
Abstract
Debaryomyces hansenii is a spoilage yeast able to grow in a variety of ecological niches, from seawater to dairy products. Results presented in this article show that (i) D. hansenii has an inherent resistance to H2O2 which could be attributed to the fact that this yeast has a basal catalase activity which is several-fold higher than that observed in Saccharomyces cerevisiae under the same culture conditions, (ii) D. hansenii has two genes (DhCTA1 and DhCTT1) encoding two catalase isozymes with a differential enzymatic activity profile which is not strictly correlated with a differential expression profile of the encoding genes.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
All aerobically growing organisms are subjected to oxidative stress which is caused by exposure to partially reduced forms of molecular oxygen, known as reactive oxygen species (ROS). These are highly reactive molecules that can damage cellular constituents such as DNA, lipids, and proteins. Exposure to ROS is a consequence of the aerobic life; organisms have evolved mechanisms to protect their components against ROS. Oxidant defense systems have been widely studied [3, 18, 40], and it has been found that after exposure to oxidative stress cells elicit a number of inducible adaptive responses which result in protection from oxidants, such as H2O2, superoxide anion, and lipid peroxidation products. The oxidative stress responses appear to be regulated, at least in part, at the transcriptional level and there is a considerable overlap between those induced by oxidative stress and those provoked by other stressful conditions, allowing the cell to integrate a combined response when cells are simultaneously affected by various environmental insults [20, 43, 48]. Yeasts can proliferate in the presence of many disturbances in its growth media, and can withstand the presence of stressors in different ways. In order to cope with the ROS that are generated during respiration when O2 is incompletely reduced and during fatty acid metabolism in the peroxisome [24, 40, 45], Saccharomyces cerevisiae displays an enzymatic and a non-enzymatic response. The enzymatic adaptation is carried out through the concerted action of superoxide dismutase and catalase proteins, which degrade ROS up to oxygen and water [5, 44]. It has been shown that when S. cerevisiae is cultured in rich media (YP) containing ethanol as a carbon source, catalase total activity is 12-fold higher than when grown in the presence of glucose (88.1 ± 21.7 and 7.27 ± 1.69, respectively) [23].
Saccharomyces cerevisiae has two catalases. CTA1-encoded catalase A that is confined to peroxisomes which is involved in the degradation of the ROS generated during fatty acid degradation [52], and the CTT-encoded catalase T, which is a cytoplasmic enzyme, that contends with the H2O2 that reaches the cytoplasm [38, 42]. It has been reported [16] that only the double null mutant devoid of Cta1 and Ctt1 is sensitive to oxidative stress, while single mutants show no stress-related phenotype, indicating that these activities are at least partially redundant.
Although S. cerevisiae has been considered to be the best known and studied eukaryotic organism, the sequencing of a large number of yeasts has put forward the possibility that other biological models could provide important information which could result in biotechnological applications which cannot be derived from studies with the budding yeast. For many organisms the exposure to high environmental osmolarity leads to dehydratation of cells and decreased viability. To overcome this, some cells have developed mechanisms to adapt the critical osmotic changes in their environments. Osmoregulation is a complex cellular response and many efforts have been made to understand the molecular mechanism of this phenomenon [5, 19, 47]. In Debaryomyces hansenii it has been shown that glycerol and arabinitol are the main osmolytes involved in salt resistance response [46]. D. hansenii is a halotolerant yeast, whose response to oxidative stress has been recently started to be analyzed [26]. Considering that, it has developed mechanisms to withstand hyperosmotic environments; the study of this yeast’s response to oxidative stress could put forward the existence of novel mechanisms that might simultaneously provide resistance to both osmotic and oxidative stress. D. hansenii is found as contaminant of brine food that displays the capacity to grow in media containing a wide range of salt concentrations, including seawater, from where it was first isolated [29]. It is also found on meat, wine, hams, dairy products, fruits, and soil. It could thus be considered that D. hansenii has evolved protection systems promoting a general adaptation to various stresses, which are not present in stress-sensitive organisms like S. cerevisiae, which is able to transiently respond to stress but not adapted to the continuous presence of high osmolyte concentration [4, 36].
Results presented in this article show that D. hansenii has two catalase-encoding genes (DhCTA1 and DhCTT1), that DhCta1 activity is selectively increased when the yeast is grown on YPEthanol (YPE), while DhCtt1 activity is only observed during stationary phase. Expression analysis showed that DhCTT1 and DhCTA1 are both expressed under fermentative conditions while DhCTA1 is most abundant under respiratory metabolism, and that NaCl regulates expression of both genes under different circumstances which not correlate with the salt-dependent modulation of catalase activity, revealing a potentially dual role for this metabolite. DhCta1 purification showed that this enzyme displays kinetic behavior similar to that of catalases purified from other microorganisms [9, 22, 25].
Materials and Methods
Strains and Growth Conditions
Debaryomyces hansenii Y7426 (kind gift of A. Peña from IFC, UNAM) and S. cerevisiae S288C wild-type strains were used throughout this study. Cells were routinely grown in rich media containing 1% yeast extract, 2% peptone, and 2% glucose (YPD) or 2% ethanol (YPE) when specified. Growth was monitored by measuring optical density at 600 nm. S. cerevisiae was routinely pre-grown overnight on YPD and D. hansenii on YPD plus 0.6 M sodium chloride (NaCl) at 30°C with shaking (180 rpm). Both strains were inoculated at 0.05 OD600nm in the specified growth media. Solid media were prepared by the addition of 2% agar. In this study, exponential growth phase was considered to be that when cultures reached an OD600nm ≈ of 1.0 (around 10 h for S. cerevisiae and 25 h for D. hansenii). Stationary phase was defined as the growth reached after 72 h of continuous culture on rich media.
