
Abstract
Bone homeostasis depends on a balance between osteoclastic bone resorption and osteoblastic bone formation. Bone cells are regulated by a variety of biochemical factors, such as hormones and cytokines, as well as various types of physical stress. The immune system affects bone, since such factors are dysregulated under pathologic conditions, including infection. The bone marrow, one of the primary lymphoid organs, provides a special microenvironment that supports the function and differentiation of immune cells and hematopoietic stem cells (HSCs). Thus, bone cells contribute to immune regulation by modulating immune cell differentiation and/or function through the maintenance of the bone marrow microenvironment. Although osteoblasts were first reported as the population that supports HSCs, the role of osteoblast-lineage cells in hematopoiesis has been shown to be more limited than previously expected. Osteoblasts are specifically involved in the differentiation of lymphoid cells under physiological and pathological conditions. It is of critical importance how bone cells are modified during inflammation and/or infection and how such modification affects the immune system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Bone as osteoimmunological tissue
The bone belongs to the skeletal system which supports the body and enables the locomotion. However, bone contains the bone marrow, in which HSCs or common progenitors are maintained. Therefore, the bone also functions as the hematopoietic or immune organ [1].
Bone homeostasis is maintained by three types of bone-constituting cells: osteoclasts, osteoblasts, and osteocytes [1, 2]. The bone tissue is constantly renewed by a dynamic remodeling process, in which bone is resorbed by osteoclasts and formed by osteoblasts. Osteocytes control both osteoclasts and osteoblasts in response to external stimuli. Since bone cells are regulated by immune regulators such as cytokines, aberrant or prolonged immune responses often affect bone metabolism.
A number of reports on the bone phenotypes of immunocompromised mice indicate that the immune and skeletal systems share a variety of molecules, including transcription factors, signaling molecules, and membrane receptors, implying an interplay between the two systems [1,2,3,4]. In this article, we discuss the changes that occur in bone cells and the bone marrow microenvironment during the course of infection from the point of osteoimmunology, including the emerging role of osteoblasts in the maintenance of early immune cell progenitors.
Hematopoietic stem cells and bone tissue
A specialized bone marrow microenvironment called a hematopoietic stem cell niche is important for both the function and maintenance of hematopoietic stem cells. Since the concept of the hematopoietic stem cell niche put forward in the late 1970s, intensive research has been carried out in an effort to elucidate the details of the bone marrow microenvironment [5].
Osteoblasts form the bone, the structural container of the space that harbors HSCs. However, do osteoblasts directly and actively support HSCs maintenance? It was initially proposed that the endosteal surface contains certain key components for supporting HSCs [6,7,8]. An increase in the hematopoietic stem cell pool was observed in genetically modified mice in which the osteoblast number was increased, raising the possibility that osteoblasts constitute the HSC niche. The in vivo importance of osteoblasts in HSC regulation was reported by two independent groups in 2003 [9, 10]. The osteoblast number correlated with the HSC number in parathyroid hormone (PTH)/PTH-related protein receptor (PPR) transgenic mice as well as bone morphogenetic protein IA (BMPRIA) conditional knockout mice. In addition, it was reported that endochondral ossification is required for ectopic hematopoietic niche formation [11]. CD105+ Thy1− skeletal progenitors isolated from fetal limb bones were shown to reconstitute ectopic HSC niche formation [12]. These observations suggested a relationship between osteoblasts and the HSCs in the bone marrow. Furthermore, osteoblasts were shown to express molecules important for the regulation of HSCs, including N-cadherin, anigipoietin-1, thrombopoietin, and osteopontin [10, 13,14,15,16].
In contrast, in the subsequent reports, it was found that the number of osteoblasts did not always correlate with the number of hematopoietic stem cells. Biglycan-deficient mice were shown to have a low osteoblast number with an osteoporotic phenotype but to nevertheless contain a normal HSC number [17, 18]. Furthermore, an increase in osteoblasts by strontium chloride administration did not affect the HSC number in the bone marrow [16]. Moreover, it has been shown that neither Ang-1 nor N-cadherin on osteoblasts was required for the maintenance of HSCs [17, 19].
Certain studies have reported that mature bone cells are not required for the maintenance of HSCs. Stem cell factor (Scf) and chemokine (CXC motif) ligand 12 (Cxcl12) are considered to be fundamental for HSC maintenance. Cell-type-specific conditional knockout mice lacking these factors indicated that HSCs were maintained even when Scf and Cxcl12 were deleted specifically in osteoblasts. LepR-expressing perivascular stromal cells were shown to be a major source of SCF in the bone marrow and to support HSCs. In addition, deletion of CXCL12 using Prx1-cre induced a decrease in the number of HSCs and immune cell progenitors in vivo [20,21,22,23]. Therefore, LepR-positive cells and CAR cells are regarded as the cells that mainly benefit the HSC niche. However, CAR cells have the ability to differentiate into osteoblasts, suggesting that at least osteoprogenitors participate in HSC maintenance [24].
