt 1437, the 1.5 kb transcript started at nt 1423 and ended at nt 2931, and the 3.3 kb transcript started at nt 6287 and ended at nt 2922. These results help us to further understand the variation between different DNV genera and its possible causes, providing clues for studying the evolutionary history of densoviruses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The family Parvoviridae includes two subfamilies: the Parvovirinae, members of which infect vertebrates, and the Densovirinae, members of which infect invertebrates [1]. Similar to other parvovirus, densoviruses (DNVs) are small icosahedral, no-enveloped particles of 18–26 nm in diameter and their genomes consist of a 4–6.5 kb single-stranded linear DNA equimolecularly encapsidated as plus and minus strands in separate virions. DNVs can infect a wide range of insects including Lepidoptera, Diptera, Orthoptera and Dictyoptrea [2] and lead to death of the host in most cases, but they are not able to infect tissues of vertebrates [3], thus making them potential biological agents for controlling main agricultural pests [4, 5].
BmDNV is a viral agent causing flacherie disease of the silkworm. Many DNV isolates have been obtained from silkworm, they were termed as BmDNV-1 (Ina isolate), BmDNV-2 (Saku isolate and Yamanashi isolate), BmDNV-3 (China isolate) and BmDNV-4 (Kenchu insolate) [6]. BmDNV-2 was parvo-like virus containing small, single-stranded linear DNA genome. Unlike BmDNV-1 and other parvovirus, BmDNV-2 has bipartite genomes and no common terminal palindromic sequences, implying that it is a new type of virus with unique replication mechanisms [7, 8]. According to ICTV Virus Taxonomy report, BmDNV-2 has not been definitely classified so far. Since the characteristic trait of BmDNV-2 was quite similar to those of densovirus, it was tentatively designated as densovirus [9, 10].
BmDNV-3 and BmDNV-2 (Yamanashi isolate) both have bipartite genomes and have the same host species, but their virulence and the symptoms of infection are different. For example, BmDNV-2 (Yamanashi isolate) almost exclusively infects the columnar cells of midgut epithelium, while BmDNV-3 can infect the columnar cells of midgut epithelium during the early stage of infection and is able to infect the cup shaped cells of midgut epithelium during late stage of infection. In this study, the genome of BmDNV-3 was completely sequenced, analyzed and compared with the genome of BmDNV-2 (Yamanashi isolate). In addition, we investigated its transcription strategy upon infection through northern blots analysis and the 3′, 5′-rapid amplification of cDNA ends (RACE), and found that BmDNV-3 VD1 has an ambisense genome like genus Densovirus [11, 12].
Materials and methods
Virus and viral DNA
Densoviruses was propagated with fifth instar larvae of BmDNV-3 susceptible silkworm strain Huaba. Midgut was taken from the larvae that showed obvious symptoms and was homogenized in TEN buffer (50 mM Tris–HCl, 1 mM EDTA, 0.1 M NaCl, pH 7.5) using a tissue homogenizer at 2,000 rpm for 2 min. To purify the viral particles, a 40% (w/w) CsCl density-gradient ultracentrifugation was performed at 45,000 rpm (170,000g) for 16 h at 4°C in a Beckman VTi50 rotor. The purified viral particles were suspended in high salt solution containing 10 mM Tris–HCl of pH 7.5, 100 mM NaCl, 15 mM MgCl2, 0.2% SDS and 0.5 mg/ml proteinase K and were incubated at 56°C for 4 h. Then, total DNAs of the viral particles were extracted with phenol/chloroform (1:1) three times and with chloroform one time, and finally recovered by means of ethanol precipitation.
Cloning of viral DNA
In live BmDNV-3 viruses, VD1 and VD2 are present in separate particles and have their own complementary strands both of which exist in single-strand status. In the extraction process, the complementary strands hybridized with each other and might left short single stranded segments at both ends. The viral DNA hybridized upon extraction was blunt ended using T4 DNA polymerase (10 U per 300 ng of DNA, and 35 μM dNTPs in 50 mM Tris–HCl, 5 mM MgCl2, and 10 mM DTT), heated for 15 min at 70°C, and then precipitated with ethanol. Endonuclease digestion, gel electrophoresis, and fragment elution from agarose gel were carried out according to the supplier’s instructions. Restriction fragments were obtained by digestion with HindIII and cloned into HindIII/SmaI-digested pUC119, or cloned into HindIII-digested pUC119. Terminal fragments were obtained and cloned into SmaI-digested pUC119. The recombinant plasmids were transformed into E. coli DH5? for replication.