Preparation of Cell-Free Extracts and Catalase Determinations
Cells were collected by centrifugation from the pertinent cultures, washed in sterile deionized water and resuspended in 0.05 M sodium phosphate buffer, pH 7.0 [6]. Cells were grinded by vortexing with glass beads and the suspension was centrifuged for 10 min at 13,000g at 4°C. The extract was kept on ice and assayed for enzyme activity, a representative sample was used for PAGE and zymograms, and the rest of the crude extract was used for enzyme purification.
H2O2 Sensitivity Assays
To test cell viability after an oxidative shock with H2O2 [8], culture tubes containing YPD media with and without 0.6 M NaCl were supplemented with 0–30 mM hydrogen peroxide concentrations, and inoculated at an OD600nm = 0.5 with yeast cells (S. cerevisiae and D. hansenii) collected from the exponential or stationary growth phases. The inoculated tubes were cultured for 180 min in the presence of H2O2 at 30°C with shaking. After the oxidative treatment, H2O2 was removed by centrifugation, cells were suspended in distilled H2O, the OD was adjusted to an OD600nm of 0.5, and the cultures were then serially diluted. Each dilution was spotted into YPD plates, and the plates were incubated at 30°C for 5 days.
RNA Isolation and Northern Blotting
The RNA extraction was performed using aurintricarboxylic acid (ATA) as nucleases inhibitor [13]. D. hansenii cells precultured in YPD-0.6 M NaCl were inoculated at OD600nm = 0.05 in YPD, YPE and YPE-0.6 M NaCl. The cultures were incubated at 30°C with shaking and yeasts were grown until they reached either exponential or stationary growth phases. 15 ml of cells was treated with 300 μl of chilled 1.0 M sodium azide (NaN3) and gently mixed. Samples were centrifuged for 7 min at 3,000 rpm. Pellets were resuspended in 1 ml of 20 mM NaN3 and transferred to a microcentrifuge tube, quickly spinned and decanted. Cells were resuspended in 100 μl of 20 mM NaN3 and 200 μl of phenol, pH 8.0, 100 μl of ATA lysis buffer [1% SDS, 2 mM ATA, 0.5× LET and 0.5 mg/μl dithiothreitol (DTT), all in H2O–DEPC treated] and 0.3 g of acid clean and sterilized glass beads were added (5× LET buffer is 50 mM LiCl, 100 mM DTT in 0.5 M Tris buffer, pH 7.4). Samples were vigorously vortexed for 3 min, 200 μl LET 1× added, mixed, and centrifuged for 15 min at 13,000g. RNAs were precipitated by adding absolute ethanol to the supernatant and freezed for at least 1 hour, washed with 70% ethanol with H2O–DEPC treated and the pellet were resuspended in 1 mM ATA. RNA was separated in a 1.5% agarose, 7% formaldehyde gel, transferred to a nylon membrane, washed, and fixed. Membrane was prehybridized for 2 h at 65°C in 7% SDS and 0.5 M Na2P04, pH 7.2. Radiolabeled probes were prepared by random primer labeling with α-32P-dCTP. Hybridization was performed at 65°C in 7% SDS and 0.5 M Na2P04, pH 7.2. The blot was washed twice in 2× SSC (1× SSC is 0.15 M NaCl and 0.015 M sodium citrate) containing 1% SDS for 30 min each and twice in 0.2× SSC and 1% SDS for 30 min each at 65°C. Filters were sequentially hybridized with the different probes for 18 h, followed by washing with 2× SSC containing 0.1% SDS at 65°C for 30 min. Signal was quantified using Typhoon 840 and ImageQuant TL.
Catalase Activity
Catalase activity was determined by a method adapted from Aebi [1]. Briefly, a sample (1–50 μl) of the crude extract or purified enzyme was transferred into 3 ml quartz cuvettes and mixed with 2.9 ml assay mixture (100 mM sodium phosphate buffer, pH 7.0 and Triton X 100 1 μl/100 ml). The reaction was triggered by addition of 100 μl of 500 mM hydrogen peroxide to the cuvette (final concentration of 16.6 mM), shaking vigorously. The cuvette was placed in the spectrophotometer cell holder and catalase activity was followed by A240nm decay for 3 min. Catalase activity was calculated based on the rate of decomposition of hydrogen peroxide, which is proportional to the reduction of the absorbance at 240 nm. Catalase activities of the extracts were normalized to total protein in the lysate or sample and expressed as units per mg of protein: Specific activity = m min/ε/mg of protein; m min = average minute slope during 3 min reaction; ε = molar extinction coefficient = 0.0394 M/cm [28].
Catalase activity was also determined by an alternative method measuring the initial rate of dioxygen production with a Clark microelectrode [37]. Reaction was started by injecting crude extract or a purified catalase, usually 5 μl or less, into a sealed chamber filled with 2 ml of 10 mM H2O2 in 10 mM phosphate buffer, pH 7.8. Units were defined as micromoles of O2 produced per min per mg of protein under these conditions.