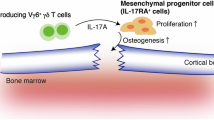
The contribution of osteoblasts to the maintenance of HSCs may be less than predicted by the earlier studies, but there are reports that osteoblasts are involved in the modulation of immune cell differentiation. The depletion of CXCL12 in osterix-expressing cells, which are mostly osteoblasts, resulted exclusively in a loss of B-lymphoid progenitors [22]. It was also reported that acute inflammation suppresses osteoblastic bone formation, which results in T and B lymphopenia through reduced interleukin-7 (IL-7) production, suggesting that osteoblasts play a role in the regulation of common lymphoid progenitors by providing IL-7 (see below) [25, 26]. One study has shown that the Delta-like 4 (DLL4) expressed on osteoblasts supports T-lineage competent cells [27]. Osteoblasts also regulate erythropoiesis by producing erythropoietin (Epo) in a hypoxia-inducible factor (HIF)-dependent manner [28]. Notably, alterations of osteoblast function induce a perturbation of HSCs. Disruption of Dicer in murine osteoblasts, which is an RNase III endonuclease essential for microRNA synthesis and processing, resulted in HSC malignancy with concomitant osteoporosis [29]. The constitutive activation of β-catenin in osteoblasts was shown to increase Jagged1 expression and induce malignant changes [30, 31]. These findings demonstrate that osteoblasts are able to produce signals that keep the HSCs from a transition into malignancy (Fig. 1).

Osteoblasts regulate the immune cell differentiation. Osteoprogenitors support the HSC maintenance. Osteoblasts produce IL-7 and DLL4 to support lymphopoiesis and T cell differentiation, respectively. Overexpression of β-catenin in osteoblasts induces Jagged1 expression, leading to hematopoietic malignancy
Osteoclasts and immune cell progenitors
Osteoclasts are large, multinucleated cells derived from monocyte/macrophage progenitor cells of hematopoietic origin. They are essential in the first place for the formation of the bone marrow cavity. Since osteoclast-deficient mice do not have sufficient space to maintain immune cell progenitors or support their differentiation within the bone marrow, extramedullary hematopoiesis occurs in the spleen and liver in osteopetrotic mice [32,33,34]. During extramedullary hematopoiesis, immature, deformed, or enlarged hematopoietic cells are typically found in the blood. In fact, it is reported that osteopetrotic patients develop anemia and are prone to severe infection due to such abnormal hematopoiesis [35, 36]. In addition, an inhibition of osteoclast function by bisphosphonate administration also induces a decrease in the HSC number and impaired engraftment [37]. It has also been shown that suppression of osteoclasts by zoledronic acid administration reduces the number of HSCs. Furthermore, HSC dysfunction is found in certain mice that have defects in osteoclast function. The frequency and absolute number of Lineage−Sca-1+c-kit+ (LSK) cells that contain HSCs are decreased in an oc/oc mouse strain bearing the mutated Tcirg1 gene, a proton pump subunit essential for bone resorption [38]. Osteoclasts may regulate HSC quiescence through degradation of the bone matrix, which results in the release of calcium ions and certain cytokines such as TGF-β [39, 40].
Mobilized peripheral blood stem cells are often used for autologous or allogeneic transplantation after myeloablative therapy in patients with myeloid malignancies. Granulocyte colony-stimulating factor (G-CSF) is the most widely used treatment for HSC mobilization in the clinic. HSC mobilization is dependent on an interaction between HSCs and bone marrow niche cells. CXCL12, which is expressed on niche cells and is an important factor for maintaining HSCs, is a ligand for CXC chemokine receptor 4 (CXCR4) on HSCs. It has been reported that receptor activator of nuclear factor κ B ligand (RANKL)-stimulated osteoclasts degrade the niche component by secreting matrix metalloproteinase (MMP) 9, resulting in impaired CXCR4–CXCL12 binding. As a result, the HSCs are unable to remain in the altered bone marrow microenvironment [41]. On the other hand, osteoclast-deficient mice, such as op/op, c-fos−/−, and Tnfsf11−/− mice, do not exhibit any defects upon G-CSF-induced HSC mobilization [42]. Subsequent studies showed that G-CSF administration does not affect the number of osteoclasts on the endosteal surface. Furthermore, G-CSF-induced HSC mobilization was observed in mice in which the osteoclast number had been decreased by zoledronate treatment.
