
Abstract
Throughout evolution, effective nutrient sensing and control of systemic energy homeostasis have relied on a close physical and functional interaction between immune and metabolically active cells. However, in today's obesogenic environment, this fine-tuned immunometabolic interface is perturbed. As a consequence, chronic inflammatory conditions and aberrant activation of immune cells have emerged as key features of obesity-related metabolic disorders, including insulin resistance, cardiovascular complications, and type 2 diabetes, whereas a major research focus has been placed on the adipocyte–macrophage interaction in the context of metabolic dysfunction; recent studies have not only expanded the scope of relevant immune cells in this setting but also highlight the impact of distinct metabolic organs, including the liver, on immunometabolic control, metabolic disease development, and potential anti-inflammatory therapeutic options in obesity-driven pathologies. This review will thus summarize recent progress in this emerging area of metabolic research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Obesity, inflammation, and insulin resistance
Epidemiological studies estimate that by the year 2030, 2.2 billion people worldwide will be overweight and 1.1 billion will be obese [1]. Obesity as such represents the core component of the so-called metabolic syndrome which is a cluster of metabolic disorders, including insulin resistance, obesity, fatty liver disease, hyperglycemia, dyslipidemia, and hypertension, mainly caused by caloric excess, sedentary lifestyle, and a genetic predisposition. Progression of the aforementioned disorders can precipitate into end-stage diseases such as type 2 diabetes, atherosclerosis, and eventually even cancer. Consequently, components of the metabolic syndrome are tightly associated with increased mortality as observed in these patients [2].
Inflammation is now recognized as a key feature of metabolic dysfunction. Dating back to the 1990s, a critical role for inflammatory mediators, i.e., tumor necrosis factor alpha (TNFα), has been described in the manifestation of obesity and associated complications most notably insulin resistance. While adipose tissue of obese humans and mice is characterized by elevated numbers of inflammatory macrophages and the induction of circulating TNFα levels [3], early studies already have shown that antibody-mediated TNFα neutralization or genetic loss-of-function mouse models for TNFα improved insulin sensitivity during diet-induced obesity [4]. Also, critical molecular effectors of inflammatory signaling such as the transcription factor nuclear factor kappa B (NFκB), its upstream kinase IKK, or the jun-N-terminal kinase (JNK) were subsequently found to be aberrantly activated under obese conditions, and prevention of NFκB/JNK signaling ameliorated substantial parts of metabolic dysfunction as associated with insulin-resistant obesity [5, 6]. Consistent with these findings, salicylate treatment indeed improved insulin sensitivity and glucose homeostasis in both humans and mice [7], again underlining the close interaction between inflammatory and metabolic cues in the control of energy homeostasis. Indeed, metabolic and inflammatory cells have acted in close physical proximity throughout evolution starting from the Drosophila fat body which combines major mammalian metabolic cell functions, including equivalents to adipocytes, hepatocytes, and various hematopoietic as well as immune cells. Over 600 million years of evolution, the intra-organ communication in flies has diverged into distinct organ compartments (i.e., adipose tissue and liver), however, still reflected by the close proximity between adipocytes, hepatocytes and dedicated tissue macrophages, and other immune cells. In fact, this tightly coupled interaction between the metabolic and immune compartments within adipose and liver tissue has been hypothesized to represent a critical interface for nutrient sensing, inter-organ communication, and metabolic control [8].
Once considered an inert energy storing depot, adipose tissue is now considered a potent endocrine organ with critical importance for overall energy homeostasis. Indeed, adipocytes secrete proteins involved in inflammation, appetite regulation, blood pressure control, and energy balance [9]. Under normal physiological conditions, adipose tissue function is tightly coupled to the systemic adaptation to varying conditions of food availability; upon fasting, stored adipose tissue lipids can be rapidly released and used for energy by peripheral organs, particularly including skeletal and cardiac muscle and liver; however, excessive adipose tissue is associated with increased risk of insulin resistance, cardiovascular disease, and cancer [10].
Given its central location within the systemic circulation, the liver serves as one of the body's critical organ for maintenance of systemic energy homeostasis [11]. Indeed, the liver represents the critical control relay in the reception of small molecules arising from food digestion or degradation of endogenous sources, their metabolic conversion or storage, and the final (re-)distribution to the periphery. Consequently, as the predominant interconversion point for energy substrates in mammals, the liver plays an essential role in the adaptive metabolic response during daily/periodic fasting–feeding cycles [11–13]. In this regard, defects in hepatic insulin signaling have been demonstrated to importantly contribute to the development of systemic peripheral insulin resistance [14]. Mice bearing a targeted disruption of the insulin receptor gene in liver display hyperglycemia, hyperinsulinemia, and impaired glucose tolerance [15]. Also, inhibition of the PI3K/Akt-dependent insulin signaling pathway in liver by the Akt-inhibitor tribbles homolog 3 leads to hyperglycemia and glucose intolerance [16].