Catalase Purification
Catalase was purified from 3 l of culture of D. hansenii grown in YPE. Cells were harvested from stationary growth phase cultures. Catalase purification was carried out using the methods described by Aebi [1] and Trindade et al. [49]. Briefly, the crude extract was subjected to ammonium sulfate (NH2SO4) precipitation, keeping the 40–70% saturation fraction in which activity was detected, for further purification. This fraction was dialyzed against 50 mM sodium phosphate buffer, pH 7.0 and subjected to hydrophobic interaction chromatography (HIC) on a phenyl–sepharose CL-4B column, using 1.7 M NH2SO4–50 mM sodium phosphate buffer to bind catalases to the column. The column was washed using a 1.7–0 M NH2SO4 gradient, and then eluted with 50 mM sodium phosphate buffer, pH 7.0, followed by spectrometric measurements at A280nm of the eluted samples to detect the protein as it emerges from the column. Catalase activity was determined in selected samples. Fractions with activity were pooled and concentrated by polyethylene glycol to ~0.2 mg/ml of protein and kept it at 4°C until further use.
Electrophoresis and Stain Activity
Non-denaturing or SDS polyacrylamide gel electrophoresis was performed on 7.5% slab gels, running slowly at 12 V for 24 h. Proteins in polyacrylamide gels were visualized with Coomassie blue. To stain for catalase activity, the non-denaturing gel was rinsed twice with tap water, incubated in 100 mM H2O2 solution for 10 min, rinsed again with tap water and soaked in 1:1 iron (III) chloride: potassium ferricyanide solution until the gel was stained. Blue color developed in the gel except at zones where H2O2 was decomposed by catalase [50].
Kinetic Determinations and Effect of Inhibitors
To measure the catalytic parameters of the enzyme, 1 μg of pure one was mixed with 2.9 ml of 50 mM phosphate buffer; the reaction was triggered by the addition of different H2O2 concentrations (0–100 mM) [33]. Catalase activity was determined spectrophotometrically measuring the rate of hydrogen peroxide degradation at 240 nm. The initial linear rate was used to calculate the specific activity.
Optimum pH was determined in 50 mM potassium phosphate buffer (pH 4.0–8.5). Kinetic constants, including V max and K M apparent values, were determined by Lineweaver–Burk and Eadie Hofstee plot graphs.
The inhibitory effects of various concentrations of NaCl, 3-amino-1,2,4-triazole (3-AT) [14, 25], NaN3, and ethanol [17, 35] on catalase activity at constant enzyme concentrations, were estimated at pH 7.0, using a H2O2 fixed concentration as a substrate. The enzyme was incubated for 5 min in the presence of the inhibitor; the reaction was triggered by the addition of 16.6 mM H2O2, and followed spectrophotometrically.
Protein Determinations
Protein was quantified using the Quick Start Bradford Protein Assay from Bio-RAD, which is based on the absorbance at 595 nm of the complex between Coomassie Brilliant Blue G-250 and protein. Bovine serum albumin was used as standard.
Tandem Mass Spectrometry (Lc/Esi–Ms/Ms)
The protein band obtained after the purification was excised from Coomassie stained SDS gel, distained, reduced, carbamidomethylated, washed, digested with modified porcine trypsin, and extracted as previously described [51]. Peptide mass spectrometric analysis was carried out using a 3,200 Q TRAP hybrid tandem mass spectrometer, equipped with a nanoelectrospray ion source (NanoSpray II) and a MicroIonSpray II head. The instrument was coupled on line to a nanoAcquity Ultra Performance LC system. Spectra were acquired in automated mode using Information Dependent Acquisition (IDA). Precursor ions were selected in Q1 using the enhanced MS mode (EMS) as survey scans. The EMS was followed by an enhanced resolution scan (ER) of the three most intense ions at the low speed of 250 amu/sec, to determine the ion charge state, and then by an enhanced product ion scan (EPI). Precursor ions were fragmented by collision-activated dissociation (CAD) in the Q2 collision cell. The fragment ions generated were captured and mass analyzed in the Q3 linear ion trap.
Protein identification was performed by searching MS/MS spectra datasets using MASCOT (version 1.6b9, available at http://www.matrixscience.com). Mass tolerances of 0.5 and 0.3 Da were used for precursor and fragment ion masses, respectively. Carbamidomethyl–cysteine was the fixed modification and one missed cleavage for trypsin was allowed. Search was conducted using the Fungi subset of the NCBInr database (http://www.ncbi.nih.gov).
Results
D. hansenii Displays a Higher H2O2 Tolerance than S. cerevisiae
Debaryomyces hansenii has been described as a halotolerant yeast [7, 12, 39]. However, we have found that the Y7426 D. hansenii strain is able to sustain a similar growth rate in either rich (μ = 0.231 h−1) or minimal (μ = 0.154 h−1) growth media with or without 1.0 M NaCl [2], in agreement with previous observations, which classify D. hansenii as an euryhaline yeast [11, 46]. This characteristic has allowed us to analyze certain aspects of the NaCl response of this yeast using salt concentrations, which do not affect growth rate, ruling out the possibility that the observed response could be due to an effect of growth rate variation. As Fig. 1 shows S. cerevisiae growth pattern is similar on either YPD or YPD plus 0.6 M NaCl, reaching the stationary phase after 30 h of incubation. D. hansenii shows a higher duplication rate as compared to that of S. cerevisiae but a similar growth kinetics on YPD and YPE in the presence and absence of 0.6 M NaCl, reaching stationary phase after 50 h.

