
Abstract
Nociception is the process of transmission of painful signals by nociceptors in the primary afferent nerve fibers, which specifically respond to noxious stimuli. These noxious stimuli are detected by nociceptors and converted into electrical signals, which are then transmitted to the spinal cord, thalamus, and the cerebral cortex, where pain is finally sensed. Transient receptor potential (TRP) ion channels have emerged as a family of evolutionarily conserved ligand-gated ion channels that function as molecular detectors of physical stimuli. Several member of this family, at least six channels from three TRP family subtypes (TRPV1–4, TRPM8, and TRPA1), are expressed in nociceptors, where they act as transducers for signals from thermal, chemical, and mechanical stimuli and play crucial roles in the generation and development of pathological pain perception. This review focuses on the increasing evidence of TRP channel involvement and contribution in nociceptive pain and the pain hypersensitivity associated with peripheral inflammation or neuropathy, and on the renewed interest in targeting TRP channels for pain relief.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Pain is defined by the International Association for the Study of Pain (IASP) as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage.” Painful signals generated by tissue damage are detected by nociceptors, which transfer the signals to the central nervous system to produce this unpleasant sensory and emotional experience. The two main types of nociceptors that detect painful signals are unmyelinated C-fibers and myelinated Aδ-fibers [1]. These nerve fibers use electrical signals, by means of action potentials, to convey noxious information rapidly to the brain. In the generation of these action potentials that carry the signal and produce the pain sensation, a crucial role is played by ion channels that are specifically expressed in the aforementioned nerve fibers. In recent years, ion channels such as transient receptor potential (TRP) channels, ATP channels, acid-sensing ion channels, and Piezo channels (whose opening causes the cation influx that depolarizes sensory neurons) have been identified as key pain receptors [2, 3]. Among these channels, certain TRP channels such as TRPV1–4, TRPA1, and TRPM8 have attracted considerable attention because they have been shown to be expressed in nociceptors, where they act as detectors and transducers for thermal, chemical, and mechanical stimuli. Activation or sensitization of these channels is deeply involved in the pathological pain condition. Pain can generally be classified as a nociceptive, inflammatory, or neuropathic condition depending on the pathogenesis. This review highlights the emerging role of TRP channels, with an emphasis on TRPV1 and TRPA1, in the peripheral mechanisms of these pain conditions.
TRPs in nociceptive pain as chemical and physical sensors
Nociceptive pain represents the normal response of somatic or visceral tissues to noxious insult or injury. This form of pain is typically the result of tissue damage caused by a noxious stimulus, but it is a critical component of the body’s defense system that protects against further damage and heals the damaged tissue.
In the elucidation of the molecular mechanism of pain, limited progress was made until the capsaicin receptor (TRPV1) was cloned in 1997. TRPV1 was first reported by Caterina et al. as a nonselective cation channel that exhibits high calcium permeability [4]. TRPV1 is expressed by the peripheral and central terminals of small-diameter sensory neurons (nociceptors) in the dorsal root ganglion (DRG), trigeminal ganglion (TG), nodose ganglion (NG), geniculate ganglion (GG), and jugular ganglion [4–8], and TRPV1 modulates pain transmission at the first sensory synapse [9–12]. Approximately 35–50 % of all DRG or TG neurons were found to be TRPV1-positive [7, 13], and these represent a large population of unmyelinated C-fibers and a small population of thinly myelinated Ad-fibers. TRPV1 expression occurs largely in association with the expression of substance P and calcitonin gene-related peptide. TRPV1 functions as a polymodal receptor [14] because it is activated not only by capsaicin but also by noxious heat (≥43 °C), low pH (protons) [4, 6, 13], and several other exogenous or endogenous agents (Table 1) such as camphor [15], allicin [16, 17], spider toxins [18, 19], anandamide [20], arachidonic acid metabolites [21], N-arachidonoyl dopamine [22], oleoylethanolamide (OEA) [23], N-OEA [24], and polyamines [25]. Intra-cutaneous administration of these chemicals in animals induces pain-related behaviors, which suggests a role of TRPV1 in nociception.
TRPV1-deficient mice show a complete loss of physiological and behavioral responses to capsaicin, partial diminution in the responses to noxious heat, and normal responses to noxious mechanical stimuli [26, 27]. In sensory neurons from mice lacking TRPV1, in vitro calcium influx or electrophysiological response to capsaicin, protons, and heat is completely absent. TRPV1 is widely recognized as a heat sensor, but, intriguingly, TRPV1 null mice in certain studies showed normal sensitivity to acute noxious heat [28, 29], which suggests that other molecules might participate in the sensation of noxious heat when TRPV1 is not expressed.
TRPA1 is another nonselective cation channel of the TRP family that is critically involved in nociception. In the sensory nervous system, TRPA1 was reported to be expressed in the DRG [7, 30], TG [7], NG [31], superior cervical ganglion (SCG) [32], and GG [8, 33]. Interestingly, TRPA1 is highly coexpressed with TRPV1 in small-diameter nociceptors [7, 30, 34], which raises the question of functional specificity. Like TRPV1, TRPA1 also functions as a polymodal receptor: TRPA1 can be activated by multiple stimuli such as chemical, thermal (≤18 °C), mechanical, and osmotic stimuli [30, 35–37], as detailed next.
First, the TRPA1 channel functions as a chemical sensor and detects a remarkably broad range of chemicals (see Table 1), including exogenous chemicals such as the pungent ingredients of mustard oil, garlic, wintergreen oil, clove oil, ginger, and cinnamon oil, all of which induce acute painful burning or pricking sensation [16, 35, 38, 39]; and endogenous chemicals that are produced during oxidative or nitrative stress, including α,β-unsaturated aldehydes such as 4-hydroxynonenal (4-HNE), cyclopentenone, prostaglandin metabolites such as 15d-PGJ2, hydrogen peroxide/hydroxyl radicals, and nitrooleic acid [40–44]. Most of these chemicals are electrophilic and have been shown to activate TRPA1 by covalently modifying cysteine residues in the channel [45, 46]. The nociception caused by these reactive chemicals is drastically reduced or eliminated in TRPA1 knockout mice [45, 47, 48]. Conversely, TRPA1 can also be activated by certain nonelectrophilic chemicals such as thymol [49], NSAIDs such as flufenamic acid and niflumic acid [50], isoflurane [51], farnesyl thiosalicylic acid [49, 52], 1,4-dihydropyridines [53], nicotine [54], and icilin [30]. The sensory qualities of most of these chemicals are clearly related to their noxious stimuli, and this supports the view that TRPA1 is a key sensor of chemical damage.
Second, TRPA1 has been suggested to serve as a thermal sensor of noxious cold stimuli [30, 38, 55], although cold-induced TRPA1 activation is substantially weaker than the activation induced by allyl isothiocyanate [56]. This finding was supported by two independent studies in which TRPA1 knockout mice displayed impaired response to cold [56, 57]. However, this property remains controversial because other studies failed to confirm direct, cold-induced TRPA1 activation, and TRPA1-deficient mice were also found to show normal cold sensitivity [31, 35, 58].
Third, TRPA1 might also detect mechanical nociceptive stimuli, although this idea requires further verification. Among TRPs, the TRPA1 channel possesses a unique structure composed of several ankyrin repeats in the N terminus; these repeats have been hypothesized to act as a spring when under mechanical stress [59]. In Drosophila and Caenorhabditis elegans, TRPA1 has been demonstrated to be involved in mechanical nociception [60, 61]. In mammals, TRPA1 expressed in mechanosensitive hair cells of the inner ear has been proposed to function as a putative mechanosensor [31, 37]. Furthermore, mice deficient in TRPA1 display impaired behavioral sensitivity to punctate mechanical stimuli [57]. In one study in which skin-nerve preparations from TRPA1-deficient mice were used, cutaneous fibers were demonstrated to exhibit a drastic reduction in their firing rate in response to mechanical stimuli [62]. In another study, under acute pharmacological inhibition of TRPA1 achieved through antagonist application to the receptive field, cutaneous fibers were shown to exhibit markedly decreased firing rates in response to mechanical stimuli [63]. Moreover, Brierley et al. used in vitro electrophysiological and pharmacological approaches and observed that TRPA1 is required for normal mechanosensory function in specific subsets of vagal, splanchnic, and pelvic afferents, and further found that behavioral responses to noxious colonic distension were substantially reduced in TRPA1-deficient mice [64]. A recent study also indicated that TRPA1 mediates mechanical currents in the plasma membrane; in this study, direct recordings of cultured mouse sensory neurons were obtained under application of a rapid, focal mechanical stimulation to the soma membrane [65]. Collectively, this emerging evidence suggests that TRPA1 plays a role in mechanosensitivity.