It is tempting to speculate that inflammation and insulin resistance have developed as adaptive traits which might provide an evolutionary advantage for organisms under specific conditions. Proper inflammatory response is extremely important for fighting invading pathogens and recognizing transformed tumor cells. It is also an energetically costly process, and therefore, insulin resistance develops in the site of inflammation to fuel immune cells. Nowadays, in obesogenic environments, an originally beneficial response has transformed to one of the key contributors to metabolic dysfunction [17]. Given the critical importance of both adipose tissue and liver for overall nutrient handling and metabolic control, intra-adipose/hepatic communication pathways between metabolic and inflammatory cells are crucial determinants of energy homeostasis and/or metabolic dysfunction, especially in the context of obesity-related insulin resistance and type 2 diabetes. Given excellent recent reviews on components of the adaptive immune system and metabolic control [18, 19], a particular focus will be placed on the innate immune system and its impact on metabolic dysfunction.
Innate immune system cells and adipose tissue (dys)function
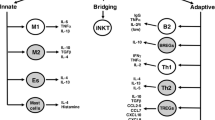
Innate or nonspecific immune response represents first line of defense against invading pathogens, and virtually, all types of innate immune cells have been identified in adipose tissue and been implicated in maintaining of adipose homeostasis or in inflammation-induced metabolic disease (Fig. 1). Most work on obesity low-grade inflammation has been focused on macrophage infiltration, and function in adipose tissue and several excellent reviews have been recently published elsewhere [19–21]. However, a role of other immune cells has recently emerged as an important topic. In addition to macrophages, cells from innate and adaptive branches of immune system seem to play a prominent role in maintaining adipose tissue homeostasis and are involved in promoting or suppressing inflammation upon obese conditions.

A schematic representation of changes in adipose tissue innate immune cells populations in obesity. Lean adipose tissue is characterized with low occurrence of innate immune cells and predominantly anti-inflammatory cytokines are secreted. With obesity, insulin sensitivity decreases proportionally to increased inflammation of the adipose tissue. Obese adipose tissue is associated with increased infiltration of immune cells and inflammatory cytokines production. IL-4 interleukin 4, IL-10 interleukin 10, TNFα tumor necrosis factor α, IFNγ interferon γ, CCL2 chemokine (C-C motif) ligand 2, IL-1β interleukin 1β
Neutrophils
In classical immune response, neutrophils are the first immune cells present at the site of inflammation and further help to recruit macrophages [22]. Similarly in obesity, they infiltrate adipose tissue as soon as 3 days after initiation of high-fat feeding and precede infiltration of macrophages [23]. Neutrophils secrete several types of proteases that are known to be involved in inflammation [24, 25]. Expression of elastase, a well-characterized peptidase, was increased in adipose of high-fat fed mice compared to chow-fed animals, and it participated on inflammation-induced insulin resistance via degradation of insulin receptor substrate 1 [24]. Upon pharmacologic or genetic loss of function of neutrophil elastase, mice showed reduced inflammation of adipose tissue with lower macrophages infiltration and improved insulin sensitivity and glucose tolerance [24]. Moreover, increased levels of neutrophils were detected in circulation of obese adults [26, 27] and children [28], which further emphasizes their function in metabolic homeostasis.
Basophils
Basophils are the rarest granulocytes in human blood and are involved in regulation and modulation of allergic inflammation. The role of basophils in obesity remains poorly understood, and their association with detrimental obesity outcomes is not clear [29–31]. To date, any reports examining basophil infiltration into adipose tissue have not been published.
Eosinophils
Eosinophils are commonly associated with allergic reactivity and parasitic infection. Despite being elevated in serum of obese humans and animals [32], their numbers in adipose tissue decline with obesity [33]. Eosinophils are a major source of interleukin (IL)-4 in adipose tissue, thereby sustaining adipose macrophages under alternatively activated M2 phenotype and protecting tissue from inflammation [33]. Eosinophil-deficient mice on high-fat diet showed augmented adiposity, impaired glucose tolerance, and increased proportion of classically activated macrophages [33], while the total number was not changed. This suggests eosinophils function in regulation of macrophages polarization and thereby influence insulin sensitivity.
Mast cells
Mast cells play a prominent role as effectors in allergic reactions and have been implicated in wound healing and tissue remodeling. Mast cells preferentially localize to mucosal and connective tissues, acting as first responders to viral and bacterial pathogens. They originate in bone marrow, but unlike the other innate immune cells, do not mature until they reach the target tissue [34]. In addition, a population of mast cells' progenitors have been previously detected in adipose tissue [35], suggesting that in addition to bone marrow, adipose tissue might be another source of mast cells. Counts of mast cells in lean adipose tissue are relatively low, but increase with fat accumulation in humans as well as in animal models [36, 37], which suggest a contribution to diet-induced obesity and insulin resistance. Indeed, mast cell-deficient mice fed a Western diet gained less body weight, showed decreased adiposity, improved glucose tolerance, and attenuated adipose tissue inflammation compared to control counterparts [36]. Administration of mast cell stabilizers, Cromolyn (DSCG) and Zaditor (ketotifen), which are used in the clinical treatment of allergies, led to analogous results [36]. Lower body weight in mast cell-depleted animals might be attributed, at least in part, to increased thermogenesis, as they showed higher resting metabolic rate and increased expression of key thermogenic protein UCP1 in brown fat [36].