Growth curve of S. cerevisiae and D. hansenii grown on YPD and YPD plus 0.6 M NaCl during 96 h. Also, the growth curve of D. hansenii shows YPE and YPE plus 0.6 M NaCl culture media
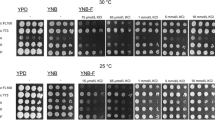
It has been shown that in S. cerevisiae, catalase activity is increased by NaCl induced stress and that D. hansenii displays high superoxide dismutase activity under these conditions [15], it could be thus considered that high H2O2 production through SOD activity could exert a positive effect on catalase activity or on the expression of the encoding gene(s). To analyze this matter, we determined catalase activity in cultures of S. cerevisiae and D. hansenii grown on rich YPD media without salt or in the presence of 0.6 M NaCl. Extracts obtained from S. cerevisiae cultures grown in the presence of salt, displayed a fivefold increased catalase activity as compared to that obtained in extracts from NaCl-free culture conditions (0.03 ± 0.009) versus 0.152 (±0.021). In extracts prepared from D. hansenii cultures, catalase activity displayed nearly a two-fold increase when it was determined from salt-grown cultures as compared to that found in the absence of salt (Table 1). In addition, D. hansenii extracts, obtained from YPD cultures, showed 24-fold higher catalase activity than those obtained from S. cerevisiae (0.03 ± 0.009 vs. 0.73 ± 0.16); accordingly, D. hansenii extracts obtained from cells grown on YPD–NaCl showed eightfold higher activity than those of S. cerevisiae grown on same media (0.152 ± 0.021 vs. 1.24 ± 0.02). It could be thus considered that the higher basal level of catalase activity shown by D. hansenii extracts obtained from YPD grown yeasts could result in a higher inherent resistance to oxidative stress as compared to that displayed by S. cerevisiae. To analyze this matter oxidative shock tolerance was assayed. Both yeast species were treated with H2O2 increasing concentrations during exponential and stationary growth phases. The results of this cell viability assay showed that under exponential growth phase, D. hansenii is able to grow in the presence of H2O2 concentrations, which inhibit S. cerevisiae growth, Fig. 2 . When yeasts were grown in the presence of 0.6 M NaCl and H2O2, resistant phenotype was evident in D. hansenii; conversely, S. cerevisiae growth was severely impaired in the presence of both stressors. When cells were collected from stationary growth phase, both yeasts improved their resistance to H2O2 treatment (Fig. 2).

S. cerevisiae and D. hansenii resistance to H2O2 stress, after grown with and without salt (0.6 M NaCl). Exponential and stationary growth phase cultures of S. cerevisiae and D. hansenii were diluted with fresh media (YPD) and exposed to 0, 2.5, 5.0, 7.5, 10, 15, 20, and 30 mM H2O2 for 3 h. After the treatment, H2O2 was removed by centrifugation. The cultures were resuspended in distilled water, serially diluted, and each dilution was spotted onto YPD plates, ensuring that the same amounts of cells were plated. Plates were incubated at 30°C for 3–5 days
Above presented results indicate that D. hansenii displays an inherently high catalase activity in either YPD or YPD-0.6 M NaCl that results in a higher H2O2 tolerance as compared to that displayed by S. cerevisiae. To further analyze the role of catalase in stress resistance, sequence genome comparison was carried out with S. cerevisiae to search the corresponding orthologous gene(s).
D. hansenii has Two Genes Encoding Catalase Isozymes
Saccharomyces cerevisiae has a CTA1-encoded peroxisomal catalase and a CTT1-encoded cytoplasmic catalase. To analyze whether D. hansenii had CTA1 and CTT1 orthologous counterparts, a computer-based analysis of the Genolevures database (available at http://www.genolevures.org) was performed [41], using D. hansenii genomic sequence. This analysis revealed that this yeast harbored two ORFs that could presumably encode for two catalase isoforms with approximate molecular weights of 54.9 and 63.1 kDa, respectively. The DhCTA1 presumed orthologous ORF was identified to be DEHA2F10582 g located in F chromosome, while DhCTT1 counterpart was found to correspond to DEHA2B16214 g located in B chromosome. Accordingly, a zymogram performed on an electrophoresed crude extract of D. hansenii, obtained from YPD grown cultures after different incubation periods, showed two bands displaying catalase activity (Fig. 3a). Worth of mention is the fact that the low molecular weight activity (DhCtt1) only appeared after 24 h of incubation and was not observed in stationary phase NaCl-treated cultures, Fig. 3a and b. Only the DhCta1 band was observed when yeasts were incubated to exponential or stationary phases in the presence of 0.6 M NaCl (Fig. 3b). Extracts from YPE-grown cells were analyzed, and only the high molecular form (DhCta1) was observed under exponential and stationary growth phases (Fig. 3b).