Osteoblasts and osteocytes also contribute to the HSC mobilization in addition to osteoclasts. Osteal tissue macrophages (osteomac), which have been shown to support osteoblast activity, are depleted by G-CSF administration. The ablation of osteomac induced disruption of both the endosteal niche and HSC mobilization [43]. Moreover, it is reported that osteoblasts and osteocytes are essential for the mobilization of HSCs in response to G-CSF [44, 45]. G-CSF administration leads to marked suppression of osteoblasts in the endosteum. A subsequent study showed that the cellular connection between the endosteal osteoblasts and bone-embedded osteocytes was disrupted after G-CSF treatment. Thus, G-CSF induces HSC mobilization through an induced alteration of the bone marrow microenvironment.
When osteoclasts degrade the bone matrix, certain factors stored in the bone matrix are released. It is possible that some of them are involved in maintaining HSCs. The local concentration of calcium ions near bone resorption sites could be considerably higher than that in the serum. HSCs lacking a calcium-sensing receptor failed to lodge at the endosteal sites near resorbing osteoclasts [39]. In addition, transforming growth factor-β (TGF-β) is deposited in bone matrix in an inactive form. During bone resorption, TGF-β is released and activated by proteolytic enzymes secreted from osteoclasts. TGF-β plays an important role in regulating HSC quiescence and self-renewal [40]. These studies raise the possibility that osteoclasts contribute to HSC regulation by releasing certain matrix-embedded factors that are able to control HSCs.
Immune regulation by bone cells during the course of infection
Sepsis is an acute host inflammatory response to severe infection associated with a high incidence of mortality. In 1989, the concept of “sepsis syndrome” was proposed. Sepsis was then described as a “systemic inflammatory response syndrome (SIRS) due to infection” in 1991 [46, 47]. However, sepsis induces not only an excessive inflammatory reaction but also an anti-inflammatory reaction. Since this definition reflects only a part of the complex condition of sepsis, the definition of sepsis was revised in 2001 [48]. Sepsis was subsequently defined as “systemic symptoms attributable to infection” and using SIRS alone as a diagnostic criterion was stopped. The definition of sepsis was further revised in 2016, almost a quarter of a century since the first definition [49]. Sepsis is a condition presenting with multiple organ failure due to a failure of the host biological response to infection with a high rate of mortality due to circulatory failure, cellular dysfunction, and metabolic abnormalities. Unfortunately, numerous therapeutic clinical trials have failed, and an effective strategy for sepsis remains elusive. Although considerable advances in antimicrobial agents have led to a marked improvement in the relative survival, long-term mortality rates in sepsis remain at just 20 to 50%. It is of critical importance to obtain a complete picture of sepsis for the development of effective treatments.
In addition to the multiple organ failure that occurs after a severe inflammatory response infection, complications resulting from the sepsis-related immunosuppression contribute to the morbidity and mortality that commonly occur. Although a vigorous and uncontrollably sustained inflammatory response to infection is indeed observed in patients in the early phase of sepsis, it is the immunosuppression that leads to life-threatening secondary infection and delay recovery that is the hallmark of severe sepsis. A key feature of resulting immunosuppression is the loss of immune cells associated with apoptosis, including CD4+ and CD8+ T and B cells [50, 51]. It is reported that circulating levels of lymphocytes decrease and remain depressed for up to 28 days [52]. Since HSCs in the bone marrow should continuously supply new immune cells to the body, it is unclear how the lymphopenia persists for a long time.
Host pathogen recognition and the subsequent initiation of the proinflammatory response are important events in the innate immune system reaction to the pathogen. The reaction leads the production of inflammatory cytokines including IL-1β, TNF-α, and G-CSF [53]. G-CSF can significantly delay neutrophil apoptosis and improve the unusual prognosis of neutropenia with severe infectious complications. However, meta-analyses of human studies using G-CSF administration for sepsis have shown no remarkable advantage in terms of the impact on mortality [54, 55].
Although it appears that the lymphopenia that occurs in the late phase of sepsis is unrelated to the failure of sepsis therapy using G-CSF administration, a better understanding of osteoblast function may provide an explanation for the two reports that suggest such a linkage.
Sepsis induces rapid and marked bone loss through the impaired osteoblastic bone formation and reduces the number of common lymphoid progenitors (CLPs) in the bone marrow, as well as the number of peripheral T and B cells. Meanwhile, the frequency and number of common myeloid progenitors (CMPs), neutrophils, and macrophages are increased during sepsis. These results suggest that osteoblasts support lymphopoiesis and suppress myeloid differentiation.
Sepsis-induced bone loss and lymphopenia are independent of the MyD88/TRIF pathway which mediates anti-bacterial defense. On the other hand, the reduced osteoblast and lymphocyte number during sepsis is recovered by treatment with a G-CSF antibody. Similar to sepsis, transient osteoblast ablation leads to a marked decrease in the CLP number and lymphopenia in the periphery, suggesting a role for osteoblasts in the regulation of lymphopoiesis. As mentioned above, it has been reported that G-CSF administration leads to the suppression of osteoblasts. These results indicate that the suppression of osteoblasts through G-CSF production causes the lymphopenia that occurs during sepsis.