Other TRP channels such as TRPV2, V3, V4, and M8 have been suggested to contribute to pathological pain in various disease states. However, the role of these channels in detecting nociceptive pain is poorly understood. Their agonists and antagonists are listed in Table 1. TRPV2 is activated by high temperature (≥52 °C) [66], which is consistent with the temperature range sensed by Ad-fibers [67, 68]. Based on this activation feature and on the expression of TRPV2 mainly in medium-sized primary afferents that do not express TRPV1, TRPV2 has been suggested to act as a high-threshold temperature sensor in Ad nociceptors [66, 69, 70]. However, evidence from studies conducted using knockout mice does not support an acute heat-sensing role for TRPV2 [29, 71].
TRPV3 is activated by warm temperature (≥34 °C) [72–74] and certain chemical agents, including camphor, menthol, carvacrol, eugenol, insensol, and 2-aminoethoxydiphenyl borate [75–81]. A role of TRPV3 in thermosensation was revealed in knockout mice, which showed strong deficits in response to innocuous and noxious heat but not in other sensory modalities [76]. TRPV3 is prominently expressed in—and functions in—skin keratinocytes, although both peripheral and central neurons, including neurons in the DRG, TG, SCG, spinal cord, and certain brain regions, are also TRPV3-positive [72–74]. In keratinocytes, TRPV3-mediated currents and calcium influx have been reported [82, 83], but evidence supporting functional TRPV3 expression in sensory neurons has not been obtained. The TRPV3 expressed in skin keratinocytes might participate in sensing physical stimuli by means of signal relay to sensory nerve endings through chemical mediators such as ATP and prostaglandin E2 [72, 84–87].
TRPV4 is another thermosensor that is activated by warm temperature (≥27 °C) [88, 89]. Unlike other thermo-TRP channels, TRPV4 was originally detected as an osmosensory channel that is activated by extracellular osmolarity (hypotonic cell swelling) and was expected to function as a putative mammalian mechanosensitive channel [90–92]. Furthermore, TRPV4 is not only activated by physical stimuli but also various stimuli ranging from exogenous to endogenous chemicals such as anandamide and arachidonic acid [93], bisandrographolide [94], 4-alpha-phorbol 12,13-didecanoate [95], acetylcholine [96], apigenin [97], and dimethylallyl pyrophosphate [98]. Therefore, like TRPV1 and AI, TRPV4 can be also defined as a polymodal receptor. Moreover, TRPV4 is expressed by a wide range of tissues, including in the sensory neurons present in the DRG, TG, and NG [99–101]. Mice lacking TRPV4 display a marked reduction in sensitivity to pressure, which suggests an essential role of TRPV4 in the normal detection of pressure and as a receptor of the high-threshold mechanosensory complex [102].
TRPM8 is defined as a sensor of cool temperatures (≤25 °C) [103, 104], and it can also be activated by cooling compounds such as menthol, icilin, and several other agents [38, 103–106]. TRPM8 is expressed in a small population of nociceptors located in the DRG and TG that do not express TRPV1 [7, 103, 104]. Given that most nociceptors are TRPV1-positive neurons, the lack of TRPM8 coexpression with TRPV1 suggests that TRPM8 is not involved in the detection of noxious stimuli. Nevertheless, a recent study involving selective cell ablation demonstrated that TRPM8-containing neurons are required for noxious cold aversion [107].
Taken together, TRP channels in peripheral sensory neurons (or in keratinocytes) act as sensors of various stimuli. Such stimuli from outside of the body (exogenous agonists) stimulate directly certain TRP channels and evoke nociceptive pain (Fig. 1). Meanwhile, endogenous agonists are often induced or upregulated following pathological conditions such as inflammation or nerve injury, which in turn stimulate their receptors (TRP channels) and contribute to the inflammatory or neuropathic pain as one potential mechanism.

TRPs in nociceptive pain
TRPs in inflammatory pain
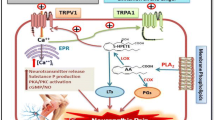
Inflammatory pain results from the activation and sensitization of the nociceptive pain pathway—characterized by a reduced threshold and an increased responsiveness of sensory neurons—by a variety of mediators released at the sites of tissue inflammation. These mediators, which include proinflammatory cytokines, chemokines, reactive oxygen species, protein kinases, vasoactive amines, lipids, ATP, acids, and other factors, might be released by infiltrating leukocytes, vascular endothelial cells, or tissue-resident mast cells after tissue injury. The mediators associated with the inflammatory response directly or indirectly stimulate or sensitize peripheral sensory neurons (nociceptors), which results in a reduction in the activation threshold of the nociceptors and an increase in their responsiveness. The activation or sensitization (or both) of TRP channels in the sensory nerve during inflammation is considered to be the major mechanism underlying inflammatory pain (Fig. 2a).

TRPs in inflammatory pain. a Inflammatory soup and oxidative products activate and/or sensitize TRPs in peripheral sensory neurons. b Intracellular mechanisms involved in TRP channels’ sensitization
Studies conducted using knockout mice have clearly demonstrated the essential roles of certain TRP channels in the initiation or maintenance of inflammatory pain. In TRPV1-deficient mice, the development of inflammatory thermal hyperalgesia was defective but mechanical hypersensitivity was unchanged [26–28, 108, 109], whereas TRPA1-deficient mice exhibited markedly reduced development of hyperalgesia in response to injections of inflammation-related chemicals, including formalin, 4-HNE, prostaglandin metabolites, H2O2, and bradykinin [41, 45, 47, 48, 58, 110]. TRPV4 is another TRP family member that might contribute to inflammatory pain; results obtained using TRPV4-deficient mice indicate that this channel is required for the development of thermal hyperalgesia induced by both cutaneous and colonic inflammation [111–115]. Conversely, TRPV2 might not be involved in inflammatory pain: as compared with wild-type mice, TRPV2 knockout mice showed no change in thermal and mechanical responses in a model of inflammation (induced by complete Freund’s adjuvant (CFA)) [71]. TRPV3 expressed in skin keratinocytes might play a role under inflammatory conditions by enhancing the peripheral input through inflammatory mediators [84, 85, 116]. TRPM8 was demonstrated to contribute the cold-induced hyperalgesia observed in inflammation produced by CFA; this hyperalgesia was impaired in TRPM8 knockout mice [117].
TRPs might participate in inflammatory pain through two mechanisms. In one mechanism, certain products released from inflamed tissues, including arachidonic acid derivatives such as anandamide, 15d-PGJ2, and 12-(S)-HPETE, and oxidative stress products such as 4-HNE and H2O2, might function as endogenous TRPV1, V4, or A1 activators and directly stimulate them and contribute to spontaneous pain [41, 48, 93, 110, 118–120]. These findings demonstrate that TRPs could contribute to inflammatory pain by acting as the terminal substrate for inflammatory mediators. Sensitization of TRPs is considered to be another potential mechanism underlying inflammatory pain. In the development of inflammatory pain, three cellular and molecular mechanisms have been established (Fig. 2b). First, inflammation might increase TRP channel expression in sensory neurons either transcriptionally or posttranslationally. For example, peripheral inflammation increases TRPV1 levels in nociceptor peripheral terminals in a transcription-independent manner and contributes to the maintenance of inflammatory heat hypersensitivity [121]. By contract, peripheral inflammation induces transcriptional upregulation of TRPA1 (but not TRPM8) in DRG neurons and thus contributes to inflammatory cold hyperalgesia [122]. Moreover, neurotrophic factors, including members of the nerve growth factor and glial cell-derived neurotrophic factor families, have been suggested to be involved in the gene regulation of both TRPV1 and TRPA1 [121–126]. Second, because TRP channels are transmembrane receptors, the number of functional TRP channels present in the plasma membrane is a critical determinant of channel function. Emerging evidence has demonstrated that inflammatory mediators induce a rapid translocation of TRP channels from the cytoplasm to the plasma membrane following the activation of second-messenger pathways and subsequent posttranslational modification such as channel phosphorylation or glycosylation [127–134]. Numerous proinflammatory agents and trophic factors have been suggested to sensitize TRP channels through posttranslational regulation; these molecules include CCL3 [135], bradykinin [136–138], serotonin [139], histamine [140], glutamate [10, 127, 141], PGE2 [142], ATP [143], trypsin/tryptase [144, 145], NGF [131], and insulin/insulin-like growth factor 1 [128, 146, 147]. Phosphorylation is one of the molecular mechanisms involved in the sensitization of TRP channels. Inflammatory mediators act on receptors that are coupled to either G proteins or tyrosine kinase pathways and thus activate phospholipase C (PLC) and/or adenylate cyclase; this, in turn, induces second-messenger pathways such as those involving protein kinase C (PKC), protein kinase A (PKA), and/or phosphoinositide 3-kinase (PI3K) activation and leads to channel phosphorylation. Previous studies have shown that PKC activation-sensitized, SNARE-dependent exocytosis, and Src-PI3K-dependent phosphorylation of TRPV1 are involved in the channel’s membrane insertion under inflammatory conditions [127, 129, 148] and that cyclin-dependent kinase 5 might regulate the transport of TRPV1 from the Golgi apparatus to the plasma membrane [149]. Third, in addition to the translocation of the channels from the cytoplasm to the membrane, channel phosphorylation and a disinhibition mechanism might alter the channel structure and functionally enhance channel sensitivity. Inflammatory mediators might sensitize TRPs by releasing the inhibition of TRP channels, which is subjected to regulation by phosphatidylinositol 4,5-bisphosphate turnover mediated by PLC activation [34, 150, 151].