a Zymogram from D. hansenii grown in rich media containing glucose as carbon source; samples were taken at different times: Lane 1, 6 h; lane 2, 12 h; lane 3, 24 h; lane 4, 48 h. b Zymogram from D. hansenii grown until exponential and stationary phases in YPD and YPE in the presence or absence of 0.6 M NaCl. Lane 1, YPD exponential (exp); lane 2, YPD stationary (sta); lane 3, YPD–NaCl exp; lane 4, YPD–NaCl sta; lane 5, YPE exp; lane 6, YPE sta; lane 7, YPE–NaCl exp; lane 8 YPE–NaCl sta; lane 9, human erythrocyte catalase. c Zymogram of the purified catalase from D. hansenii: Lane 1, human erythrocyte catalase; lane 2, 0.1 μg of purified D. hansenii catalase; lane 3, Coomasie’s stain PAGE 10 μg of purified D. hansenii catalase
D. hanseniiCTA1-Encoded Catalase Activity is Positively Modulated by Growth Phase and Non-Fermentable Carbon Sources
It has been previously reported, that when S. cerevisiae is grown in 2% ethanol as carbon source, the activity of the primary antioxidant enzymes (SOD and catalase) as well as the levels of carbonyl-proteins and TBARS (thiobarbituric acid-reactive substances), which accumulate as a result of oxidative damage to proteins and lipids, are increased with respect to glucose containing media [24]. To analyze whether D. hansenii showed a similar response when grown in the presence of 2% ethanol, D. hansenii catalase activity was determined during exponential and stationary phases on YPD (glucose) and YPE (ethanol) growth medium. Catalase activity determined as described by Aebi [1], on extracts prepared from cultures grown on YPD, showed a twofold increased activity in stationary growth phase as compared to those prepared from exponential growth cultures, which could be afforded by the combined action of DhCta1 and DhCtt1, since both activities are evident under stationary phase (Fig. 3a and b, Table 1). DhCtt1 zymogram-determined activity was not observed in extracts obtained from stationary growth-phase cultures carried out in the presence of NaCl, thus indicating that in the presence of salt activity could only be provided by DhCta1. These results also indicate that DhCTT1 could have a differential expression pattern that may result in expression repression under exponential growth phase and in the presence of NaCl. In addition, it was found that catalase activity determined in extracts prepared from either exponential or stationary YPE cultures, showed a four to sevenfold increase as compared to activity determined in equivalent extracts prepared from YPD cultures, which was not observed when yeast was grown on YPE in the presence of 0.6 M NaCl. These results indicate that DhCTA1-dependent catalase activity is positively regulated under stationary phase, this effect is enhanced in the presence of ethanol as sole carbon source; these positive effects are hindered in the presence of 0.6 M NaCl.
D. hansenii Catalase-Encoding Paralogues are Differentially Regulated by the Carbon Source
In order to analyze whether increased catalase activity in the presence of ethanol corresponded to increased expression level, Northern blot analysis was performed with total RNA obtained from D. hansenii grown in YPD, YPD-0.6 M NaCl; YPE and YPE-0.6 M NaCl, and samples were taken from exponential and stationary growth phases. Results show that DhCTA1 and DhCTT1 are differentially expressed. DhCTA1 expression levels are slightly higher under respiratory conditions as compared to those found on glucose, in agreement with the results obtained in the zymogram analysis. In addition, DhCTA1 expression was repressed when the yeast was grown in glucose and NaCl, this effect was not observed in RNA samples obtained from ethanol grown cells. These results indicate that DhCTA1 is transcriptionally regulated by the carbon source and by NaCl. Conversely, DhCTT1 expression was highest under fermentative conditions and repressed when the yeast was grown on non-fermentable substrates like ethanol. In the presence of 0.6 M NaCl, ethanol-dependent expression was repressed (Fig. 4). These will be further discussed.

Northern blot of D. hansenii RNA from cells grown on either YPD or YPE and in the presence or absence of salt. Total RNA was isolated from exponential (exp) and stationary (sta) growth phase cultures. a Northern blot using DhCTA1, DhCTT1, and DhrDNA 18S as a probe. The latter was used as internal control. Three sets of deoxyoligonucleotides were used to PCR-amplify three fragments of 2,607 bp; 3,174 and 1,800 bp those were used as probes for sequential hybridization of DhCTA1, DhCTT1, and Dh18S rDNA. b Normalized expression levels of DhCTA1 and DhCTT1 using data obtained from the Northern blot
Catalase Purification from Ethanol-Growing Cells and Identification of the Peroxisomal Catalase (DhCta1) by Tandem Mass Spectrometry
Considering that on stationary phase ethanol grown cultures a large single catalase band was observed, we purified this isoform through ammonium sulfate precipitation followed by a chromatographic HIC column. A single band was obtained as seen on SDS/PAGE (Fig. 3c). The purified protein was analyzed by MS/MS spectrometry. It was found that the protein corresponded to a presumed peroxisomal catalase (gi∣50424473), DhCta1 present in D. hansenii. The global score was 1,182 with 26 matching peptides that covered 63% of the sequence. The protein had a nominal mass of 54,836 Da with a calculated pI of 6.47 (Fig. 5). Thus it can be presumed that the peroxisomal catalase (DhCta1) is involved in hydrogen peroxide detoxification generated during the oxidative stress induced by ethanol in D. hansenii.