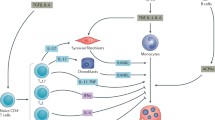
Furthermore, specific deletion of IL-7 in osteoblasts recapitulates the lymphopenic phenotype together with a lower CLP number, indicating that the osteoblast-derived IL-7 maintains CLPs in the bone marrow. In the study in which this was demonstrated, a reciprocal interaction between the immune and bone systems was evident, as acute inflammation induced a defect in bone cells resulting in immunosuppression. In addition, osteoblast activation by PTH administration provided an amelioration of sepsis-related lymphopenia. These findings suggest that osteoblasts are a potential treatment target in infectious diseases [26] (Fig. 2).

The role of osteoblasts in the maintenance of common lymphoid progenitors and lymphocyte differentiation in the bone marrow. The IL-7 produced by osteoblasts plays an important role in supporting common lymphoid progenitors in the bone marrow. When systemic inflammation ablates osteoblasts, the amount of IL-7 in the bone marrow also decreases. The decrease in the number of lymphoid common precursors exerts an impact on peripheral B and T cells. In addition, osteoblasts suppress myeloid cell differentiation
The immune regulation effected by osteoblasts is important not only for bacterial but also parasitic infection. Malaria is a life-threatening disease with serious complications, but most patients present with relatively weak symptoms. However, incomplete recovery from infection can result in chronic symptoms. It has been reported that products derived from the malaria parasite induce bone loss and growth retardation. Both resorption and formation are reduced in early-stage malaria infection. After eliminating the malaria parasite, the malaria parasite–derived product remains and accumulates in the bone marrow, resulting in chronic inflammation dependent on MyD88. It is shown that this response not only promotes inflammatory cytokine expression and osteoclastogenesis but also stimulates RANKL expression in osteoblasts. The administration of a vitamin D3 analog suppresses the bone loss and inflammation that occur in mice infected with malaria parasites [56]. These results highlight the risk of bone loss in malaria-infected patients and suggest the potential benefit of bone loss treatment in malaria.
Infection and bone tissue
There are other infectious diseases that are also involved in the defects of bone metabolism. It has been reported that osteoclastogenesis is affected by the proinflammatory cytokines and chemokines produced during immune responses. Therefore, immunological responses against a bacteria or virus may influence osteoclast function hence bone mass.
Most human immunodeficiency virus (HIV)-infected patients manifest abnormalities of the skeletal system. The prevalence of reduced bone mineral density (BMD) and increased fracture incidence is observed in HIV-infected patients compared with uninfected individuals. There are reports of enhanced osteoclastogenesis in HIV infection. HIV proteins can shift the OPG/RANKL ratio in favor of RANKL-mediated osteoclastogenesis [57]. The gp120 component of the HIV envelope can stimulate TNF-α production via the immune response [58, 59]. In addition, HIV viral protein R, a factor needed for viral replication that is required for the nuclear import of the HIV-1 pre-integration complex, upregulates RANKL [60]. C–C chemokine receptor 5 (CCR5) is a co-receptor of HIV that is important for HIV transmission. Ccr5-deficient (Ccr5−/−) mice present with dysfunctional osteoclasts and are resistant to RANKL-induced osteoporosis. Both a CCR5-antagonist and an anti-hCCR5 neutralizing antibody significantly reduced the expression levels of CTSK, NFATC1, and ACP5 in human monocytic U-937 cells [61]. Moreover, gp120 influences not only osteoclastogenesis but also osteoblastogenesis. TNF-α induced by gp120 elicits apoptosis of both osteoblast cell lines and primary cells [62]. HIV-1 Gag p55, which is a precursor protein that forms the core structure of the HIV virus and is indispensable to its reproduction, also inhibits osteoblastogenesis [63]. These results suggest that the HIV infection modulates bone metabolism. Considering the importance of osteoblast function in T cell differentiation, it is likely that the impaired bone marrow microenvironment affects the acquired immunodeficiency that develops from HIV infection [64].
Osteomyelitis is known as an infectious disease that causes bone loss and inflammation [65]. Most osteomyelitis patients are infected with Staphylococcus aureus (S. aureus). Although bone is normally resistant to bacterial colonization, S. aureus is able to colonize bone tissues via the bloodstream, e.g., by direct inoculation following trauma and subsequent spread of a contiguous infection. The pathophysiological significance of the bone remodeling that takes place in this disease has been elucidated using a mouse model of osteomyelitis [66, 67]. Osteoclast differentiation is promoted by S. aureus-stimulated proinflammatory cytokines, including IL-1β, IL-6, and TNF-α [68, 69]. CCL3 and CXCL2, which are observed in osteomyelitis, also induce osteoclast differentiation [70]. Staphylococcus aureus infection not only promotes osteoclastogenesis but also stimulates osteoblast apoptosis [71].