Sequencing and sequence analysis
The cloned viral DNAs were sequenced from both directions using the GenomelabTM DTCS-QUICK START KIT (BECKMAN COULTER) in CEQTM 8000 Genetic Analysis System according to the manufacturer’s instructions. Multiple clones were sequenced to verify the sequences. In order to guarantee the accuracy, sequencing was performed to clones that had a 5- to 7-fold coverage to the whole genome. The nucleotide sequence was processed with MegAlign program of DNAStar (LagerGene, USA). ORF characterization and amino acid sequence deduction were performed using the SeqBuilder program of DNAStar. Comparative analysis including multiple alignments of nucleotide and amino acid sequences was performed with clustalW method. Homology searches were conducted against the NCBI nucleotide and protein databases. Transcriptional control signals were assessed by Neural Network Promoter Prediction (NNPP) software [13, 14].
Northern blots
Total RNA was isolated from midgut of the infected larvae of BmDNV-3 susceptible silkworm strain Huaba at 72 h posterior to virus treatment. The midgut was submerged in a fresh 0.5% diethyipyrocarbonate (DEPC) solution for 30–60 min before being frozen in liquid nitrogen, and subjected to RNA purification using an RNA isolation kit (QIAGEN). The total RNA was submitted to electrophoresis in 1.2% formaldehyde agarose gels and blotted onto positively charged nylon membranes (Roche, USA). The blotted membranes were incubated overnight at 42°C with two 32P-labeled probes obtained using a probe preparation kit (Takara) based on the sequence from nts 1116 to 2573 and the sequence from nts 4451 to 5414 of the BmDNV-3 VD1 genome.
Mapping the 3′ and 5′ ends of viral transcripts
A 3′, 5′-RACE kit (BD Biosciences Clontech, USA) was used to characterize the 3′-end and 5′-end of the transcripts. From the open reading frames obtained through sequence analysis, the most probable locations of the transcript were predicted. Using DNV-specific primers and the 3′-RACE and 5′-RACE universal primers, PCR amplification was carried out according to manufacturer’s instruction of the RACE kit. The PCR products were subject to cloning and sequencing analyses as described above.
Results and discussion
Analysis of the viral genome
The complete nucleotide sequence of the viral genome has been determined. The sequence data was assembled into two contiguous sequences named VD1 (6,543 nts) and VD2 (6,022 nts). The GenBank accession numbers are DQ017268 and DQ017269, respectively. They were in accordance with a previous estimate of 6.6 and 6.0 kb, respectively, based on the DNA agarose gel electrophoresis and restriction enzyme digestion experiment (unpublished data). Base composition of VD1 was 68.2% A+T, and that of VD2 was 72.1% A+T. Both VD1 and VD2 had a high content of A+T.
VD1 had ITRs of 224 nts, and VD2 had ITRs of 524 nts. In addition, VD1 and VD2 had an identical 53 nts sequence in their ITRs ends. However, the terminal palindromic sequence commonly seen in many other densoviruses was not found. Structural analysis of both VD1 and VD2 ITRs suggested that the single stranded viral DNA molecules could form a “panhandle structure”. The 5′ terminal sequences of 224 nucleotides of each single strand VD1 DNA and the 224 nucleotides at its 3′ end were complementary. This also applied for 524 nucleotides at the termini of VD2 DNA. The GC content of BmDNV-3 ITRs (VD1, 40.5%; VD2, 42.4%) is higher than that of the entire virus genome (VD1, 31.8%; VD2, 27.9%), which may contribute to the stability of the ITRs secondary structure.