The exact mechanism by which mast cells contribute to pathophysiology of obesity is not yet fully understood, but secretion of pro-inflammatory cytokines IL-6 and interferon γ (IFNγ) are likely to contribute. Furthermore, mast cells promote obesity via stimulation of adipogenesis [38]. Mast cells produce a vast spectrum of bioactive molecules including prostaglandin D2 [39], which is metabolized to 15-deoxy-δ-PGJ2, the major endogenous ligand of peroxisome proliferator-activated receptor γ (PPARγ) [40, 41], a well-known inducer of adipocyte differentiation [40, 42]. Therefore, mast cells might be able to induce adipogenesis by prostanoids production. It has been proposed that mast cells also promote angiogenesis in adipose tissue by production of certain proteases [36, 43]. This notion would be further supported by mast cell localization next to the microvasculature and positive correlation of mast cells and microvessels number during obesity development [36].
Natural killer cells
Natural killer (NK) cells are large granular lymphocytes with potent ability to activate antigen-independent cytotoxic response upon viral infection and participate in antitumor response. Their function in adipose tissue has not been clearly defined. NK cells are abundant in adipose tissue forming about 30 % of the cells in stromal vascular fraction [44]; however, the results concerning quantity of NK cells in adipose tissue and its changes upon obesity are inconsistent showing decrease [44–46] or no change [47] in animal and human studies or even increase in human visceral adipose tissue from obese subjects [48]. Their counts seem to be regulated by other leukocytes, as B and T cells deficiency resulted in a pronounced increase in NK cell number in adipose tissue [47]. Furthermore, an important regulator of NK cells function, IL-15, was suggested to influence NK cells number in adipose tissue. Transgenic mice overexpressing IL-15 have more NK cells in adipose tissue and showed lean phenotype, while IL-15 deficiency was associated with increased body weight and less NK cells in adipose tissue [45, 49].
Leptin, a hormone secreted specifically by adipocytes, is implicated in body weight and appetite regulation. Its levels are increased in obesity accompanied by leptin resistance [50]. Moreover, leptin plays a prominent role in immunity [51] as it enhances inflammatory response during infection, promotes production of many cytokines, e.g., IL-1β, IFNγ, IL-6, TNFα [52–54], activation of macrophages [55] and proliferative and anti-apoptotic effects [56], and thus provides plausible link between obesity and immune dysfunction.
Indeed, obesity leads to decreased cytotoxicity of NK cells [57] decreasing their ability to fight against infected and transformed cells. Leptin might be likely candidate involved in the process as long leptin receptor was found to be expressed on subpopulation of human circulating NK cells. Proportion and also total number of NK cells in leptin receptor deficient db/db mice were declined as well as their cytotoxic activity [58], suggesting leptin signaling to be involved in NK cells development and activation.
It has been shown that short-term leptin stimulation of primary human NK cells increased a secretion of IFNγ and cell-dependent cytotoxic lysis of tumor cells [59]. However, the opposite is true for a long-term leptin stimulation, which better mimics obese conditions, as it led to dampened secretion of inflammatory and cytotoxic agents by NK cells [59]. Therefore, leptin resistance might impair central immune function and possibly mediates cancer susceptibility of obese people.
Natural killer T cells
Family of natural killer T (NKT) cells represents a bridge between innate and adaptive immunity and comprises several types of NKT cells. Best characterized are type I NKT cells, so-called invariant NKT cells, which are also most abundant in adipose tissue. NKT cells share characteristics of NK cells and simultaneously express invariant T cells receptor and CD3—T lymphocyte markers. They are able to skew immune response to inflammatory or anti-inflammatory direction, depending on stimuli provided under specific conditions either nutritional or immunological. NKT cells recognize and are activated by lipid antigens presented by MHC class I-like molecule CD1d on antigen-presenting cells [60]. Thus, not surprisingly, they have been found enriched in adipose tissue [44, 61]; however, function of NKT and their influence of inflammatory process in adipose have not been well characterized yet. Model of diet-induced obesity showed inconsistent results. While significant enrichment of adipose tissue with NKT cells upon high-fat feeding was detected [62, 63]; the others showed completely opposite results demonstrating negative correlation of NKT cells amount with adiposity [61, 64], BMI, and insulin resistance [65] and percentage of NKT cells was reduced also in ob/ob mice, a well-established genetic obesity model [61, 66]. Additionally, in some studies, number of NKT cells remains comparable between high-fat and low-fat feeding [67], but cells were shown to be more active with high-fat feeding and produce more pro-inflammatory cytokines [67]. Contrast to mice, NKT cells are scarce in human adipose tissue [68, 69] and are found even in lower frequencies with obesity [68]. Using mouse models lacking NKT cells have not shed more light on the problematic. β2 microglobulin knockout mice lacking NKT cells fed high-fat diet demonstrated ameliorated glucose tolerance and polarization of macrophages towards M2 phenotype when compared to high-fat fed controls [62]. In contrast, opposite results have been achieved with another mouse strain with depleted NKT cells, namely the CD1d knockout mouse [61, 66], where positive correlation between insulin sensitivity and NKT cell abundance was demonstrated [65]. Furthermore, in this study, a link between NKT cells and improved glucose homeostasis has been further supported with experiments encompassing NKT cells activation with α-galactosylceramide (αGalCer, a potent glycolipid agonist derived from marine sponge not found in mammals). After αGalCer injection and NKT cells activation, obese mice fed high-fat diet showed improved glucose tolerance and macrophages polarization shifted to M2 phenotype [65]. Interestingly, αGalCer injection did not have any effect in lean mice [65], which contrasted results gained by Wu et al. showing expansion of NKT cells in adipose after αGalCer treatment and raised pro-inflammatory cytokines secretion [67].