TRPs in neuropathic pain
Neuropathic pain is initiated or caused by a primary lesion or disease in the somatosensory nervous system; this type of pain includes the pain in diabetic neuropathy or chemotherapy-related peripheral neuropathy, postherpetic neuralgia, spinal cord injury pain, and phantom limb pain. Pathophysiological changes in primary sensory neurons induced by nerve injury and disease and the consequent changes in signal processing in the central nervous system constitute the underlying mechanism of neuropathic pain. Following peripheral nerve damage, TRP channel expression changes dynamically in sensory neurons. First, the expression of TRP channels, including TRPV1, TRPA1, TRPV3, and TRPM8, decreases in the injured neuron in response to peripheral axonal damage; this downregulation of TRP channels might be a result of the loss of trophic support following injury [8, 13, 122, 152–154]. Concurrently, however, certain TRP channels, such as TRPV1 and TRPA1, are upregulated in nearby spared neurons following nerve injury, and this increase correlates closely with behavioral change [13, 122, 152–154]. In studies conducted using human tissue, TRPV1 and TRPV3 levels were found to be decreased in the skin of patients with diabetic or other painful neuropathies but increased in intact nerve fibers in certain patients with pain hypersensitivity, whereas the level of TRPV4 was observed to remain unchanged [99, 155]. One potential mechanism has been suggested for these phenotypic changes: the damaged neurons might release growth factors and neurotransmitters into the surrounding area and, consequently, cause an increase in the excitability of surrounding neurons [13, 153, 156, 157]. These dynamic changes contrast the changes detected in inflammation, in which TRP channels are generally upregulated (see above); this reflects the notion that inflammatory pain simply features a “surplus” of nociceptive signaling, whereas the sensory abnormalities of neuropathies include not only hypersensitivity (hyperalgesia or allodynia), but also paresthesias, hypoesthesia, or complete deficits perceived as numbness [158] (Fig. 3).

TRPs in neuropathic pain
In addition to the drastic phenotypic change observed as alterations in TRPV1 and A1 channel expression patterns, knockdown or pharmacological inhibition of these TRPs has been widely demonstrated to reduce pain behavior in several animal models of neuropathic pain [122, 154, 159–178]. Interestingly, however, studies conducted using knockout mice have provided little evidence supporting the notion that these TRPs (TRPV1 and TRPA1) are essential contributors to the development of neuropathic pain. For example, knockout of TRPV1 or TRPA1 typically exerts no effect on pain behaviors induced by nerve injury [26, 27, 57] or diabetes [179]. By contrast, mechanical hyperalgesia induced by paclitaxel, vincristine, or diabetes was strongly reduced in TRPV4 knockout mice, and knockdown or pharmacological inhibition of TRPV4 reduced chemotherapy-induced mechanical or hypotonic hyperalgesia [173, 180]; this suggests a critical role of TRPV4 in mechanotransduction in the setting of nerve injury [181]. Conversely, TRPM8 null mice display a marked reduction in injury-induced responsiveness to acetone cooling [117], and pharmacological blockade of TRPM8 leads to a reduction in nerve injury-induced cold hypersensitivity [182], which indicates that TRPM8 might play a role in cooling-induced neuropathic pain. Furthermore, recent studies have suggested the involvement of TRPM2 in nerve injury-induced mechanical allodynia [183–185].
In conclusion, TRPs act as chemical, thermal, and mechanical sensors in nociceptors and detect nociceptive pain. Certain TRPs—particularly TRPV1 and TRPA1—are essential for the development of hyperalgesia and allodynia under inflammatory conditions, whereas TRPV4 and TRPM8 might contribute to neuropathic hypersensitivity to mechanical and cold stimuli, respectively. However, the essential role of all such TRP channels in the development of neuropathic pain remains to be further identified, although antagonists of certain TRPs have been shown to reduce the pain behavior following nerve injury.
TRPs as targets of analgesics
The roles of TRP channels in mediating pathological pain make them potential targets for analgesics. TRP channels are located in nociceptors where pain is generated, and thus the simplest access for analgesics would involve blocking the channels directly. Two main approaches have been proposed to inhibit TRP channels: blocking the channel activity using antagonists and, paradoxically, stimulating the channels to desensitize them. Drug development studies have focused on TRPV1, TRPV3, TRPM8, and TRPA1, which have been clearly demonstrated to be involved in pathological pain (see above). Currently, at least seven antagonists of TRPV1, two of TRPA1, and one of TRPV3 are being clinically tested by pharmaceutical industries [186–188]. However, as discussed in the preceding section, several TRP channels perform dual functions: they act as sensors/detectors of nociceptive signals under normal conditions, which helps prevent tissue damage, and they also act as contributors to inflammatory or neuropathic pain under pathological conditions, and this can pose a risk of noxious perception being blunted when the channels’ activity is blocked. In accord, in healthy human volunteers, increase in heat pain threshold was observed after the administration of TRPV1 antagonists, including SB-705498 [189], MK-2295 [190], ABT-102 [191], and AZD1386 [192]. Hyperthermia is another adverse effect of TRPV1 antagonists; almost all TRPV1 antagonists undergoing clinical development, such as AMG517 (Amgen) [193], ABT-102 (Abbott) [191], and AZD1386 (AstraZeneca) [192], caused hyperthermia in human volunteers that in certain cases lasted for 1–4 days, with core body temperatures rising up to 40.2 °C [193].
In contrast to the challenges associated with developing antagonists, in the case of agonists that have been applied to locally desensitize TRP channels and have been used clinically, severe adverse effects have not been reported, although the agonists might produce initial irritation or even degeneration of sensory nerves. This notion of a paradoxical use of agonists might be derived from the experience gained with certain herbal remedies used in traditional medicine. For example, preparations or prescriptions containing agonists of TRP channels, such as menthol (TRPM8), cinnamaldehyde (TRPA1), or shogaol (TRPV1), have long been used topically or orally to relieve neuralgia, arthralgia, menstrual pain, and headache in traditional Chinese medicine. Moreover, capsaicin-containing creams, occlusive patches, and liquid formulations have been developed and used for treating chronic painful conditions such as diabetic neuropathy, postherpetic neuralgia, and other painful disorders [186, 194–196]. A recent study analyzed several related human studies involving a total of over 2000 participants, and the results indicated that high-concentration capsaicin patches were effective in the treatment of postherpetic neuralgia and HIV neuropathy [197]. As an ultrapotent analog of capsaicin, resiniferatoxin has been developed preclinically for treating intractable cancer pain through intrathecal administration [186, 198]. Furthermore, clinical trials have shown beneficial effects of topically administered peppermint oil in alleviating thermally elicited pain and postherpetic neuralgia [199, 200]; in accord, the results of animal experiments have shown that oral menthol administration can cause short-term analgesia [201]. Icilin and menthol might also reduce mechanical and thermal hypersensitivity caused by peripheral nerve injury [202]. The minor side effects observed with these agonists provide useful information for developing TRP-targeted analgesics.
As discussed in preceding sections, the sensitization of TRPs contributes to inflammatory and neuropathic pain. In addition to simply blocking TRP channels by using antagonists, an alternative strategy for developing analgesics that target TRPs involves preventing or reducing TRP sensitization. Targeting the channels by using endogenous modulators is an attractive approach for controlling TRP activity. As already noted, numerous intracellular and extracellular agents might activate/sensitize or inhibit TRP channels, and thus interfering with these processes might result in analgesia. For example, inhibition of PKCε has been reported to completely block both inflammatory mediator-induced TRPV1 sensitization and heat hyperalgesia [203, 204]. Another study recently reported that disrupting the interaction between TRPV1 and A-kinase-anchoring protein 79/150 using inhibitory peptides prevents TRPV1 sensitization and inflammatory hyperalgesia [205]. Furthermore, endogenous inhibitors of TRPV1/A1, such as resolvins [206] and artemin [207], have been proposed for use in the treatment of pain conditions. These attempts might yield novel ideas for maximizing the reduction of the activity of sensitized TRPs while concurrently minimizing any disruption of their sensor functions.