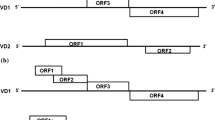
Using methionine-initiated codon (ATG), stop codons (TAA, TAG and TGA), at least 100 aa long and having minimal overlap as the criteria for searching ORFs, seven putative ORF regions were identified in BmDNV-3 genome. The locations, orientations, and sizes of the predicted ORFs are shown in Fig. 1A. It was discovered that VD1 contained four ORFs (VD1 ORF1, VD1 ORF2, VD1 ORF3, and VD1 ORF4) and VD2 had three ORFs (VD2 ORF1a, VD2 ORF1b, and VD2 ORF2). The overall organization of VD1 genome is similar to that of JcDNV and MlDNV [12, 15], and that of VD2 genome is similar to that of BmDNV-1 [16, 17].

Comparison of genome organization of (A) BmDNV-3 and (B) BmDNV-2 (Yamanashi isolate). The horizontal bar represents the genomic DNA molecules. Open boxes above the horizontal bars represent the ORFs found in the genome sequence and those below the horizontal bars represent ORFs found in the complementary
Comparison of BmDNV-3 and BmDNV-2 (Yamanashi isolate)
According to previous reports [7, 8], the genome of BmDNV-2 (Yamanashi isolate) has six ORFs in all (shown in Fig. 1B), and consists of 6542 nts in VD1, 6031 nts in VD2 (GenBank AB033596). The analysis of the BmDNV-3 DNA genome showed that this virus was closely related to BmDNV-2 (Yamanashi isolate). The two virus genomes shared 98.4% identity in VD1 nucleotide sequence, and 97.7% identity in VD2 nucleotide sequence, with a total of 228 bp substitutions, 3 bp insertions, and 11 bp deletions in BmDNV-3. The major difference between BmDNV-3 and BmDNV-2 (Yamanashi isolate) in sequence is shown in Table 1. BmDNV-3 genome is 1 bp larger than that of BmDNV-2 (Yamanashi isolate) in VD1, and 9 bp smaller than that of BmDNV-2 (Yamanashi isolate) in VD2. Among 228 bp substitutions, 176 bp substitutions occurred in the coding regions of seven ORFs and resulted in a total number of 43 aa residue changes in five ORFs (VD1 ORF3, VD1 ORF4, VD2 ORF1a, VD2 ORF1b, VD2 ORF2). 127 bp substitutions were silent among the 176 bp substitutions in ORFs. A single nucleotide “A” deletion in VD2 ORF1a (position nt 1583) in BmDNV-3 resulted in a frame shift mutation that brought about a premature stop codon. Therefore, BmDNV-3 VD2 has two ORFs (VD2 ORF1a and ORF1b) in this region compared to VD2 of BmDNV-2 (Yamanashi isolate), which has only one ORF (VD2 ORF1) in the corresponding region. In order to clarify whether the frame shifts mutation was caused by sequencing errors, the region was further PCR amplified and sequenced. The result confirmed that our VD2 ORF1a sequence was correct. In addition, there are many variations in the ITRs, for example, VD2 of BmDNV-3 contains 524 nts, while that of BmDNV-2 (Yamanashi isolate) contains 537 nts.
Comparison of the BmDNV-3 and BmDNV-2 (Yamanashi isolate) genomes showed that there were highly variable regions (VD1 ORF3, VD1 ORF4, VD2 ORF1b, and ITRs) and the frame shift mutation, being potential contributors for the variation between BmDNV-3 and BmDNV-2 (Yamanashi isolate). These variations in sequence and length might result from different evolutionary environments. On the other hand, VD1 ORF1 and VD1 ORF2 were found to be highly conserved in both genomes. Their deduced amino acid sequences showed an identity of 100%.
Transcript mapping
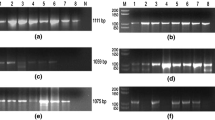
The transcripts of the virus were mapped to examine its expression strategy. Northern blots analysis revealed that the left-half of VD1 plus strand contained two transcripts with 1.1 and 1.5 kb, respectively, and the right-half of VD1 minus strand contained one transcript of 3.3 kb long (Fig. 2B). Sequencing of their 3′ and 5′ ends showed that transcription of the 1.1 kb transcript started at nt 290 and ended at nt 1437. This transcript consisted of two ORFs (VD1 ORF1 and VD1 ORF2). VD1 ORF1 started at nt 331 and ended at nt 691. VD1 ORF2 started at nt 474 and ended at nt 1424. Both VD1 ORF1 and VD1 ORF2 were contained completely within this transcript. There was no splicing being observed to this transcript. The 1.5 kb transcript was found to started at nt 1423 and ended at nt 2931, and the 3.3 kb transcript started at nt 6287 and ended at nt 2922 (Fig. 2A). Therefore, the 1.5 kb transcript and the 3.3 kb transcript overlapped for 10 nts at the 3′ ends (antisense RNAs) at 50 map units. These results indicate that this genus employs a radically different transcription strategy from that of the other reported DNVs.