The question remains why some studies demonstrated NKT cells to increase production of pro-inflammatory cytokines upon lipid activation accompanied with worsen obesity-associated pathogenesis while the others showed improved obesity-associated conditions. One of the reasons might be the usage of different mouse strains, different lipid content, and composition of diets or feeding period. Future investigations will be necessary to clarify NKT cells role in adipose tissue function.
Dendritic cells
Dendritic cells (DCs) are a heterogeneous antigen-presenting cell population containing conventional or myeloid, plasmacytoid, and newly characterized inflammatory DCs. They contribute to pathogen defense and are involved in recruitment of macrophages into the site of inflammation and priming of naïve CD4+ T cells [70–72]. However, there are not many data available about DCs and their function in obesity-mediated low-grade adipose tissue inflammation and associated insulin resistance. DCs were detected in adipose tissue of mice and humans [73–75], and their number was increased with obesity [74, 75]. Further, DCs are implicated in recruitment of macrophages into adipose during overnutrition [75], and mice lacking DCs showed to be resistant to high-fat induced obesity [75]. DCs are capable of presenting antigens to CD4+ T cells, which then become effector cells showing Th1 immune response in lean animals. In contrast, adipose tissue of obese or type 2 diabetic mice display a switch from Th1 to Th17 response [74]. Th17 immune response has been recently shown to deteriorate autoimmunity [76] and might play an important role in metabolic process; however, this notion would need further examination.
The liver: an immune organ
A coordinated network of cells, tissues, and organs comprise the innate immune system, which remains the first line of defense against pathogenic and or damaged “self” signals. Anatomical location and dual blood supply of the human liver (20 % oxygenated blood from the hepatic artery and 80 % nutrient rich blood from the portal vein) ensures constant exposure to various antigens, pathogenic stimuli, and toxins; underscoring the organ's relevance in immunological response [77]. Innate immune cells of the liver include both phagocytic and lymphocytic subsets. Kupffer cells (resident hepatic macrophages) in concerted effort with lymphocytic natural killer cells, dendritic cells, and natural killer T cells modulate liver immune status. Pathogen-associated molecular patterns or damage-associated molecular patterns are received by pattern recognition receptors (PRRs) [e.g., Toll-like receptors (TLRs)] expressed by cells of the innate immune system to compliment nonspecific killing mechanisms and other innate immune barriers [77, 78]. Experimental animal models accompanied by human clinical studies demonstrate involvement of the aforementioned immune cells (amongst others) during basal and injurious conditions within the hepatic microenvironment. Specifically, cytokine balance/imbalance controlled by cells of the innate hepatic immune system critically regulates disease pathogenesis, including those evolving from metabolic syndrome [79].
Systemic metabolic disturbances and innate hepatic immune activation
Indices of metabolic syndrome are known causative factors for development of liver steatosis or nonalcoholic fatty liver disease (NAFLD). Excessive fat accumulation and increased hepatic triglycerides can arise from delivery of free fatty acids from lipolysis of visceral fat, from de novo lipogenesis, or from dietary consumption of high-fat high-sugar foods/beverages [80]. Portal vein delivery of fatty acids impairs hepatic extraction of insulin and stimulates gluconeogenesis/triglyceride synthesis. Metabolic syndrome/obesity has become a global pandemic as such occurrence rates of NAFLD are alarmingly high in the USA and abroad, where NAFLD currently ranks as the most common liver disease in the Western world [81]. Classic chronic liver disease pathogenesis includes progression from NAFLD to nonalcoholic steatohepatitis (NASH), a more severe state of injury characterized by steatosis, increased inflammation, and elevated parenchymal cell damage [82]. Increases in lipid peroxidation and reactive oxygen species continually evoke cytokine overproduction, further propelling cellular dysfunction in fatty liver disease. As such, approximately 20 % of NASH patients will progress to end-stage liver disease (cirrhosis), highlighting the importance of hepatic manifestations of metabolic syndrome [81]. As basic science research in the field has expanded, the immunological component has received much attention resulting in numerous studies that clearly define contributions of the innate immune response in NAFLD and NASH [83] (Fig. 2).