The purified protein was found to correspond to peroxisomal catalase of D. hansenii (gi|50424473). The global score was 1,182 with 26 matching peptides that covered 63% of sequence. The protein had a nominal mass of 54,836 Da and a calculated pI of 6.47
Kinetic analysis of the purified enzyme preparation under various H2O2 concentrations, using the Lineweaver–Burk and Eadie Hofstee plotting methods allowed to get the apparent K M and V max values which average of 44 mM and 106 mmol/min/mg of protein, respectively, Fig. 6.

Kinetics parameters of catalase purified from YPE of D. hansenii. The apparent K M and V max of catalase were calculated using two methods
The effect of ionic strength on the activity of the purified catalase was also tested with NaCl, showing virtually no inhibition at any NaCl concentration assessed (0.1–1,000 mM). In addition, we examined the effect of some catalase inhibitors, including NaN3, which is a very potent inhibitor that binds irreversibly to the heme cofactor [17]. This inhibitor impaired catalase activity at a concentration as low as 0.5 μM, and reached 80% enzyme inhibition at a concentration lower than 0.2 mM. Conversely, the specific catalase inhibitor 3AT [14] showed no effect even at a 5 mM concentration. The effect of pH was also assayed. As expected, a bell-shaped curve for catalase activity was obtained for the pure DhCTA1 preparation. The optimum pH for this purified catalase was 7.0 (data not shown).
Discussion
When the yeast S. cerevisiae is grown in rich media in the presence of 2% ethanol, it exhibits a strong activation of antioxidant enzymes as compared to those found on rich media containing glucose [23]. This article reports that, as opposed to what has been found for S. cerevisiae, D. hansenii shows a strong catalase activity in rich media containing glucose, which increases significantly when the yeast is grown in non-fermentable carbon sources like ethanol. Catalase activity is further augmented in ethanol cultures during stationary growth phase. However, the positive effect exerted trough ethanol and stationary phase is not observed in the presence of NaCl, suggesting that under this condition, oxidative stress could be dampened thus diminishing the stress response that triggers catalase increase. This could also be considered as a NaCl protective role, which has also been observed for other xenobiotic injuries [10, 30, 34].
Saccharomyces cerevisiae has two genes CTT1 and CTA1 that encode two proteins with catalase enzymatic activity. Encoded enzymes have been studied in detail [38, 42, 52]. By gene sequence comparison, we identified two putative ORFs in the D. hansenii genome, DEHA2B16214 g and DEHA2F10582 g, respectively, localized in the B and F chromosomes. In addition, we identified two proteins of different size with catalase activity, using PAGE and dye analyses. When we evaluated the specific activity using spectrophotometrical and oxymetrical methods, we found a higher level of catalase activity in this euryhaline yeast as compared to that detected in S. cerevisiae (Table 1). Zymogram analysis revealed DhCta1 activity is increased when D. hansenii is grown on YPE; the effect is enhanced during stationary phase and hindered in the presence of NaCl. Increased activity corresponded to increased DhCTA1 specific mRNA; however, the negative effect exerted by NaCl was not correlated with reduced expression on YPE–NaCl, suggesting post transcriptional regulation. In spite of this, in glucose grown cultures DhCTA1 expression was repressed in the presence of NaCl. In regard to DhCtt1, although zymogram analysis indicated that this enzyme is only evident in stationary phase, expression analysis indicated that the encoding gene is not regulated by growth phase since similar DhCTT1 mRNA levels were detected under exponential or stationary phase. These results indicate that as well as for DhCTA1, posttranscriptional mechanisms could play a key role determining DhCtt1 levels. In the presence of ethanol, DhCTT1 expression was repressed suggesting that this gene expression could be regulated by the nature or quality of the carbon source. The presence of NaCl in the growth media further decreased DhCTT1 expression. Therefore, as opposed to what has been observed in Saccharomyces, NaCl does not trigger the mechanism that results in increased catalase activity in D. hansenii. NaCl could have a protective role against oxidative stress in D. hansenii, as has been reported recently by Navarrete et al. [26]. We thus propose that D. hansenii growing in ethanol and salt is refractory to oxidative stress and consequently does not trigger a stress responsive reaction.
Catalase post transcriptional regulation has been observed in the extremely halotolerant black yeast Hortaea werneckii [31]. Author showed increased hydrogen peroxide degradation, indicating increased catalase activity when this yeast was grown in 17% NaCl, however, no change in oxidative responsive genes expression was observed [32], thus it was concluded that increased activity could be due to a post transcriptional mechanism. Conversely, in Aspergillus nidulans the two catalase-encoding genes, catA and catB were transcribed under different stress conditions although only CatB activity was present in all conditions [27].
Debaryomyces hansenii has been classified as marine yeast, mainly due to the fact that it was isolated from marine water [29], where the average NaCl content is around 0.6 M. However, this non-conventional yeast has been largely recognized as a common contaminant of sausages, chesses, brines, and dairy products, and even soft drinks and fruit syrups [36]. Other yeast species also found in fruits, soil and food, are most likely not native to estuaries and seas, even if they are very frequently isolated from such areas [21]. D. hansenii has been defined as a Na+ includer organism, since some reports have shown that sodium is not toxic to this yeast and it can even be stored inside the cell [34]. Some authors have classified D. hansenii as a halotolerant yeast, others as a halophilic one, and more recently as a salt loving yeast [36]. We think that a more suitable classification may be euryhaline yeast [11, 46], because it can adapt to a wide range of salinities as described above.