Paget’s disease is characterized by accelerated bone remodeling in which excessive bone resorption is followed by robust bone formation, resulting in a mosaic of woven bone that is characterized by significantly increased risk for fracture and deformity [72]. Although the osteoclast precursors from Paget’s disease patients are known to express measles virus nucleocapsid protein (MVNP), which enhances osteoclast formation, the mechanism by which MVNP stimulates abnormal osteoclastogenesis is unclear at present. A recent study reported that MVNP expression in osteoclasts increases the NFAT-activating protein with an ITAM motif (NFAM) 1, and NFAM1 inhibition suppresses MVNP-induced osteoclastogenesis. MVNP modulation of the NFAM1 signaling axis might therefore play a role in the enhanced osteoclast differentiation observed in Paget’s disease patients [73].
Periodontitis is an oral inflammatory disease characterized by periodontal pocket formation and alveolar bone resorption [74, 75]. RANKL plays a critical role in periodontal bone resorption. Innate immune cells stimulated by oral bacteria produce a number of inflammatory cytokines including IL-1β, IL-6, and TNF-α [76, 77]. These cytokines contribute to the expansion of Th17 and exFoxp3Th17 cells, which in turn triggers osteoclastogenesis through increased RANKL expression [78, 79].
Osteonecrosis of the jaw is associated with bone antiresorptive drugs, such as bisphosphonate and denosumab. Although the mechanism has not been fully elucidated, it has been suggested that infection triggers osteonecrosis due to reduced bone turnover [80].
Since chronic inflammation is associated with bone loss, it is possible that the disturbance of bone homeostasis by bacterial or viral infection influences the host defense during the course of infectious diseases.
Conclusion
As assay technologies and mouse genetics have progressed, the more our understanding of the bone marrow microenvironment has advanced. Although most of the studies have been conducted in the steady state, analyses of mice under pathological conditions have led to the novel recognition of reciprocal interactivity between bone and immune cells. In the sepsis model, osteoblasts maintain lymphopoiesis, which is suppressed by sepsis-induced osteoblast ablation. Furthermore, malaria, HIV, or S. aureus infection leads to a decrease in bone mass, a result which is linked to the production of inflammatory cytokines. It has been found that pharmacological activation of osteoblasts not only increases bone mass but also ameliorates certain immune deficits. Although classical osteoimmunology has been studied mainly under steady-state condition, recent studies have shed light on the mutual relationship between the bone and immune systems in the acute as well as chronic immune response. Further studies are expected to lead to novel therapeutic strategies against infection by targeting bone cells.
References
Takayanagi H (2012) New developments in osteoimmunology. Nat Rev Rheumatol 8:684–689
Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, Sawa S, Nitta T, Takayanagi H (2017) Osteoimmunology: the conceptual framework unifying the immune and skeletal systems. Physiol Rev 97:1295–1349
Takayanagi H (2007) Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol 7:292–304
Terashima A, Takayanagi H (2018) Overview of osteoimmunology. Calcif Tissue Int 102:503–511
Morrison SJ, Scadden DT (2014) The bone marrow niche for haematopoietic stem cells. Nature 505:327–334
Lord BI, Testa NG, Hendry JH (1975) The relative spatial distributions of CFUs and CFUc in the normal mouse femur. Blood 46:65–72
Gong JK (1978) Endosteal marrow: a rich source of hematopoietic stem cells. Science 199:1443–1445
Taichman RS, Emerson SG (1994) Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J Exp Med 179:1677–1682
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, Milner LA, Kronenberg HM, Scadden DT (2003) Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425:841–846
Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, Harris S, Wiedemann LM, Mishina Y, Li L (2003) Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425:836–841
Chan CK, Chen CC, Luppen CA, Kim JB, DeBoer AT, Wei K, Helms JA, Kuo CJ, Kraft DL, Weissman IL (2009) Endochondral ossification is required for haematopoietic stem-cell niche formation. Nature 457:490–494
Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, Frenette PS (2013) PDGFRα and CD51 mark human Nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J Exp Med 210:1351–1367 %1357 2013/1306/1319
Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, Suda T (2004) Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 118:149–161
Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, Gomei Y, Iwasaki H, Matsuoka S, Miyamoto K, Miyazaki H, Takahashi T, Suda T (2007) Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell 1:685–697
Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grunewald E, Cheng T, Dombkowski D, Calvi LM, Rittling SR, Scadden DT (2005) Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med 201:1781–1791
Zhou BO, Ding L, Morrison SJ (2015) Hematopoietic stem and progenitor cells regulate the regeneration of their niche by secreting Angiopoietin-1. Elife 4:e05521
Kiel MJ, Radice GL, Morrison SJ (2007) Lack of evidence that hematopoietic stem cells depend on N-cadherin-mediated adhesion to osteoblasts for their maintenance. Cell Stem Cell 1:204–217
Lymperi S, Horwood N, Marley S, Gordon MY, Cope AP, Dazzi F (2008) Strontium can increase some osteoblasts without increasing hematopoietic stem cells. Blood 111:1173–1181
Bromberg O, Frisch BJ, Weber JM, Porter RL, Civitelli R, Calvi LM (2012) Osteoblastic N-cadherin is not required for microenvironmental support and regulation of hematopoietic stem and progenitor cells. Blood 120:303–313
Ding L, Saunders TL, Enikolopov G, Morrison SJ (2012) Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481:457–462
Ding L, Morrison SJ (2013) Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495:231–235
Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC (2013) CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495:227–230
Omatsu Y, Seike M, Sugiyama T, Kume T, Nagasawa T (2014) Foxc1 is a critical regulator of haematopoietic stem/progenitor cell niche formation. Nature 508:536–540
Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, Nagasawa T (2010) The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 33:387–399
Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, Scadden DT, Kronenberg HM (2008) Osteoblastic regulation of B lymphopoiesis is mediated by Gsa-dependent signaling pathways. Proc Natl Acad Sci U S A 105:16976–16981
Terashima A, Okamoto K, Nakashima T, Akira S, Ikuta K, Takayanagi H (2016) Sepsis-induced osteoblast ablation causes immunodeficiency. Immunity 44:1434–1443
Yu VWC, Saez B, Cook C, Lotinun S, Pardo-Saganta A, Wang YH, Lymperi S, Ferraro F, Raaijmakers M, Wu JY, Zhou L, Rajagopal J, Kronenberg HM, Baron R, Scadden DT (2015) Specific bone cells produce DLL4 to generate thymus-seeding progenitors from bone marrow. J Exp Med 212:759–774
Rankin EB, Wu C, Khatri R, Wilson TL, Andersen R, Araldi E, Rankin AL, Yuan J, Kuo CJ, Schipani E, Giaccia AJ (2012) The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell 149:63–74
Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, Ebert BL, Al-Shahrour F, Hasserjian RP, Scadden EO, Aung Z, Matza M, Merkenschlager M, Lin C, Rommens JM, Scadden DT (2010) Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 464:852–857
Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R, Brum A, Park D, Galili N, Mukherjee S, Teruya-Feldstein J, Raza A, Rabadan R, Berman E, Kousteni S (2014) Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 506:240–244
Kode A, Mosialou I, Manavalan SJ, Rathinam CV, Friedman RA, Teruya-Feldstein J, Bhagat G, Berman E, Kousteni S (2015) FoxO1-dependent induction of acute myeloid leukemia by osteoblasts in mice. Leukemia 30:1–13
Lowell CA, Niwa M, Soriano P, Varmus HE (1996) Deficiency of the Hck and Src tyrosine kinases results in extreme levels of extramedullary hematopoiesis. Blood 87:1780–1792
Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J (1999) RANK is essential for osteoclast and lymph node development. Genes Dev 13:2412–2424
Begg SK, Radley JM, Pollard JW, Chisholm OT, Stanley ER, Bertoncello I (1993) Delayed hematopoietic development in osteopetrotic (op/op) mice. J Exp Med 177:237–242
Sreehari S, Naik DR, Eapen M (2011) Osteopetrosis: a rare cause of anemia. Hematol Rep 3:e1
Gerritsen EJ, Vossen JM, van Loo IH, Hermans J, Helfrich MH, Griscelli C, Fischer A (1994) Autosomal recessive osteopetrosis: variability of findings at diagnosis and during the natural course. Pediatrics 93:247–253
Lymperi S, Ersek A, Ferraro F, Dazzi F, Horwood NJ (2011) Inhibition of osteoclast function reduces hematopoietic stem cell numbers in vivo. Blood 117:1540–1549
Mansour A, Abou-Ezzi G, Sitnicka E, Jacobsen SE, Wakkach A, Blin-Wakkach C (2012) Osteoclasts promote the formation of hematopoietic stem cell niches in the bone marrow. J Exp Med 209:537–549
Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC, Kos CH, Pollak MR, Brown EM, Scadden DT (2006) Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 439:599–603
Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, Taketo MM, Karlsson S, Iwama A, Nakauchi H (2011) Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 147:1146–1158
Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, Tesio M, Samstein RM, Goichberg P, Spiegel A, Elson A, Lapidot T (2006) Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med 12:657–664
Miyamoto K, Yoshida S, Kawasumi M, Hashimoto K, Kimura T, Sato Y, Kobayashi T, Miyauchi Y, Hoshi H, Iwasaki R, Miyamoto H, Hao W, Morioka H, Chiba K, Yasuda H, Penninger JM, Toyama Y, Suda T, Miyamoto T (2011) Osteoclasts are dispensable for hematopoietic stem cell maintenance and mobilization. J Exp Med 208:2175–2181
Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, Levesque JP (2010) Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 116:4815–4828
Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS (2006) Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124:407–421
Asada N, Katayama Y, Sato M, Minagawa K, Wakahashi K, Kawano H, Kawano Y, Sada A, Ikeda K, Matsui T, Tanimoto M (2013) Matrix-embedded osteocytes regulate mobilization of hematopoietic stem/progenitor cells. Cell Stem Cell 12:737–747
Bone RC, Sprung CL, Sibbald WJ (1992) Definitions for sepsis and organ failure. Crit Care Med 20:724–726
Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ (1992) Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee American College of Chest Physicians/Society of Critical Care Medicine. Chest 101:1644–1655
Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G, SCCM/ESICM/ACCP/ATS/SIS (2003) 2001 SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Crit Care Med 31:1250–1256
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC (2016) The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 315:801–810
Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O’Malley KA, Wynn JL, Antonenko S, Al-Quran SZ, Swan R, Chung CS, Atkinson MA, Ramphal R, Gabrilovich DI, Reeves WH, Ayala A, Phillips J, Laface D, Heyworth PG, Clare-Salzler M, Moldawer LL (2007) MyD88-dependent expansion of an immature GR-1+CD11b+ population induces T cell suppression and Th2 polarization in sepsis. J Exp Med 204:1463–1474
Davey MS, Morgan MP, Liuzzi AR, Tyler CJ, Khan MWA, Szakmany T, Hall JE, Moser B, Eberl M (2014) Microbe-specific unconventional T cells induce human neutrophil differentiation into antigen cross-presenting cells. J Immunol 193:3704–3716
Monserrat J, de Pablo R, Reyes E, Díaz D, Barcenilla H, Zapata MR, De la Hera A, Prieto A, Alvarez-Mon M (2009) Clinical relevance of the severe abnormalities of the T cell compartment in septic shock patients. Crit Care 13:R26
Chaudhry H, Zhou J, Zhong Y, Ali MM, McGuire F, Nagarkatti PS, Nagarkatti M (2013) Role of cytokines as a double-edged sword in sepsis. In Vivo 27:669–684
Bo L, Wang F, Zhu J, Li J, Deng X (2011) Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: a meta-analysis. Crit Care 15:R58
Mohammad RA (2010) Use of granulocyte colony-stimulating factor in patients with severe sepsis or septic shock. Am J Health Syst Pharm 67:1238–1245
Lee MSJ, Maruyama K, Fujita Y, Konishi A, Lelliott PM, Itagaki S, Horii T, Lin JW, Khan SM, Kuroda E, Akira S, Ishii KJ, Coban C (2017) Products persist in the bone marrow and promote chronic bone loss. Sci Immunol 2:eaam8093
Gibellini D, Borderi M, De Crignis E, Cicola R, Vescini F, Caudarella R, Chiodo F, Re MC (2007) RANKL/OPG/TRAIL plasma levels and bone mass loss evaluation in antiretroviral naive HIV-1-positive men. J Med Virol 79:1446–1454
Clouse KA, Cosentino LM, Weih KA, Pyle SW, Robbins PB, Hochstein HD, Natarajan V, Farrar WL (1991) The HIV-1 gp120 envelope protein has the intrinsic capacity to stimulate monokine secretion. J Immunol 147:2892–2901
Fakruddin JM, Laurence J (2003) HIV envelope gp120-mediated regulation of osteoclastogenesis via receptor activator of nuclear factor kappa B ligand (RANKL) secretion and its modulation by certain HIV protease inhibitors through interferon-gamma/RANKL cross-talk. J Biol Chem 278:48251–48258
Fakruddin JM, Laurence J (2005) HIV-1 Vpr enhances production of receptor of activated NF-kappaB ligand (RANKL) via potentiation of glucocorticoid receptor activity. Arch Virol 150:67–78
Lee JW, Hoshino A, Inoue K, Saitou T, Uehara S, Kobayashi Y, Ueha S, Matsushima K, Yamaguchi A, Imai Y, Iimura T (2017) The HIV co-receptor CCR5 regulates osteoclast function. Nat Commun 8:2226
Gibellini D, De Crignis E, Ponti C, Cimatti L, Borderi M, Tschon M, Giardino R, Re MC (2008) HIV-1 triggers apoptosis in primary osteoblasts and HOBIT cells through TNFalpha activation. J Med Virol 80:1507–1514
Cotter EJ, Malizia AP, Chew N, Powderly WG, Doran PP (2007) HIV proteins regulate bone marker secretion and transcription factor activity in cultured human osteoblasts with consequent potential implications for osteoblast function and development. AIDS Res Hum Retrovir 23:1521–1530