Immune cell-mediated NAFLD pathogenesis. The pathogenesis of NAFLD is often described by the “two-hit” hypothesis. Excessive consumption of dietary fats along with potential underlying metabolic disruption, genetic or otherwise, stimulates an increase in fatty acid synthesis with concurrent increases in triglycerides, β-oxidation, and disruption of glucose homeostasis. Simple steatosis coupled with additional injury or a “second-hit” may progress the state of injury to NASH marked by tissue inflammation. Immune cell activation observed in NAFLD, and elevated inflammatory cell infiltrate observed in NASH, marres the hepatic microenvironment with inflammatory cytokines, with subsequent increases in lipoapotosis, oxidative stress, and insulin resistance. Innate immune cells are critical mediators of this process; those highlighted in this review are diagrammed above with notable secretory factors and receptors. DC dendritic cell, KC Kupffer cell, NK natural killer cell, NKT natural killer T cell
Kupffer cells
Nonspecific phagocytosis in the liver is predominantly mediated by Kupffer cells (KCs), which account for over 80 % of fixed-tissue macrophages within the human body and approximately 20 % of nonparenchymal hepatic cells [77]. Zonal distribution analyses indicate KCs possessing increased lysosomal activity and greater phagocytic capacity are sequestered to the periportal tract, where blood flow from the portal vein delivers bacterial products transported from the gut [84]. KCs are directly linked to the liver's response to infection, toxins as well as other stressors and are known to display characteristic macrophage polarization. As active phagocytes, KCs secrete various inflammatory cytokines in response to intravascular debris and serve to eliminate bacterial cells and other particulates via cell surface receptor complexes, notably complement receptors (ICAM-1 binding adhesion receptors and TLRs facilitating recognition of polysaccharides) [77, 78, 85]. Overproduction of these inflammatory mediators by KCs, including TNFα, IL-1 and 6, MIP1α, TGFβ, and RANTES can lead to parenchymal injury/necrosis and subsequently activate inflammatory cascades in neighboring cells, including upregulation of pro-inflammatory/pro-fibrotic signaling with the hepatic stellate cell population. Concrete links have previously been established between KCs activation and the pathogenesis of NAFLD and NASH, indicating reduction of this cell population attenuates histological signs of steatosis, inflammation, and necrosis [86–89]. More recently, elegant studies by Tosello-Trampont et al. demonstrate depletion of KCs (either pre- or post-methionine-choline deficient [MCD] diet) triggers a significant hepatic influx of CD11bintLy6Chi pro-inflammatory blood monocytes during development and progression of NASH [90]. Interestingly, in this rodent model of NASH development, KCs are polarized to the M1 macrophage phenotype and, along with the blood-derived monocytes, account for the main cellular source of TNFα. Elevation in hepatic TNFα promotes recruitment of Th1 inflammatory cells through subsequent elevation in MCP-1 levels [90]. Additionally, TNFα is known to regulate intrahepatic lipid metabolism through various mechanisms (e.g., insulin resistance), further amplifying NASH pathology [81]. Visceral adiposity observed in clinical NAFLD and NASH is often accompanied by increased gut permeability [91]. Thus, elevated gut-derived endotoxin levels trigger KC activation (via TLR4), which may also contribute to the classical M1 phenotype often observed [91]. While KC numbers have been shown to increase as a result of liver injury, it is clear from the recent work of Leroux and colleagues that differential phenotype is also observed in the setting of increased lipogenesis [92]. KCs isolated from high-fat diet fed mice were characterized by increased cell size and lipid droplet retention. Specifically, lipid droplets enriched with ceramides and diacylglycerols were reported, the former already known to stimulate macrophage activation and apoptosis. In this same study, increased inflammatory signaling was accompanied by an induction of lipogenic gene expression in KCs from fatty livers, which was attenuated by inhibiting the first step in lipogenesis (via suppression of acetyl-CoA carboxylase) [92]. In contrast to the deleterious M1 KC phenotype, previous studies have shown alternatively polarized M2 KCs to ameliorate insulin resistance in diet-induced obesity [93, 94]. Additionally, while the mechanism of KC activation/polarization remains unclear, recent studies have shown that neurotransmitter serotonin and cognate receptors may skew macrophage polarization. Specifically, KCs preferentially express serotonin receptor 5HT2B, and upon agonist activation, KCs display the characteristic M2 phenotype (anti-inflammatory, pro-cell growth, and tissue repair) [95]. Serotonin levels are decreased in patients with metabolic syndrome [96]; thus, it would reason to speculate that the pronounced M1 pro-inflammatory phenotype of KCs observed in NAFLD/NASH may be in some way linked to serotonin levels in the peripheral blood supply. Additionally, oxidative stress due to elevated ROS has been implicated as the secondary insult in the two-hit hypothesis of NAFLD [79]. KCs are a significant source of hepatic redox disruption via both direct and indirect ROS generation, and these effects on disease pathology and activation of adaptive immunity have been reviewed previously [79].