Conclusions
Eighteen years have passed since the first pain sensor, TRPV1, was cloned [4]. During this period, numerous pain-related TRP channels have been discovered and comprehensively investigated. The rapid progress in the identification and characterization of TRP channels has enhanced our understanding of both nociceptive and pathological pain. Based on the growing knowledge of the TRP channels and their involvement in distinct pain conditions, we can expect pharmacological interventions targeting TRPV1/A1 channels to be effective in the treatment of chronic inflammatory syndromes that involve thermal hyperalgesia; conversely, targeting TRPV4 or TRPM8 should be effective for treating neuropathic conditions that include mechanical or cold hypersensitivity, respectively, in diseases that involve nerve injury or neuropathy. Moreover, several TRPs are expressed in a board range of tissue and organs that are outside the somatosensory system; thus, in addition to functioning as pain sensors or enhancers, these channels might perform other functions in organisms and might be involved in various diseases. Consequently, pharmacological inhibition of TRP channels might produce both clinical benefits for analgesia and unexpected adverse effects, such as the hyperthermia noted in the preceding section. To overcome these challenges, further investigation must be conducted using newly devised approaches, such as the discovery of second-generation antagonists or selective delivery of drugs through topical application or local injection instead of systemic administration.
References
Woolf CJ, Ma Q (2007) Nociceptors–noxious stimulus detectors. Neuron 55:353–364
Hwang SW, Oh U (2007) Current concepts of nociception: nociceptive molecular sensors in sensory neurons. Curr Opin Anaesthesiol 20:427–434
Volkers L, Mechioukhi Y, Coste B (2015) Piezo channels: from structure to function. Pflugers Arch 467:95–99
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389:816–824
Yamamoto Y, Sato Y, Taniguchi K (2007) Distribution of TRPV1- and TRPV2-immunoreactive afferent nerve endings in rat trachea. J Anat 211:775–783
Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D (1998) The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21:531–543
Kobayashi K, Fukuoka T, Obata K, Yamanaka H, Dai Y, Tokunaga A, Noguchi K (2005) Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with adelta/c-fibers and colocalization with trk receptors. J Comp Neurol 493:596–606
Tatsumi E, Katsura H, Kobayashi K, Yamanaka H, Tsuzuki K, Noguchi K, Sakagami M (2015) Changes in transient receptor potential channels in the rat geniculate ganglion after chorda tympani nerve injury. Neuroreport 26:856–861
Jeffry JA, Yu SQ, Sikand P, Parihar A, Evans MS, Premkumar LS (2009) Selective targeting of TRPV1 expressing sensory nerve terminals in the spinal cord for long lasting analgesia. PLoS One 4:e7021
Sikand P, Premkumar LS (2007) Potentiation of glutamatergic synaptic transmission by protein kinase C-mediated sensitization of TRPV1 at the first sensory synapse. J Physiol 581:631–647
Baccei ML, Bardoni R, Fitzgerald M (2003) Development of nociceptive synaptic inputs to the neonatal rat dorsal horn: glutamate release by capsaicin and menthol. J Physiol 549:231–242
Nakatsuka T, Furue H, Yoshimura M, Gu JG (2002) Activation of central terminal vanilloid receptor-1 receptors and alpha beta-methylene-ATP-sensitive P2X receptors reveals a converged synaptic activity onto the deep dorsal horn neurons of the spinal cord. J Neurosci 22:1228–1237
Fukuoka T, Tokunaga A, Tachibana T, Dai Y, Yamanaka H, Noguchi K (2002) VR1, but not P2X(3), increases in the spared L4 DRG in rats with L5 spinal nerve ligation. Pain 99:111–120
Clapham DE (2003) TRP channels as cellular sensors. Nature 426:517–524
Xu H, Blair NT, Clapham DE (2005) Camphor activates and strongly desensitizes the transient receptor potential vanilloid subtype 1 channel in a vanilloid-independent mechanism. J Neurosci 25:8924–8937
Macpherson LJ, Geierstanger BH, Viswanath V, Bandell M, Eid SR, Hwang S, Patapoutian A (2005) The pungency of garlic: activation of TRPA1 and TRPV1 in response to allicin. Curr Biol 15:929–934
Macpherson LJ, Hwang SW, Miyamoto T, Dubin AE, Patapoutian A, Story GM (2006) More than cool: promiscuous relationships of menthol and other sensory compounds. Mol Cell Neurosci 32:335–343
Siemens J, Zhou S, Piskorowski R, Nikai T, Lumpkin EA, Basbaum AI, King D, Julius D (2006) Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 444:208–212
Bohlen CJ, Priel A, Zhou S, King D, Siemens J, Julius D (2010) A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell 141:834–845
Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED (1999) Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400:452–457
Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U (2000) Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci U S A 97:6155–6160
Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SN, Geppetti P, Walker JM, Di Marzo V (2002) An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci U S A 99:8400–8405
Ahern GP (2003) Activation of TRPV1 by the satiety factor oleoylethanolamide. J Biol Chem 278:30429–30434
Chu CJ, Huang SM, De Petrocellis L, Bisogno T, Ewing SA, Miller JD, Zipkin RE, Daddario N, Appendino G, Di Marzo V, Walker JM (2003) N-Oleoyldopamine, a novel endogenous capsaicin-like lipid that produces hyperalgesia. J Biol Chem 278:13633–13639
Ahern GP, Wang X, Miyares RL (2006) Polyamines are potent ligands for the capsaicin receptor TRPV1. J Biol Chem 281:8991–8995
Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288:306–313
Bolcskei K, Helyes Z, Szabo A, Sandor K, Elekes K, Nemeth J, Almasi R, Pinter E, Petho G, Szolcsanyi J (2005) Investigation of the role of TRPV1 receptors in acute and chronic nociceptive processes using gene-deficient mice. Pain 117:368–376
Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA (2000) Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405:183–187
Woodbury CJ, Zwick M, Wang S, Lawson JJ, Caterina MJ, Koltzenburg M, Albers KM, Koerber HR, Davis BM (2004) Nociceptors lacking TRPV1 and TRPV2 have normal heat responses. J Neurosci 24:6410–6415
Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A (2003) ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112:819–829
Nagata K, Duggan A, Kumar G, Garcia-Anoveros J (2005) Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J Neurosci 25:4052–4061
Smith MP, Beacham D, Ensor E, Koltzenburg M (2004) Cold-sensitive, menthol-insensitive neurons in the murine sympathetic nervous system. Neuroreport 15:1399–1403
Katsura H, Tsuzuki K, Noguchi K, Sakagami M (2006) Differential expression of capsaicin-, menthol-, and mustard oil-sensitive receptors in naive rat geniculate ganglion neurons. Chem Senses 31:681–688
Dai Y, Wang S, Tominaga M, Yamamoto S, Fukuoka T, Higashi T, Kobayashi K, Obata K, Yamanaka H, Noguchi K (2007) Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J Clin Invest 117:1979–1987
Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D (2004) Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427:260–265
Zhang XF, Chen J, Faltynek CR, Moreland RB, Neelands TR (2008) Transient receptor potential A1 mediates an osmotically activated ion channel. Eur J Neurosci 27:605–611
Corey DP, Garcia-Anoveros J, Holt JR, Kwan KY, Lin SY, Vollrath MA, Amalfitano A, Cheung EL, Derfler BH, Duggan A, Geleoc GS, Gray PA, Hoffman MP, Rehm HL, Tamasauskas D, Zhang DS (2004) TRPA1 is a candidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature 432:723–730
Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, Patapoutian A (2004) Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41:849–857
Bautista DM, Movahed P, Hinman A, Axelsson HE, Sterner O, Hogestatt ED, Julius D, Jordt SE, Zygmunt PM (2005) Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci U S A 102:12248–12252
Bessac BF, Sivula M, von Hehn CA, Escalera J, Cohn L, Jordt SE (2008) TRPA1 is a major oxidant sensor in murine airway sensory neurons. J Clin Invest 118:1899–1910