References
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Alba–Lois L, Segal C, Rodarte B et al (2004) NADP–glutamate dehydrogenase activity is increased under hyperosmotic conditions in the halotolerant yeast Debaryomyces hansenii. Curr Microbiol 48:68–72
Aguirre J, Ríos-Momberg M, Hewitt D, Hansberg W (2005) Reactive oxygen species and development in microbial eukaryotes. Trends Microbiol 13:111–118
Bansal PK, Mondal AK (2000) Isolation and sequence of the HOG1 homologue from Debaryomyces hansenii by complementation of the hog1 strain of Saccharomyces cerevisiae. Yeast 16:81–88
Bartosz G (2005) Superoxide dismutases and catalase. In: The handbook of environmental chemistry, vol 2, Part O. Springer, Heidelberg, pp 109–149
Beers RF, Sizer IW (1952) A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem 195:133–140
Breuer U, Harms H (2006) Debaryomyces hansenii–an extremophilic yeast with biotechnological potential. Yeast 23:415–437
Cuéllar-Cruz M, Briones-Martin-del-Campo M, Cañas-Villamar I et al (2008) High resistance to oxidative stress in the fungal pathogen Candida glabrata is mediated by a single catalase, Cta1p, and is controlled by the transcription factors Yap1p, Skn7p, Msn2p, and Msn4p. Eukaryot Cell 7:814–825
Fernandes R, Melo A, Flávia K et al (2010) Purification of Paracoccidioides brasiliensis catalase P: subsequent kinetic and stability Studies. J Biochem 147:345–351
Gori K, Hébraud M, Chambon C et al (2007) Proteomic changes in Debaryomyces hansenii upon exposure to NaCl stress. FEMS Yeast Res 7:293–303
Govind NS, McNally KL, Trench RK (1992) Isolation and sequence analysis of the small subunit ribosomal RNA gene from the euryhaline yeast Debaryomyces hansenii. Curr Genet 22:191–195
Guerrero C, Aranda C, DeLuna A et al (2005) Salt-dependent expression of ammonium assimilation genes in the halotolerant yeast, Debaryomyces hansenii. Curr Genet 47:163–171
Hallick RB, Chelm BK, Gray WP, Orozco EM (1977) Use of aurintricarboxylic acid as an inhibitor of nucleases during nucleic acid isolation. Nucleic Acids Res 4:3055–3064
Havir EA (2003) The in vivo and in vitro inhibition of catalase from leaves of Nicotiana sylvestris by 3-amino-1,2,4-triazole. J App Microbiol 95:364–371
Hernández-Saavedra Y, Ochoa JL (1999) Copper–zinc superoxide dismutase from the marine yeast Debaryomyces hansenii. Yeast 15:657–668
Izawa S, Inoue Y, Kimura A (1996) Importance of catalase in the adaptive response to hydrogen peroxide: analysis of acatalasemic Saccharomyces cerevisiae. Biochem J 320:61–67
Johnston MA, Delwiche EA (1965) Isolation and characterization of the cyanide-resistant and azide-resistant catalase of Lactobacillus plantarum. J Bacteriol 90:352–356
Jones DP (2008) Radical-free biology of oxidative stress. Am J Physiol 295:C849–C868
Koleva DI, Petrova VY, Kujumdzieva V (2008) Comparison of enzymatic antioxidant defense systems in different metabolic types of yeasts. Can J Microbiol 54:957–963
Krawiec Z, Bilinski T, Schüller C, Ruis H (2000) Reactive oxygen species as second messengers? Induction of the expression of yeast catalase T gene by heat and hyperosmotic stress does not require oxygen. Acta Biochim Pol 47:201–207
Kutty SN, Philip R (2008) Marine yeast–a review. Yeast 25:465–483
Levy E, Eyal Z, Hochmant A (1992) Purification and characterization of a catalase–peroxidase from the fungus Septoria tritici. Arch Biochem Biophys 296:321–327
Lushchak VI, Gospodaryov DV (2005) Catalases protect cellular proteins from oxidative modification in Saccharomyces cerevisiae. Cell Biol Intern 29:187–192
Lushchak VI (2006) Budding yeast Saccharomyces cerevisiae as a model to study oxidative modification of proteins in eukaryotes. Acta Biochim Pol 53:679–684
Mozaffar S, Ueda M, Kitatsuji K et al (1986) Properties of catalase purified from a methanol-grown yeast, Kloeckera sp. 2201. Eur J Biochem 155:527–531
Navarrete C, Siles A, Martinez JL et al (2009) Oxidative stress sensitivity in Debaryomyces hansenii. FEMS Yeast Res 9:582–590
Navarro RE, Aguirre J (1998) Posttranscriptional control mediates cell type-specific localization of catalase A during Aspergillus nidulans development. J Bact 180:5733–5738
Nelson DP, Kiesow LA (1972) Enthalpy of decomposition of hydrogen peroxide by catalase at 25°C (with molar extinction coefficients of H2O2 solutions in the UV). Anal Biochem 49:474–478
Norkrans B (1966) Studies on marine occurring yeasts: growth related to pH, NaCl concentration and temperature. Arch Mikrobiol 54:374–392
Papouska K, Sychrova H (2007) The co-action of osmotic and high temperature stresses results in a growth improvement of Debaryomyces hansenii cells. Int J Food Microbiol 118:1–7