Natural killer cells
Originally termed Pit cells, NK cells reside within the hepatic sinusoid functioning as hepatic lymphocytes with qualities distinct from that of peripheral NK cells, T and B cells, due to antigen receptor expression deficiency [97]. Re-population rates remain high under normal physiological conditions, with cellular turnover every ~1–2 weeks. Extrahepatic replenishment of NK cells is controversial, but likely attributable to bone marrow-derived stem cell, with tissue-enrichment of NK cells attributed to sinusoidal endothelial cell–NK cell adhesion. Hepatic NK cells have the ability to directly and/or indirectly kill pathogens, stressed parenchymal and nonparenchymal cells, and tumor cells within the hepatic microenvironment [78]. Regulatory functions of NK cells have been reported with regard to Kupffer cells, T and B cells, as well as dendritic cells via production of various chemokines, cytokines, and growth factors. NK–target cell interactions are dictated by the presence of NK cell receptors, notably NKG2D, and target cell ligand expression [98, 99]. In addition to cell-activating ligands, activation of KCs is induced by various cytokines and chemokines, predominantly IFNα/β and CCL2 [100]. Several studies have reported key regulatory functions of NK cells in various liver pathologies, including HBV, HCV, and alcoholic liver disease (recently reviewed here [97, 101]); however, far less has been reported on the involvement of this cell type in NAFLD and NASH. Significant increases in NK cell presence has been reported in diabetes and obesity with corresponding increases in NK cell ligand expression. MIC A/B, ligands for NK cell receptor NKG2D, are increased in response to liver injury and have recently been reported to be upregulated along with NKG2D and other NK cell-associated mediators in livers of obese patients [102]. Expression of NK cell death receptors (TRAIL, CD95/FASL) was also significantly increased in livers of NASH patients compared to NAFLD and healthy controls, indicating an associating between activated hepatic NK cells and NASH pathogenesis [102]. Conflictingly, O'Shea and colleagues reported obese patients to have significantly fewer circulating NK cells compared to healthy controls [103]; however, peripheral blood NK cell counts may not accurately reflect the hepatic compartment. While the specific role of NK cells in NAFLD/NASH remains unclear, inferences may be drawn from studies examining NK contributions in liver fibrosis and HCV, both of which commonly present with underlying steatosis [97]. Production of cytotoxic mediators by NK cells can induce hepatic injury, but can also inhibit fibrogenesis through direct killing of the collagen-secreting hepatic stellate cell and production of IFNγ. These findings were recapitulated in the setting of HCV, wherein NK cells isolated from HCV-infected patients were able to induce apoptosis in activated HSCs, pointing to a beneficial role of this cells subset in progressive disease [104]. Paradoxically, a recent study from Gomez-Santos et al. indicates that NK cells actively promote a classical Th1 (pro-inflammatory) response in early stages of NAFLD as evidenced by increased TRAIL expression and cytotoxic activity [105]. Overall, these reports indicate divergent roles of hepatic NK in early vs. late stage disease, which should be a primary consideration when developing therapeutic strategies.
Messengers across the divide: dendritic cells and NKT cells
Dendritic cells
In concert with Kupffer cells, DCs are the first to detect invading pathogens and are classified as professional antigen-presenting cells regulating immunity and tolerance. DCs also provide an important link between the innate and adaptive immune systems through priming of T cell responses. DCs are subdivided into two major populations: plasmacytoid (IFN producing CD123+) and myeloid (CD11c+), both of which are present in human liver [78, 106]. Interestingly, properties and functions of dendritic cells vary among tissue types and to a greater degree among species (e.g., human DCs vs. rat), making it difficult to identify DC populations by cell surface markers. DCs initiate innate immune responses to eliminate foreign microbes, similarly to KCs, dendritic cells express several PRRs, including TLRs. Due to constant exposure to bacterial LPS, downregulation of TLR4 is observed; thus, somewhat limited direct responsiveness to danger signals and increased tolerogenic properties through expression of IL-10 is observed [107]. Upon detection and uptake of invading microbes, activated DCs have the capability to migrate to draining lymph nodes to promote NK cell activation in addition to their ability to modulate T cell response and activate neighboring macrophages [106]. In experimental models of cholestatic disease, accumulation of DCs has been reported; however, less is known concerning the function of DC in hepatic metabolic syndrome and progression to NASH. High-fat diet alone has been reported to increase hepatic DCs [108], and in a recent study, DC-depleted mice present with decreased liver macrophages as well as resistance of weight gain and metabolic disturbances from high-fat diet [75]. Henning et al. has recently reported NASH-associated dendritic cells present a more mature phenotype characterized by increased expression of co-stimulatory molecules and cytokines [109]. Depletion of CD11c+ DCs in a murine model of NASH markedly increased intrahepatic inflammation, with expansion of activated Kupffer and monocyte populations observed. Additionally, DCs blunted expansion of CD8+T cells and inflammatory monocytes. These elegant studies by Henning and colleagues indicate a positive role for DCs in NASH potentially via clearance of necrotic and apoptotic cell debris [109]. These data support previous studies examining a role for DCs in a fibrotic milieu [107], which suggests this cell type may be important in the progression of liver disease.
Natural killer T cells
Similar to hepatic DCs, NKT cells provide an important line of communication between the innate and adaptive arms of the immune response [97, 110]. NKT cells, which express NK cell characteristics, express a variable pattern of T cell receptor and NK cell markers and recognize lipid antigen CD1d [97]. Intrahepatic NKT cells (reported to develop in the thymus) are positive for both CD3 and CD56 and are classically divided as types I, II, or III (CD1-independent), with all subtypes providing critical intravascular immune surveillance. Hepatic antigen-presenting cells will present microbial glycolipid antigens to NKT cells, stimulating secretion of Th1 or Th2 cytokines, which subsequently activates neighboring innate immune cells and adaptive T cells [97, 101]. A distant role for NKT cells in NAFLD progression is beginning to take shape, with NKT cell depletion reported in fatty liver, but high accumulation in NASH-related fibrosis [111]. This reduction in hepatic NKT cell content is reportedly mirrored in the periphery in patients with NAFLD [112]. High-fat diet and obesity are known to trigger apoptosis of NKT cells through induction IL-12 and increases in immature myeloid cells, which alone can direct NKT cell death. Invariant NKT cells (iNKT, type I cells) have been widely studied in adipose tissue derived from lean and obese mice [113]. Lynch et al. have shown high enrichment of iNKT cells in human and murine adipose and liver tissue. In an obese model, depletion of iNKT cells was observed, which correlated with the presence of macrophage infiltration [61]. Mice deficient in iNKT cells presented with enhanced weight loss, fatty liver, and insulin resistance. Hepatic NKT cells also have the ability to secrete osteopontin (OPN) and sonic hedgehog (Hh), both of which are known to promote NAFLD/NASH and fibrosis progression. Elegant studies from Syn et al. reported attenuated Hh and OPN expression and dramatically blunted liver damage in mice depleted of NKT and fed the MCD diet, indicating that hepatic NKT cells drive the progression of NASH via production of OPN and Hh ligands [114]. These studies support previous evidence that type II NKT cells initiate hepatic inflammation and exacerbate obesity leading to insulin resistance [110]. In addition to being influenced by Hh signals, NFκB signaling may also be critical to NKT regulation. NFκB1-deficient mice, which develop enhanced NASH, displayed increased hepatic NKT recruitment compared to wild-type mice [115]. NKT cell recruitment was associated with increased IL-15 expression, a cytokine that influences NKT maturation and survival. Interestingly, while NFκB loss increased NKT cell recruitment and subsequently enhanced IFNγ production, liver fibrosis (which should be attenuated by IFNγ) was still observed indicating the appreciable complexity of innate NKT-mediated responses [115].