Andersson DA, Gentry C, Moss S, Bevan S (2008) Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci 28:2485–2494
Taylor-Clark TE, Ghatta S, Bettner W, Undem BJ (2009) Nitrooleic acid, an endogenous product of nitrative stress, activates nociceptive sensory nerves via the direct activation of TRPA1. Mol Pharmacol 75:820–829
Miyamoto T, Dubin AE, Petrus MJ, Patapoutian A (2009) TRPV1 and TRPA1 mediate peripheral nitric oxide-induced nociception in mice. PLoS One 4:e7596
Sawada Y, Hosokawa H, Matsumura K, Kobayashi S (2008) Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur J Neurosci 27:1131–1142
Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A (2007) Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445:541–545
Hinman A, Chuang HH, Bautista DM, Julius D (2006) TRP channel activation by reversible covalent modification. Proc Natl Acad Sci U S A 103:19564–19568
McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM (2007) TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci U S A 104:13525–13530
Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, Imamachi N, Andre E, Patacchini R, Cottrell GS, Gatti R, Basbaum AI, Bunnett NW, Julius D, Geppetti P (2007) 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A 104:13519–13524
Xiao B, Dubin AE, Bursulaya B, Viswanath V, Jegla TJ, Patapoutian A (2008) Identification of transmembrane domain 5 as a critical molecular determinant of menthol sensitivity in mammalian TRPA1 channels. J Neurosci 28:9640–9651
Hu H, Tian J, Zhu Y, Wang C, Xiao R, Herz JM, Wood JD, Zhu MX (2010) Activation of TRPA1 channels by fenamate nonsteroidal anti-inflammatory drugs. Pflugers Arch 459:579–592
Matta JA, Cornett PM, Miyares RL, Abe K, Sahibzada N, Ahern GP (2008) General anesthetics activate a nociceptive ion channel to enhance pain and inflammation. Proc Natl Acad Sci U S A 105:8784–8789
Maher M, Ao H, Banke T, Nasser N, Wu NT, Breitenbucher JG, Chaplan SR, Wickenden AD (2008) Activation of TRPA1 by farnesyl thiosalicylic acid. Mol Pharmacol 73:1225–1234
Fajardo O, Meseguer V, Belmonte C, Viana F (2008) TRPA1 channels: novel targets of 1,4-dihydropyridines. Channels (Austin) 2:429–438
Talavera K, Gees M, Karashima Y, Meseguer VM, Vanoirbeek JA, Damann N, Everaerts W, Benoit M, Janssens A, Vennekens R, Viana F, Nemery B, Nilius B, Voets T (2009) Nicotine activates the chemosensory cation channel TRPA1. Nat Neurosci 12:1293–1299
Sawada Y, Hosokawa H, Hori A, Matsumura K, Kobayashi S (2007) Cold sensitivity of recombinant TRPA1 channels. Brain Res 1160:39–46
Karashima Y, Talavera K, Everaerts W, Janssens A, Kwan KY, Vennekens R, Nilius B, Voets T (2009) TRPA1 acts as a cold sensor in vitro and in vivo. Proc Natl Acad Sci U S A 106:1273–1278
Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, Corey DP (2006) TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron 50:277–289
Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D (2006) TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124:1269–1282
Sotomayor M, Corey DP, Schulten K (2005) In search of the hair-cell gating spring elastic properties of ankyrin and cadherin repeats. Structure 13:669–682
Tracey WD Jr, Wilson RI, Laurent G, Benzer S (2003) painless, a Drosophila gene essential for nociception. Cell 113:261–273
Kindt KS, Viswanath V, Macpherson L, Quast K, Hu H, Patapoutian A, Schafer WR (2007) Caenorhabditis elegans TRPA-1 functions in mechanosensation. Nat Neurosci 10:568–577
Kwan KY, Glazer JM, Corey DP, Rice FL, Stucky CL (2009) TRPA1 modulates mechanotransduction in cutaneous sensory neurons. J Neurosci 29:4808–4819
Kerstein PC, del Camino D, Moran MM, Stucky CL (2009) Pharmacological blockade of TRPA1 inhibits mechanical firing in nociceptors. Mol Pain 5:19
Brierley SM, Hughes PA, Page AJ, Kwan KY, Martin CM, O'Donnell TA, Cooper NJ, Harrington AM, Adam B, Liebregts T, Holtmann G, Corey DP, Rychkov GY, Blackshaw LA (2009) The ion channel TRPA1 is required for normal mechanosensation and is modulated by algesic stimuli. Gastroenterology 137:2084–95 e3
Vilceanu D, Stucky CL (2010) TRPA1 mediates mechanical currents in the plasma membrane of mouse sensory neurons. PLoS One 5:e12177
Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D (1999) A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398:436–441
Nagy I, Rang H (1999) Noxious heat activates all capsaicin-sensitive and also a sub-population of capsaicin-insensitive dorsal root ganglion neurons. Neuroscience 88:995–997
Meyer RA, Campbell JN (1981) Myelinated nociceptive afferents account for the hyperalgesia that follows a burn to the hand. Science 213:1527–1529
Greffrath W, Binzen U, Schwarz ST, Saaler-Reinhardt S, Treede RD (2003) Co-expression of heat sensitive vanilloid receptor subtypes in rat dorsal root ganglion neurons. Neuroreport 14:2251–2255
Ahluwalia J, Rang H, Nagy I (2002) The putative role of vanilloid receptor-like protein-1 in mediating high threshold noxious heat-sensitivity in rat cultured primary sensory neurons. Eur J Neurosci 16:1483–1489
Park U, Vastani N, Guan Y, Raja SN, Koltzenburg M, Caterina MJ (2011) TRP vanilloid 2 knock-out mice are susceptible to perinatal lethality but display normal thermal and mechanical nociception. J Neurosci 31:11425–11436
Peier AM, Reeve AJ, Andersson DA, Moqrich A, Earley TJ, Hergarden AC, Story GM, Colley S, Hogenesch JB, McIntyre P, Bevan S, Patapoutian A (2002) A heat-sensitive TRP channel expressed in keratinocytes. Science 296:2046–2049
Xu H, Ramsey IS, Kotecha SA, Moran MM, Chong JA, Lawson D, Ge P, Lilly J, Silos-Santiago I, Xie Y, DiStefano PS, Curtis R, Clapham DE (2002) TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature 418:181–186
Smith GD, Gunthorpe MJ, Kelsell RE, Hayes PD, Reilly P, Facer P, Wright JE, Jerman JC, Walhin JP, Ooi L, Egerton J, Charles KJ, Smart D, Randall AD, Anand P, Davis JB (2002) TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 418:186–190
Colton CK, Zhu MX (2007) 2-Aminoethoxydiphenyl borate as a common activator of TRPV1, TRPV2, and TRPV3 channels. Handb Exp Pharmacol 173–87
Moqrich A, Hwang SW, Earley TJ, Petrus MJ, Murray AN, Spencer KS, Andahazy M, Story GM, Patapoutian A (2005) Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science 307:1468–1472
Sherkheli MA, Vogt-Eisele AK, Weber K, Hatt H (2013) Camphor modulates TRPV3 cation channels activity by interacting with critical pore-region cysteine residues. Pak J Pharm Sci 26:431–438
Moussaieff A, Rimmerman N, Bregman T, Straiker A, Felder CC, Shoham S, Kashman Y, Huang SM, Lee H, Shohami E, Mackie K, Caterina MJ, Walker JM, Fride E, Mechoulam R (2008) Incensole acetate, an incense component, elicits psychoactivity by activating TRPV3 channels in the brain. FASEB J 22:3024–3034
Vogt-Eisele AK, Weber K, Sherkheli MA, Vielhaber G, Panten J, Gisselmann G, Hatt H (2007) Monoterpenoid agonists of TRPV3. Br J Pharmacol 151:530–540
Sherkheli MA, Benecke H, Doerner JF, Kletke O, Vogt-Eisele AK, Gisselmann G, Hatt H (2009) Monoterpenoids induce agonist-specific desensitization of transient receptor potential vanilloid-3 (TRPV3) ion channels. J Pharm Pharm Sci 12:116–128
Xu H, Delling M, Jun JC, Clapham DE (2006) Oregano, thyme and clove-derived flavors and skin sensitizers activate specific TRP channels. Nat Neurosci 9:628–635
Chung MK, Lee H, Caterina MJ (2003) Warm temperatures activate TRPV4 in mouse 308 keratinocytes. J Biol Chem 278:32037–32046
Chung MK, Lee H, Mizuno A, Suzuki M, Caterina MJ (2004) TRPV3 and TRPV4 mediate warmth-evoked currents in primary mouse keratinocytes. J Biol Chem 279:21569–21575
Mandadi S, Sokabe T, Shibasaki K, Katanosaka K, Mizuno A, Moqrich A, Patapoutian A, Fukumi-Tominaga T, Mizumura K, Tominaga M (2009) TRPV3 in keratinocytes transmits temperature information to sensory neurons via ATP. Pflugers Arch 458:1093–1102