Beaupere C, Garcia M, Larghero J, Fève B, Capeau J, Lagathu C (2015) The HIV proteins Tat and Nef promote human bone marrow mesenchymal stem cell senescence and alter osteoblastic differentiation. Aging Cell 14:534–546
Lew DP, Waldvogel FA (2004) Osteomyelitis. Lancet 364:369–379
Cassat JE, Hammer ND, Campbell JP, Benson MA, Perrien DS, Mrak LN, Smeltzer MS, Torres VJ, Skaar EP (2013) A secreted bacterial protease tailors the Staphylococcus aureus virulence repertoire to modulate bone remodeling during osteomyelitis. Cell Host Microbe 13:759–772
Loughran AJ, Gaddy D, Beenken KE, Meeker DG, Morello R, Zhao H, Byrum SD, Tackett AJ, Cassat JE, Smeltzer MS (2016) Impact of sarA and phenol-soluble modulins on the pathogenesis of osteomyelitis in diverse clinical isolates of Staphylococcus aureus. Infect Immun 84:2586–2594
Yoshii T, Magara S, Miyai D, Nishimura H, Kuroki E, Furudoi S, Komori T, Ohbayashi C (2002) Local levels of interleukin-1β, -4, -6 and tumor necrosis factor alpha in an experimental model of murine osteomyelitis due to staphylococcus aureus. Cytokine 19:59–65
Zwerina J, Hayer S, Tohidast-Akrad M, Bergmeister H, Redlich K, Feige U, Dunstan C, Kollias G, Steiner G, Smolen J, Schett G (2004) Single and combined inhibition of tumor necrosis factor, interleukin-1, and RANKL pathways in tumor necrosis factor-induced arthritis: effects on synovial inflammation, bone erosion, and cartilage destruction. Arthritis Rheum 50:277–290
Dapunt U, Maurer S, Giese T, Gaida MM, Hänsch GM (2014) The macrophage inflammatory proteins MIP1α (CCL3) and MIP2α (CXCL2) in implant-associated osteomyelitis: linking inflammation to bone degradation. Mediat Inflamm 2014:728619
Young AB, Cooley ID, Chauhan VS, Marriott I (2011) Causative agents of osteomyelitis induce death domain-containing TNF-related apoptosis-inducing ligand receptor expression on osteoblasts. Bone 48:857–863
Singer FR (2015) Paget’s disease of bone-genetic and environmental factors. Nat Rev Endocrinol 11:662–671
Sambandam Y, Sundaram K, Saigusa T, Balasubramanian S, Reddy SV (2017) NFAM1 signaling enhances osteoclast formation and bone resorption activity in Paget’s disease of bone. Bone 101:236–244
Moutsopoulos NM, Madianos PN (2006) Low-grade inflammation in chronic infectious diseases: paradigm of periodontal infections. Ann N Y Acad Sci 1088:251–264
Sokos D, Everts V, de Vries TJ (2015) Role of periodontal ligament fibroblasts in osteoclastogenesis: a review. J Periodontal Res 50:152–159
Wada N, Maeda H, Yoshimine Y, Akamine A (2004) Lipopolysaccharide stimulates expression of osteoprotegerin and receptor activator of NF-kappa B ligand in periodontal ligament fibroblasts through the induction of interleukin-1 beta and tumor necrosis factor-α. Bone 35:629–635
Morandini AC, Sipert CR, Gasparoto TH, Greghi SL, Passanezi E, Rezende ML, Sant’ana AP, Campanelli AP, Garlet GP, Santos CF (2010) Differential production of macrophage inflammatory protein-1alpha, stromal-derived factor-1, and IL-6 by human cultured periodontal ligament and gingival fibroblasts challenged with lipopolysaccharide from P. gingivalis. J Periodontol 81:310–317
Tsukasaki M, Komatsu N, Nagashima K, Nitta T, Pluemsakunthai W, Shukunami C, Iwakura Y, Nakashima T, Okamoto K, Takayanagi H (2018) Host defense against oral microbiota by bone-damaging T cells. Nat Commun 9:701
Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H (2013) Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 20:62–68
Woo SB, Hellstein JW, Kalmar JR (2006) Narrative [corrected] review: bisphosphonates and osteonecrosis of the jaws. Ann Intern Med 144:753–761
Funding
This work was supported in part by a grant for Practical Research Project for Rare/Intractable Diseases (JP19ek0109379) from the Japan Agency for Medical Research and Development.
Author information
Authors and Affiliations
Contributions
AT and HT wrote the manuscript. HT also edited it.
Corresponding author
Ethics declarations
Conflict of interest
AT declare that she belongs to an endowment department, Department of Osteoimmunology, supported with an unrestricted grant from AYUMI Pharmaceutical Corporation, Chugai Pharmaceutical Co., Ltd., MIKI HOUSE Co., Ltd., and Noevir Co., Ltd.
Additional information
This article is a contribution to the special issue on Osteoimmunology - Guest Editor: Mary Nakamura
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Terashima, A., Takayanagi, H. The role of bone cells in immune regulation during the course of infection. Semin Immunopathol 41, 619–626 (2019). https://doi.org/10.1007/s00281-019-00755-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-019-00755-2