Outlook
In light of the vast complexity of cell phenotypes/functionality, which is highly dependent on etiology and disease state, it is evident that future work is needed to validate whether (innate) immune cell subsets can be exploited for therapeutic intervention in metabolic disorders. Classical anti-inflammatory strategies to treat insulin resistance and other obesity-mediated disorders include high-dose salicylate administration [7], TNFα neutralization [116], and inhibition of interleukin-1 signaling [117, 118], however, associated with varying effectiveness and degree of clinical improvements. In this respect, also the main class of anti-diabetic drugs, PPARγ agonists of the TZD family, has been found to exert anti-inflammatory effects, but was also associated with severe (cardiovascular) side effects [119]. Novel approaches have begun to characterize the inflammatory infiltrate in NAFLD and NASH patients to uncover markers of inflammatory cells that correlate to disease severity. Unsurprisingly, recent assessment of immune cell profiles in pediatric NAFLD indicates increased CD136+ Kupffer cell number correlates to severity of disease, while the opposite effect is observed in CD3+ cells [120]. Additionally, in adult patients with NASH, distribution of naive, memory, LT helper, and cytotoxic subpopulations in the periphery are also skewed revealing a distinct profile [121]. While these studies are small in number, expansion is expected also into other relevant organ entities, including adipose tissue and skeletal muscle, which may provide more substantial data from larger patient cohorts with appropriate controls. It is clear that each immune subpopulation is unique in function within the corresponding microenvironment, and that depletion and/or expansion of certain cell types can alter disease progression. Thus, future research in immunometabolism can be expected to further define avenues to anti-obesity and anti-diabetic therapies.
References
Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, Singh GM, Gutierrez HR, Lu Y, Bahalim AN, Farzadfar F, Riley LM, Ezzati M (2011) National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet 377(9765):557–567. doi:10.1016/S0140-6736(10)62037-5
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414(6865):813–820
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Investig 112(12):1796–1808
Hotamisligil GS, Budavari A, Murray D, Spiegelman BM (1994) Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes, central role of tumor necrosis factor-alpha. J Clin Investig 94(4):1543–1549
Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, Ma JT, Zhou J, Qi N, Westcott D, Delproposto JB, Blackwell TS, Yull FE, Saltiel AR (2009) The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 138(5):961–975. doi:10.1016/j.cell.2009.06.046
Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS (2002) A central role for JNK in obesity and insulin resistance. Nature 420(6913):333–336. doi:10.1038/nature01137 nature01137
Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE (2001) Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 293(5535):1673–1677
Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444(7121):860–867
Farooqi IS (2011) Genetic, molecular and physiological insights into human obesity. Eur J Clin Investig 41(4):451–455. doi:10.1111/j.1365-2362.2010.02468.x
Rohm M, Sommerfeld A, Strzoda D, Jones A, Sijmonsma TP, Rudofsky G, Wolfrum C, Sticht C, Gretz N, Zeyda M, Leitner L, Nawroth PP, Stulnig TM, Diaz MB, Vegiopoulos A, Herzig S (2013) Transcriptional cofactor TBLR1 controls lipid mobilization in white adipose tissue. Cell metabolism 17(4):575–585. doi:10.1016/j.cmet.2013.02.010
Casteels K, Mathieu C (2003) Diabetic ketoacidosis. Rev Endocr Metab Disord 4(2):159–166
Sommerfeld A, Krones-Herzig A, Herzig S (2011) Transcriptional co-factors and hepatic energy metabolism. Mol Cell Endocrinol 332(1–2):21–31. doi:10.1016/j.mce.2010.11.020
Kulozik P, Jones A, Mattijssen F, Rose AJ, Reimann A, Strzoda D, Kleinsorg S, Raupp C, Kleinschmidt J, Muller-Decker K, Wahli W, Sticht C, Gretz N, von Loeffelholz C, Stockmann M, Pfeiffer A, Stohr S, Dallinga-Thie GM, Nawroth PP, Berriel Diaz M, Herzig S (2011) Hepatic deficiency in transcriptional cofactor TBL1 promotes liver steatosis and hypertriglyceridemia. Cell metabolism 13(4):389–400. doi:10.1016/j.cmet.2011.02.011
Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI (2000) Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem 275(12):8456–8460
Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR (2000) Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 6(1):87–97