Huang SM, Lee H, Chung MK, Park U, Yu YY, Bradshaw HB, Coulombe PA, Walker JM, Caterina MJ (2008) Overexpressed transient receptor potential vanilloid 3 ion channels in skin keratinocytes modulate pain sensitivity via prostaglandin E2. J Neurosci 28:13727–13737
Lumpkin EA, Caterina MJ (2007) Mechanisms of sensory transduction in the skin. Nature 445:858–865
Lee H, Caterina MJ (2005) TRPV channels as thermosensory receptors in epithelial cells. Pflugers Arch 451:160–167
Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M (2002) Heat-evoked activation of the ion channel, TRPV4. J Neurosci 22:6408–6414
Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nilius B (2002) Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem 277:47044–47051
Wissenbach U, Bodding M, Freichel M, Flockerzi V (2000) Trp12, a novel Trp related protein from kidney. FEBS Lett 485:127–134
Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S (2000) Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103:525–535
Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD (2000) OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol 2:695–702
Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B (2003) Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 424:434–438
Smith PL, Maloney KN, Pothen RG, Clardy J, Clapham DE (2006) Bisandrographolide from Andrographis paniculata activates TRPV4 channels. J Biol Chem 281:29897–29904
Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, Vriens J, Cairns W, Wissenbach U, Prenen J, Flockerzi V, Droogmans G, Benham CD, Nilius B (2002) Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem 277:13569–13577
Adapala RK, Talasila PK, Bratz IN, Zhang DX, Suzuki M, Meszaros JG, Thodeti CK (2011) PKCalpha mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. Am J Physiol Heart Circ Physiol 301:H757–H765
Ma X, He D, Ru X, Chen Y, Cai Y, Bruce IC, Xia Q, Yao X, Jin J (2012) Apigenin, a plant-derived flavone, activates transient receptor potential vanilloid 4 cation channel. Br J Pharmacol 166:349–358
Bang S, Yoo S, Yang TJ, Cho H, Hwang SW (2012) Nociceptive and pro-inflammatory effects of dimethylallyl pyrophosphate via TRPV4 activation. Br J Pharmacol 166:1433–1443
Facer P, Casula MA, Smith GD, Benham CD, Chessell IP, Bountra C, Sinisi M, Birch R, Anand P (2007) Differential expression of the capsaicin receptor TRPV1 and related novel receptors TRPV3, TRPV4 and TRPM8 in normal human tissues and changes in traumatic and diabetic neuropathy. BMC Neurol 7:11
Gopinath P, Wan E, Holdcroft A, Facer P, Davis JB, Smith GD, Bountra C, Anand P (2005) Increased capsaicin receptor TRPV1 in skin nerve fibres and related vanilloid receptors TRPV3 and TRPV4 in keratinocytes in human breast pain. BMC Womens Health 5:2
Chung MK, Jung SJ, Oh SB (2011) Role of TRP channels in pain sensation. Adv Exp Med Biol 704:615–636
Suzuki M, Mizuno A, Kodaira K, Imai M (2003) Impaired pressure sensation in mice lacking TRPV4. J Biol Chem 278:22664–22668
McKemy DD, Neuhausser WM, Julius D (2002) Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416:52–58
Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, Patapoutian A (2002) A TRP channel that senses cold stimuli and menthol. Cell 108:705–715
Sherkheli MA, Vogt-Eisele AK, Bura D, Beltran Marques LR, Gisselmann G, Hatt H (2010) Characterization of selective TRPM8 ligands and their structure activity response (S.A.R) relationship. J Pharm Pharm Sci 13:242–253
Behrendt HJ, Germann T, Gillen C, Hatt H, Jostock R (2004) Characterization of the mouse cold-menthol receptor TRPM8 and vanilloid receptor type-1 VR1 using a fluorometric imaging plate reader (FLIPR) assay. Br J Pharmacol 141:737–745
Pogorzala LA, Mishra SK, Hoon MA (2013) The cellular code for mammalian thermosensation. J Neurosci 33:5533–5541
Keeble J, Russell F, Curtis B, Starr A, Pinter E, Brain SD (2005) Involvement of transient receptor potential vanilloid 1 in the vascular and hyperalgesic components of joint inflammation. Arthritis Rheum 52:3248–3256
Barton NJ, McQueen DS, Thomson D, Gauldie SD, Wilson AW, Salter DM, Chessell IP (2006) Attenuation of experimental arthritis in TRPV1R knockout mice. Exp Mol Pathol 81:166–170
Cruz-Orengo L, Dhaka A, Heuermann RJ, Young TJ, Montana MC, Cavanaugh EJ, Kim D, Story GM (2008) Cutaneous nociception evoked by 15-delta PGJ2 via activation of ion channel TRPA1. Mol Pain 4:30
Alessandri-Haber N, Joseph E, Dina OA, Liedtke W, Levine JD (2005) TRPV4 mediates pain-related behavior induced by mild hypertonic stimuli in the presence of inflammatory mediator. Pain 118:70–79
Brierley SM, Page AJ, Hughes PA, Adam B, Liebregts T, Cooper NJ, Holtmann G, Liedtke W, Blackshaw LA (2008) Selective role for TRPV4 ion channels in visceral sensory pathways. Gastroenterology 134:2059–2069
Sipe WE, Brierley SM, Martin CM, Phillis BD, Cruz FB, Grady EF, Liedtke W, Cohen DM, Vanner S, Blackshaw LA, Bunnett NW (2008) Transient receptor potential vanilloid 4 mediates protease activated receptor 2-induced sensitization of colonic afferent nerves and visceral hyperalgesia. Am J Physiol Gastrointest Liver Physiol 294:G1288–G1298
Alessandri-Haber N, Dina OA, Joseph EK, Reichling D, Levine JD (2006) A transient receptor potential vanilloid 4-dependent mechanism of hyperalgesia is engaged by concerted action of inflammatory mediators. J Neurosci 26:3864–3874
Todaka H, Taniguchi J, Satoh J, Mizuno A, Suzuki M (2004) Warm temperature-sensitive transient receptor potential vanilloid 4 (TRPV4) plays an essential role in thermal hyperalgesia. J Biol Chem 279:35133–35138
Miyamoto T, Petrus MJ, Dubin AE, Patapoutian A (2011) TRPV3 regulates nitric oxide synthase-independent nitric oxide synthesis in the skin. Nat Commun 2:369
Colburn RW, Lubin ML, Stone DJ Jr, Wang Y, Lawrence D, D'Andrea MR, Brandt MR, Liu Y, Flores CM, Qin N (2007) Attenuated cold sensitivity in TRPM8 null mice. Neuron 54:379–386
Taylor-Clark TE, Undem BJ, Macglashan DW Jr, Ghatta S, Carr MJ, McAlexander MA (2008) Prostaglandin-induced activation of nociceptive neurons via direct interaction with transient receptor potential A1 (TRPA1). Mol Pharmacol 73:274–281
Ross RA (2003) Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol 140:790–801
Singh Tahim A, Santha P, Nagy I (2005) Inflammatory mediators convert anandamide into a potent activator of the vanilloid type 1 transient receptor potential receptor in nociceptive primary sensory neurons. Neuroscience 136:539–548
Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ (2002) p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36:57–68
Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Tominaga M, Noguchi K (2005) TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest 115:2393–2401
Elitt CM, McIlwrath SL, Lawson JJ, Malin SA, Molliver DC, Cornuet PK, Koerber HR, Davis BM, Albers KM (2006) Artemin overexpression in skin enhances expression of TRPV1 and TRPA1 in cutaneous sensory neurons and leads to behavioral sensitivity to heat and cold. J Neurosci 26:8578–8587
Amaya F, Shimosato G, Nagano M, Ueda M, Hashimoto S, Tanaka Y, Suzuki H, Tanaka M (2004) NGF and GDNF differentially regulate TRPV1 expression that contributes to development of inflammatory thermal hyperalgesia. Eur J Neurosci 20:2303–2310
Ikeda-Miyagawa Y, Kobayashi K, Yamanaka H, Okubo M, Wang S, Dai Y, Yagi H, Hirose M, Noguchi K (2015) Peripherally increased artemin is a key regulator of TRPA1/V1 expression in primary afferent neurons. Mol Pain 11:8
Anand U, Otto WR, Casula MA, Day NC, Davis JB, Bountra C, Birch R, Anand P (2006) The effect of neurotrophic factors on morphology, TRPV1 expression and capsaicin responses of cultured human DRG sensory neurons. Neurosci Lett 399:51–56
Morenilla-Palao C, Planells-Cases R, Garcia-Sanz N, Ferrer-Montiel A (2004) Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J Biol Chem 279:25665–25672
Van Buren JJ, Bhat S, Rotello R, Pauza ME, Premkumar LS (2005) Sensitization and translocation of TRPV1 by insulin and IGF-I. Mol Pain 1:17
Zhang X, Huang J, McNaughton PA (2005) NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J 24:4211–4223