Characteristics of BmDNV-3 VD1 genome structure. (A) Genome organization of BmDNV-3 VD1, the horizontal bar represents the genomic DNA molecules VD1, shaded boxes above the horizontal bars show the ORFs found in the genome sequence and the shaded box below the bar represents the ORF found in the complementary sequence. Abbreviated sequence of the BmDNV-3 VD1 genome illustrated its characteristic properties. The functional promoters are indicated with TATA boxes. (B) RNA blot was hybridized against the single-stranded probes. Lanes 1–2: Northern blots with 1.458 kb and 0.964 kb probes, respectively
To further understand the genomic structure of VD1, we also analyzed the distribution of initiation signals and polyadenylation signals on VD1 sequence. Using NNPP algorithm for predicting putative transcription regulatory sequences, we found that one TATA box (TATATAA) was located between nt 260 and nt 266, 21 nts upstream of the start codon of VD1 ORF1. This putative promoter was believed to control the transcription of VD1 ORF1 and VD1 ORF2. Another TATA box was located between nt 1402 and nt 1408, which possibly controlled the transcription of VD1 ORF3. A third TATA box was located on the minus strand between nt 6301 and nt 6307, which possibly controlled the transcription of VD1 ORF1. Polyadenylation signals (AATAAA) were found among nt 1406–1411 and nt 2911–2916 on the plus strand, respectively. The complementary strand contained a polyadenylation signal at nt 2943–2938.
Homology among parvovirus and densovirus amino acid
In VD1, BLAST analyses of the predicted 126 aa gene of VD1 ORF1 and 316 aa gene of VD1 ORF2 revealed some homology with replication initiator protein and putative major nonstructural protein of parvovirus, respectively. A stretch of 60 aa (aa position 52–121) of VD1 ORF1 aligned and shared 27% identity with erythrocyte binding protein of Plasmodium falciparum (GenBank accession No. 34495238), 87 aa (aa position 25–111) shared 25% identity with chromosomal replication initiator protein of Pseudomonas putida. A stretch of 226 aa (aa position 89–314) of VD1 ORF2 shared 21% identity with the nonstructural protein of Penaeus stylirostris densovirus (PstDNV) (GenBank accession No. AF273215), 123 aa (aa position 144–266) shared 27% identity with the nonstructural protein of Porcine parvovirus (GenBank accession No. M37899), and 241 aa (aa position 58–298) shared 22% identity with NTP-binding and helicase domains of adeno-associated virus (GenBank accession No. AF043303). Amino acids 178–296 of VD1 ORF2 contained a stretch of conserved sequence characteristic of NTP-binding and helicase domains of the nonstructural protein (NS-1) shared by all parvovirus [18] (shown in Fig. 3). In vertebrate parvovirus, NS-1 was found to be covalently bound to the 5′ terminus of viral DNA [19] and to influence packaging of the viral DNAs [20]. It was believed that NTP-binding and helicase domains play a central role in NS-1 [21]. Based on these sequence similarities, we suggested that VD1 ORF1 and VD1 ORF2 encoded the viral nonstructural proteins. The ORF4 was presumed to encode nonstructural protein, since the ORF4 contained sequences conserved among various DNA polymerases, and showed an evolutionary relationship with the DNA polymerases involved in protein-primed replication [10]. Judging from the results of peptide mapping of major structural proteins, and amino acid sequencing of BmDNV-2 capsid proteins [10], it was obvious that BmDNV-3 VD1 ORF3 coded for major structural proteins of the virus.