Petrovič U (2006) Role of oxidative stress in the extremely salt-tolerant yeast Hortaea werneckii. FEMS Yeast Res 6:816–822
Petrovič U, Gunde-Cimerman N, Plemenitaŝ A (2002) Cellular responses to environmental salinity in the halophilic black yeast Hortaea werneckii. Mol Microbiol 45:665–672
Price VE, Rechcigl M, Hartley RW (1961) Methods for determining the rates of catalase synthesis and destruction in vivo. Nature 189:62–63
Prista C, Almagro A, Loureiro-Diaz MC, Ramos J (1997) Physiological basis for the high salt tolerance of Debaryomyces hansenii. Appl Enviromen Microbiol 63:4005–4009
Putnam CD, Arvai AS, Bourne Y, Tainer JA (2000) Active and inhibited human catalase structures: ligand and NADPH binding and catalytic mechanism. J Mol Biol 296:295–309
Ramos J (2005) Introducing Debaryomyces hansenii, a salt loving yeast. In: Gunde-Cimerman N, Oren A, Plemenitaš A (eds) Adaptation to life at high salt concentrations in Archaea, Bacteria, and Eukarya. Springer, Netherlands, pp 441–451
Rørth M, Jensen PK (1976) Determination of catalase activity by means of the Clark oxygen electrode. Biochim Biophys Acta 139:171–173
Ruis H, Köller F (1997) Biochemistry, molecular and cell biology of yeast and fungal catalases. In: Scandalios JG (ed) Oxidative stress and the molecular biology of antioxidant defences. Cold Spring Harbor, New York, pp 309–342
Sánchez NS, Arreguín R, Calahorra M, Peña A (2008) Effects of salts on aerobic metabolism of Debaryomyces hansenii. FEMS Yeast Res 8:1303–1312
Scandalios JG (2002) The rise of ROS. Trends Biochem Sci 27:483–486
Sherman DJ, Martin T, Nikolski M, et al. Génolevures Consortium (2009) Génolevures: protein families and synteny among complete hemiascomycetous yeast proteomes and genomes. Nucl Acid Res 37, Database issue: D550–D554
Schuller C, Brewster JL, Alexander MR et al (1994) The HOG pathway controls osmotic regulation of transcription via the stress response element (STRE) of the Saccharomyces cerevisiae CTT1 gene. EMBO J 13:4382–4389
Steels EL, Learmonth RP, Watson K (1994) Stress tolerance and membrane lipid unsaturation in Saccharomyces cerevisiae grown aerobically or anaerobically. Microbiol 140:576–596
Switala J, Loewen PC (2002) Diversity of properties among catalases. Arch Biochem Biophys 401:145–154
Tanghe A, Prior B, Thevelein JM (2006) Yeast responses to stress. In: Rosa CA, Peter G (eds) Biodiversity and ecophysiology of yeasts. Springer, Berlin, pp 175–195
Thome PE, Trench RK (1999) Osmoregulation and the genetic induction of glycerol-3-phosphate dehydrogenase by NaCl in the euryhaline yeast Debaryomyces hansenii. Mar Biotechnol 1:230–238
Thome-Ortiz PE, Peña A, Ramirez J (1998) Monovalent cation fluxes and physiological changes of Debaryomyces hansenii grown at high concentrations of KCl and NaCl. Yeast 14:1355–1371
Toledano M, Delaunay A, Biteau B et al (2003) Oxidative stress responses in yeast. In: Hohmann S, Mager WH (eds) Topics current genetics, vol 1. Springer, Berlin, pp 241–303
Trindade H, Karmali A, Pais MS (1988) One-step purification and properties of catalase from leaves of Zandedeschia aetgiopica. Biochimie 70:1759–1763
Woodbury W, Spencer AK, Stahman MA (1971) An improved procedure using ferricyanide for detecting catalase isozymes. Anal Biochem 44:301–305
Xolalpa W, Vallecillo AJ, Lara M et al (2007) Identification of novel bacterial plasminogen-binding proteins in the human pathogen Mycobacterium tuberculosis. Proteomics 7:3332–3341
Zimniak P, Hartter E, Woloszczuk W, Ruis H (1976) Catalase biosynthesis in yeast: formation of catalase A and catalase T during oxygen adaptation of Saccharomyces cerevisiae. Eur J Biochem 71:393–398
Acknowledgments
The authors are deeply indebted with the advices of René Cárdenas throughout the work, of Facultad de Ciencias, UNAM and to the technical assistance of Cristina Aranda and Pablo Rangel, from Instituto de Fisiología Celular, and Alfonso Vilchis of Facultad de Ciencias, UNAM. We are grateful to L. Ongay and M. Mora (Unidad de Biología Molecular, Instituto de Fisiología Celular, UNAM) for the synthesis of deoxyoligonucleotides. Claudia Segal-Kischinevzky is grateful to the Posgrado en Ciencias Biológicas, UNAM, for the support during PhD studies. This work was supported partially by the Dirección General de Asuntos del Personal Académico, Universidad Nacional Autónoma de México (IN241602).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Segal-Kischinevzky, C., Rodarte-Murguía, B., Valdés-López, V. et al. The Euryhaline Yeast Debaryomyces hansenii has Two Catalase Genes Encoding Enzymes with Differential Activity Profile. Curr Microbiol 62, 933–943 (2011). https://doi.org/10.1007/s00284-010-9806-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-010-9806-z