Cuajungco MP, Grimm C, Oshima K, D'Hoedt D, Nilius B, Mensenkamp AR, Bindels RJ, Plomann M, Heller S (2006) PACSINs bind to the TRPV4 cation channel. PACSIN 3 modulates the subcellular localization of TRPV4. J Biol Chem 281:18753–18762
Stein AT, Ufret-Vincenty CA, Hua L, Santana LF, Gordon SE (2006) Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J Gen Physiol 128:509–522
Schmidt M, Dubin AE, Petrus MJ, Earley TJ, Patapoutian A (2009) Nociceptive signals induce trafficking of TRPA1 to the plasma membrane. Neuron 64:498–509
D'Hoedt D, Owsianik G, Prenen J, Cuajungco MP, Grimm C, Heller S, Voets T, Nilius B (2008) Stimulus-specific modulation of the cation channel TRPV4 by PACSIN 3. J Biol Chem 283:6272–6280
Xu H, Fu Y, Tian W, Cohen DM (2006) Glycosylation of the osmoresponsive transient receptor potential channel TRPV4 on Asn-651 influences membrane trafficking. Am J Physiol Ren Physiol 290:F1103–F1109
Zhang N, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ (2005) A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc Natl Acad Sci U S A 102:4536–4541
Premkumar LS, Ahern GP (2000) Induction of vanilloid receptor channel activity by protein kinase C. Nature 408:985–990
Sugiura T, Tominaga M, Katsuya H, Mizumura K (2002) Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. J Neurophysiol 88:544–548
Wang YY, Chang RB, Waters HN, McKemy DD, Liman ER (2008) The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. J Biol Chem 283:32691–32703
Ohta T, Ikemi Y, Murakami M, Imagawa T, Otsuguro K, Ito S (2006) Potentiation of transient receptor potential V1 functions by the activation of metabotropic 5-HT receptors in rat primary sensory neurons. J Physiol 576:809–822
Kajihara Y, Murakami M, Imagawa T, Otsuguro K, Ito S, Ohta T (2010) Histamine potentiates acid-induced responses mediating transient receptor potential V1 in mouse primary sensory neurons. Neuroscience 166:292–304
Hu HJ, Bhave G, Gereau RW (2002) Prostaglandin and protein kinase A-dependent modulation of vanilloid receptor function by metabotropic glutamate receptor 5: potential mechanism for thermal hyperalgesia. J Neurosci 22:7444–7452
Zhang X, Li L, McNaughton PA (2008) Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron 59:450–461
Tominaga M, Wada M, Masu M (2001) Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc Natl Acad Sci U S A 98:6951–6956
Amadesi S, Nie J, Vergnolle N, Cottrell GS, Grady EF, Trevisani M, Manni C, Geppetti P, McRoberts JA, Ennes H, Davis JB, Mayer EA, Bunnett NW (2004) Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J Neurosci 24:4300–4312
Dai Y, Moriyama T, Higashi T, Togashi K, Kobayashi K, Yamanaka H, Tominaga M, Noguchi K (2004) Proteinase-activated receptor 2-mediated potentiation of transient receptor potential vanilloid subfamily 1 activity reveals a mechanism for proteinase-induced inflammatory pain. J Neurosci 24:4293–4299
Li Y, Cai J, Han Y, Xiao X, Meng XL, Su L, Liu FY, Xing GG, Wan Y (2014) Enhanced function of TRPV1 via up-regulation by insulin-like growth factor-1 in a rat model of bone cancer pain. Eur J Pain 18:774–784
Miura M, Sasaki M, Mizukoshi K, Shibasaki M, Izumi Y, Shimosato G, Amaya F (2011) Peripheral sensitization caused by insulin-like growth factor 1 contributes to pain hypersensitivity after tissue injury. Pain 152:888–895
Camprubi-Robles M, Planells-Cases R, Ferrer-Montiel A (2009) Differential contribution of SNARE-dependent exocytosis to inflammatory potentiation of TRPV1 in nociceptors. FASEB J 23:3722–3733
Xing BM, Yang YR, Du JX, Chen HJ, Qi C, Huang ZH, Zhang Y, Wang Y (2012) Cyclin-dependent kinase 5 controls TRPV1 membrane trafficking and the heat sensitivity of nociceptors through KIF13B. J Neurosci 32:14709–14721
Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411:957–962
Kim D, Cavanaugh EJ, Simkin D (2008) Inhibition of transient receptor potential A1 channel by phosphatidylinositol-4,5-bisphosphate. Am J Physiol Cell Physiol 295:C92–C99
Hudson LJ, Bevan S, Wotherspoon G, Gentry C, Fox A, Winter J (2001) VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur J Neurosci 13:2105–2114
Fukuoka T, Noguchi K (2002) Contribution of the spared primary afferent neurons to the pathomechanisms of neuropathic pain. Mol Neurobiol 26:57–67
Katsura H, Obata K, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Sakagami M, Noguchi K (2006) Antisense knock down of TRPA1, but not TRPM8, alleviates cold hyperalgesia after spinal nerve ligation in rats. Exp Neurol 200:112–123
Lauria G, Morbin M, Lombardi R, Capobianco R, Camozzi F, Pareyson D, Manconi M, Geppetti P (2006) Expression of capsaicin receptor immunoreactivity in human peripheral nervous system and in painful neuropathies. J Peripher Nerv Syst 11:262–271
Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K (2001) Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci 21:4891–4900
Sexton JE, Vernon J, Wood JN (2014) TRPs and pain. Handb Exp Pharmacol 223:873–897
Devor M (2013) Clinical states/neuropathic pain. In: McMahon SB, Koltzenburg M, Tracey I, Turk DC (eds) Wall and Melzack's textbook of pain. Elsevier/Saunders, Philadelphia, pp 861–888
Hong S, Wiley JW (2005) Early painful diabetic neuropathy is associated with differential changes in the expression and function of vanilloid receptor 1. J Biol Chem 280:618–627
Jhaveri MD, Elmes SJ, Kendall DA, Chapman V (2005) Inhibition of peripheral vanilloid TRPV1 receptors reduces noxious heat-evoked responses of dorsal horn neurons in naive, carrageenan-inflamed and neuropathic rats. Eur J Neurosci 22:361–370
Christoph T, Gillen C, Mika J, Grunweller A, Schafer MK, Schiene K, Frank R, Jostock R, Bahrenberg G, Weihe E, Erdmann VA, Kurreck J (2007) Antinociceptive effect of antisense oligonucleotides against the vanilloid receptor VR1/TRPV1. Neurochem Int 50:281–290
Hong S, Agresta L, Guo C, Wiley JW (2008) The TRPV1 receptor is associated with preferential stress in large dorsal root ganglion neurons in early diabetic sensory neuropathy. J Neurochem 105:1212–1222
Hara T, Chiba T, Abe K, Makabe A, Ikeno S, Kawakami K, Utsunomiya I, Hama T, Taguchi K (2013) Effect of paclitaxel on transient receptor potential vanilloid 1 in rat dorsal root ganglion. Pain 154:882–889
Culshaw AJ, Bevan S, Christiansen M, Copp P, Davis A, Davis C, Dyson A, Dziadulewicz EK, Edwards L, Eggelte H, Fox A, Gentry C, Groarke A, Hallett A, Hart TW, Hughes GA, Knights S, Kotsonis P, Lee W, Lyothier I, McBryde A, McIntyre P, Paloumbis G, Panesar M, Patel S, Seiler MP, Yaqoob M, Zimmermann K (2006) Identification and biological characterization of 6-aryl-7-isopropylquinazolinones as novel TRPV1 antagonists that are effective in models of chronic pain. J Med Chem 49:471–474
Kanai Y, Nakazato E, Fujiuchi A, Hara T, Imai A (2005) Involvement of an increased spinal TRPV1 sensitization through its up-regulation in mechanical allodynia of CCI rats. Neuropharmacology 49:977–984
Walker KM, Urban L, Medhurst SJ, Patel S, Panesar M, Fox AJ, McIntyre P (2003) The VR1 antagonist capsazepine reverses mechanical hyperalgesia in models of inflammatory and neuropathic pain. J Pharmacol Exp Ther 304:56–62
Chen J, Joshi SK, DiDomenico S, Perner RJ, Mikusa JP, Gauvin DM, Segreti JA, Han P, Zhang XF, Niforatos W, Bianchi BR, Baker SJ, Zhong C, Simler GH, McDonald HA, Schmidt RG, McGaraughty SP, Chu KL, Faltynek CR, Kort ME, Reilly RM, Kym PR (2011) Selective blockade of TRPA1 channel attenuates pathological pain without altering noxious cold sensation or body temperature regulation. Pain 152:1165–1172
Zhang W, Liu Y, Zhao X, Gu X, Ma Z (2014) The effect of intrathecal administration TRPA1 antagonists in a rat model of neuropathic pain. Anesth Analg 119:179–185
Miyakawa T, Terashima Y, Takebayashi T, Tanimoto K, Iwase T, Ogon I, Kobayashi T, Tohse N, Yamashita T (2014) Transient receptor potential ankyrin 1 in spinal cord dorsal horn is involved in neuropathic pain in nerve root constriction rats. Mol Pain 10:58