Conserved sequence motifs in viral proteins of vertebrate and invertebrate parvoviruses. Region of NS-1 with highly conserved 119-aa sequence that was aligned using clustalW. NTP-binding and helicase domains[ A], [B], and [C] are also present in the BmDNV-3 VD1 ORF2. Numbers in parentheses indicate the position of sequence for each viral protein shown in the figure. GenBank accession numbers are shown in brackets
It was reported that the synthesis of GmDNV and MlDNV capsid proteins might be initiated from one of four codons within one ORF with a mechanism termed leaky scanning [11, 12], and the translation products contained a conserved region of phospholipase A2 domain. Although BmDNV-3 VD1 ORF3 coded for structural protein, the leaky scanning mechanism and the conserved region of phospholipase A2 domain were not present in VD1 ORF3 (data not shown). So BmDNV-3 VD1 should be different feature in capsid proteins.
References
G. Fédière, Contrib. Microbiol. 4, 1 (2000)
M. Bergoin, P. Tijssen, The Insect Viruses. (Plenum, New York, 1998) p. 141
M. El-Far, Y. Li, G. Fédière, S. Abol-Ela, P. Tijssen, Virus Res. 99, 17 (2004)
J. Corsini, B. Afanasiev, I.H. Maxwell, J.O. Carlson, Adv. Virus Res. 47, 303 (1996)
J. Carlson, B. Afanasiev, E. Suchman, Insect Transgenesis: Methods and Applications. (CRC Press, NY, 2000) p. 139
P. Tijssen, M. Bergoin, Semin. Virol. 6, 347 (1995)
T. Hayakawa, S. Asano, K. Sahara, T. Iizuka, H. Bando, Arch. Virol. 142, 393 (1997)
T. Hayakawa, K. Kojima, K. Nonaka, M. Nakagaki, K. Sahara, S.I. Asano T. Iizuka, H. Bando, Virus Res. 66, 101 (2000)
H. Bando, H. Choi, Y. Ito, M. Nakagaki, S. Kawase, Arch. Virol. 124, 187 (1992)
H. Bando, T. Hayakawa, S. Asano, K. Sahara, M. Nakagaki, T. Iizuka, Arch. Virol. 140, 1147 (1995)
P. Tijssen, Y. Li, M. EI-Far, J. Szelei, M. Letarte, Z. Zádori, J. Virol. 77, 10357 (2003)
G. Fédière, M. Ei-Far, Y. Li, M. Bergoin, P. Tijssen, Virology 320, 181 (2004)
U. Ohler, S. Harbeck, H. Niemann, E. Noth, M.G. Peese, Bioinfoprmatics 15, 362 (1999)
M.G. Reese, F.H. Eeckman, D. Kulp, D. Haussler, J. Comput. Biol. 4, 311 (1997)
B. Dumas, M. Jourdan, A.M. Pascaud, M. Bergoin, Virology 191, 202 (1992)
H. Bando, J. Kusuda, T. Gojobori, T. Maruyama, S. Kawase, J. Virol. 61, 553 (1987)
Y. Li, Z. Zádori, H. Bando, R. Dubue, G. Fédière, J. Szelei, P. Tijssen, J. Gen. Virol. 82, 2821 (2001)
B.N. Afanasiev, E.E. Galyov, L.P. Buchatsky, Y.V. Kozlov, Virology 185, 323 (1991)
S.F. Cotmore, P. Tattersall, J. Virol. 62, 851 (1988)
S.F. Cotmore, P. Tattersall, J. Virol. 63, 3902 (1989)
H. Shike, A.K. Dhar, J.C. Burns, C. Shimizu, F.X. Jousset, K.R. Klimpel M. Bergoin, Virology 277, 167 (2000)
Acknowledgments
We are grateful to Professor K. W. Huang at Institute of Sericulture, Chinese Academy of Agricultural Sciences for providing the virus. This work was supported by a grant from Academy Natural Science Foundation of Jiangsu Province, China (project no. BK2006074) and National Basic Research Program of China (project no. 2005CB121000).
Author information
Authors and Affiliations
Corresponding author
Additional information
Yong Jie Wang and Qin Yao have contributed equally to this study.
Rights and permissions
About this article
Cite this article
Wang, Y.J., Yao, Q., Chen, K.P. et al. Characterization of the genome structure of Bombyx mori densovirus (China isolate). Virus Genes 35, 103–108 (2007). https://doi.org/10.1007/s11262-006-0034-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-006-0034-3