Pinheiro Fde V, Villarinho JG, Silva CR, Oliveira SM, Pinheiro Kde V, Petri D, Rossato MF, Guerra GP, Trevisan G, Antonello Rubin M, Geppetti P, Ferreira J, Andre E (2015) The involvement of the TRPA1 receptor in a mouse model of sympathetically maintained neuropathic pain. Eur J Pharmacol 747:105–113
Eid SR, Crown ED, Moore EL, Liang HA, Choong KC, Dima S, Henze DA, Kane SA, Urban MO (2008) HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol Pain 4:48
Wei H, Hamalainen MM, Saarnilehto M, Koivisto A, Pertovaara A (2009) Attenuation of mechanical hypersensitivity by an antagonist of the TRPA1 ion channel in diabetic animals. Anesthesiology 111:147–154
Materazzi S, Fusi C, Benemei S, Pedretti P, Patacchini R, Nilius B, Prenen J, Creminon C, Geppetti P, Nassini R (2012) TRPA1 and TRPV4 mediate paclitaxel-induced peripheral neuropathy in mice via a glutathione-sensitive mechanism. Pflugers Arch 463:561–569
Zhao M, Isami K, Nakamura S, Shirakawa H, Nakagawa T, Kaneko S (2012) Acute cold hypersensitivity characteristically induced by oxaliplatin is caused by the enhanced responsiveness of TRPA1 in mice. Mol Pain 8:55
Wei H, Viisanen H, Amorim D, Koivisto A, Pertovaara A (2013) Dissociated modulation of conditioned place-preference and mechanical hypersensitivity by a TRPA1 channel antagonist in peripheral neuropathy. Pharmacol Biochem Behav 104:90–96
Trevisan G, Materazzi S, Fusi C, Altomare A, Aldini G, Lodovici M, Patacchini R, Geppetti P, Nassini R (2013) Novel therapeutic strategy to prevent chemotherapy-induced persistent sensory neuropathy by TRPA1 blockade. Cancer Res 73:3120–3131
Salat K, Filipek B (2015) Antinociceptive activity of transient receptor potential channel TRPV1, TRPA1, and TRPM8 antagonists in neurogenic and neuropathic pain models in mice. J Zhejiang Univ Sci B 16:167–178
Eid SR (2011) Therapeutic targeting of TRP channels--the TR(i)P to pain relief. Curr Top Med Chem 11:2118–2130
Pabbidi RM, Yu SQ, Peng S, Khardori R, Pauza ME, Premkumar LS (2008) Influence of TRPV1 on diabetes-induced alterations in thermal pain sensitivity. Mol Pain 4:9
Alessandri-Haber N, Dina OA, Yeh JJ, Parada CA, Reichling DB, Levine JD (2004) Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J Neurosci 24:4444–4452
Alessandri-Haber N, Dina OA, Joseph EK, Reichling DB, Levine JD (2008) Interaction of transient receptor potential vanilloid 4, integrin, and SRC tyrosine kinase in mechanical hyperalgesia. J Neurosci 28:1046–1057
Knowlton WM, Daniels RL, Palkar R, McCoy DD, McKemy DD (2011) Pharmacological blockade of TRPM8 ion channels alters cold and cold pain responses in mice. PLoS One 6:e25894
So K, Haraguchi K, Asakura K, Isami K, Sakimoto S, Shirakawa H, Mori Y, Nakagawa T, Kaneko S (2015) Involvement of TRPM2 in a wide range of inflammatory and neuropathic pain mouse models. J Pharmacol Sci 127:237–243
Isami K, Haraguchi K, So K, Asakura K, Shirakawa H, Mori Y, Nakagawa T, Kaneko S (2013) Involvement of TRPM2 in peripheral nerve injury-induced infiltration of peripheral immune cells into the spinal cord in mouse neuropathic pain model. PLoS One 8:e66410
Haraguchi K, Kawamoto A, Isami K, Maeda S, Kusano A, Asakura K, Shirakawa H, Mori Y, Nakagawa T, Kaneko S (2012) TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J Neurosci 32:3931–3941
Kaneko Y, Szallasi A (2014) Transient receptor potential (TRP) channels: a clinical perspective. Br J Pharmacol 171:2474–2507
Brederson JD, Kym PR, Szallasi A (2013) Targeting TRP channels for pain relief. Eur J Pharmacol 716:61–76
Moran MM, McAlexander MA, Biro T, Szallasi A (2011) Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov 10:601–620
Chizh BA, O'Donnell MB, Napolitano A, Wang J, Brooke AC, Aylott MC, Bullman JN, Gray EJ, Lai RY, Williams PM, Appleby JM (2007) The effects of the TRPV1 antagonist SB-705498 on TRPV1 receptor-mediated activity and inflammatory hyperalgesia in humans. Pain 132:132–141
Kort ME, Kym PR (2012) TRPV1 antagonists: clinical setbacks and prospects for future development. Prog Med Chem 51:57–70
Rowbotham MC, Nothaft W, Duan WR, Wang Y, Faltynek C, McGaraughty S, Chu KL, Svensson P (2011) Oral and cutaneous thermosensory profile of selective TRPV1 inhibition by ABT-102 in a randomized healthy volunteer trial. Pain 152:1192–1200
Krarup AL, Ny L, Astrand M, Bajor A, Hvid-Jensen F, Hansen MB, Simren M, Funch-Jensen P, Drewes AM (2011) Randomised clinical trial: the efficacy of a transient receptor potential vanilloid 1 antagonist AZD1386 in human oesophageal pain. Aliment Pharmacol Ther 33:1113–1122
Gavva NR, Treanor JJ, Garami A, Fang L, Surapaneni S, Akrami A, Alvarez F, Bak A, Darling M, Gore A, Jang GR, Kesslak JP, Ni L, Norman MH, Palluconi G, Rose MJ, Salfi M, Tan E, Romanovsky AA, Banfield C, Davar G (2008) Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 136:202–210
Bley K, Boorman G, Mohammad B, McKenzie D, Babbar S (2012) A comprehensive review of the carcinogenic and anticarcinogenic potential of capsaicin. Toxicol Pathol 40:847–873
Szallasi A, Sheta M (2012) Targeting TRPV1 for pain relief: limits, losers and laurels. Expert Opin Investig Drugs 21:1351–1369
Knotkova H, Pappagallo M, Szallasi A (2008) Capsaicin (TRPV1 agonist) therapy for pain relief: farewell or revival? Clin J Pain 24:142–154
Derry S, Sven-Rice A, Cole P, Tan T, Moore RA (2013) Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst Rev 2:CD007393
Brown DC, Iadarola MJ, Perkowski SZ, Erin H, Shofer F, Laszlo KJ, Olah Z, Mannes AJ (2005) Physiologic and antinociceptive effects of intrathecal resiniferatoxin in a canine bone cancer model. Anesthesiology 103:1052–1059
Davies SJ, Harding LM, Baranowski AP (2002) A novel treatment of postherpetic neuralgia using peppermint oil. Clin J Pain 18:200–202
Gobel H, Schmidt G, Soyka D (1994) Effect of peppermint and eucalyptus oil preparations on neurophysiological and experimental algesimetric headache parameters. Cephalalgia 14:228–234, discussion 182
Green BG, McAuliffe BL (2000) Menthol desensitization of capsaicin irritation. Evidence of a short-term anti-nociceptive effect. Physiol Behav 68:631–639
Proudfoot CJ, Garry EM, Cottrell DF, Rosie R, Anderson H, Robertson DC, Fleetwood-Walker SM, Mitchell R (2006) Analgesia mediated by the TRPM8 cold receptor in chronic neuropathic pain. Curr Biol 16:1591–1605
Zhu W, Xu P, Cuascut FX, Hall AK, Oxford GS (2007) Activin acutely sensitizes dorsal root ganglion neurons and induces hyperalgesia via PKC-mediated potentiation of transient receptor potential vanilloid I. J Neurosci 27:13770–13780
Zhang H, Cang CL, Kawasaki Y, Liang LL, Zhang YQ, Ji RR, Zhao ZQ (2007) Neurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCepsilon: a novel pathway for heat hyperalgesia. J Neurosci 27:12067–12077
Fischer MJ, Btesh J, McNaughton PA (2013) Disrupting sensitization of transient receptor potential vanilloid subtype 1 inhibits inflammatory hyperalgesia. J Neurosci 33:7407–7414
Park CK, Xu ZZ, Liu T, Lu N, Serhan CN, Ji RR (2011) Resolvin D2 is a potent endogenous inhibitor for transient receptor potential subtype V1/A1, inflammatory pain, and spinal cord synaptic plasticity in mice: distinct roles of resolvin D1, D2, and E1. J Neurosci 31:18433–18438
Yoshida N, Kobayashi K, Yu L, Wang S, Na R, Yamamoto S, Noguchi K, Dai Y (2011) Inhibition of TRPA1 channel activity in sensory neurons by the glial cell line-derived neurotrophic factor family member, artemin. Mol Pain 7:41
Acknowledgments
The author thanks Dr. Shenglan Wang at Hyogo University of Health Sciences for helpful discussions and providing images. This study was supported by JSPS KAKENHI Grant Numbers 26460713.
Conflict of interest
The author declares that he has no conflict of interest
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the Special Issue on the Role of TRP Ion Channels in Physiology and Pathology - Guest Editor: Armen Akopian
Rights and permissions
About this article

Cite this article
Dai, Y. TRPs and pain. Semin Immunopathol 38, 277–291 (2016). https://doi.org/10.1007/s00281-015-0526-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-015-0526-0