Du K, Herzig S, Kulkarni RN, Montminy M (2003) TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 300(5625):1574–1577
Odegaard JI, Chawla A (2013) Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 339(6116):172–177. doi:10.1126/science.1230721
Sell H, Habich C, Eckel J (2012) Adaptive immunity in obesity and insulin resistance. Nat Rev Endocrinol 8(12):709–716. doi:10.1038/nrendo.2012.114
Shu CJ, Benoist C, Mathis D (2012) The immune system's involvement in obesity-driven type 2 diabetes. Semin Immunol 24(6):436–442. doi:10.1016/j.smim.2012.12.001
Biswas SK, Mantovani A (2012) Orchestration of metabolism by macrophages. Cell Metab 15(4):432–437. doi:10.1016/j.cmet.2011.11.013
Dalmas E, Clement K, Guerre-Millo M (2011) Defining macrophage phenotype and function in adipose tissue. Trends Immunol 32(7):307–314. doi:10.1016/j.it.2011.04.008
Mantovani A, Cassatella MA, Costantini C, Jaillon S (2011) Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11(8):519–531. doi:10.1038/nri3024
Elgazar-Carmon V, Rudich A, Hadad N, Levy R (2008) Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res 49(9):1894–1903. doi:10.1194/jlr.M800132-JLR200
Talukdar S, da Oh Y, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, Ofrecio J, Lin M, Brenner MB, Olefsky JM (2012) Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18(9):1407–1412. doi:10.1038/nm.2885
Pham CT (2006) Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 6(7):541–550. doi:10.1038/nri1841
Nijhuis J, Rensen SS, Slaats Y, van Dielen FM, Buurman WA, Greve JW (2009) Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity (Silver Spring) 17(11):2014–2018. doi:10.1038/oby.2009.113
Herishanu Y, Rogowski O, Polliack A, Marilus R (2006) Leukocytosis in obese individuals: possible link in patients with unexplained persistent neutrophilia. Eur J Haematol 76(6):516–520. doi:10.1111/j.1600-0609.2006.00658.x
Zaldivar F, McMurray RG, Nemet D, Galassetti P, Mills PJ, Cooper DM (2006) Body fat and circulating leukocytes in children. Int J Obes (Lond) 30(6):906–911. doi:10.1038/sj.ijo.0803227
Suzukawa M, Nagase H, Ogahara I, Han K, Tashimo H, Shibui A, Koketsu R, Nakae S, Yamaguchi M, Ohta K (2011) Leptin enhances survival and induces migration, degranulation, and cytokine synthesis of human basophils. J Immunol 186(9):5254–5260. doi:10.4049/jimmunol.1004054
Laurson KR, McCann DA, Senchina DS (2011) Age, sex, and ethnicity may modify the influence of obesity on inflammation. Journal of investigative medicine : the official publication of the American Federation for Clinical Research 59 (1): 27–31. doi:10.231/JIM.0b013e318200151a
Johannsen NM, Priest EL, Dixit VD, Earnest CP, Blair SN, Church TS (2010) Association of white blood cell subfraction concentration with fitness and fatness. Br J Sports Med 44(8):588–593. doi:10.1136/bjsm.2008.050682
Vasudevan AR, Wu H, Xydakis AM, Jones PH, Smith EO, Sweeney JF, Corry DB, Ballantyne CM (2006) Eotaxin and obesity. J Clin Endocrinol Metab 91(1):256–261. doi:10.1210/jc.2005-1280
Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, Locksley RM (2011) Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332(6026):243–247. doi:10.1126/science.1201475
Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M (2005) Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol 23:749–786. doi:10.1146/annurev.immunol.21.120601.141025
Poglio S, De Toni-Costes F, Arnaud E, Laharrague P, Espinosa E, Casteilla L, Cousin B (2010) Adipose tissue as a dedicated reservoir of functional mast cell progenitors. Stem Cells 28(11):2065–2072. doi:10.1002/stem.523
Liu J, Divoux A, Sun J, Zhang J, Clement K, Glickman JN, Sukhova GK, Wolters PJ, Du J, Gorgun CZ, Doria A, Libby P, Blumberg RS, Kahn BB, Hotamisligil GS, Shi GP (2009) Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med 15(8):940–945. doi:10.1038/nm.1994
Altintas MM, Azad A, Nayer B, Contreras G, Zaias J, Faul C, Reiser J, Nayer A (2011) Mast cells, macrophages, and crown-like structures distinguish subcutaneous from visceral fat in mice. J lipid Res 52(3):480–488. doi:10.1194/jlr.M011338
Tanaka A, Nomura Y, Matsuda A, Ohmori K, Matsuda H (2011) Mast cells function as an alternative modulator of adipogenesis through 15-deoxy-delta-12, 14-prostaglandin J2. Am J Physiol Cell Physiol 301(6):C1360–1367. doi:10.1152/ajpcell.00514.2010
Murakami M, Tada K, Nakajima K, Kudo I (1997) Cyclooxygenase-2-dependent delayed prostaglandin D2 generation is initiated by nerve growth factor in rat peritoneal mast cells: its augmentation by extracellular type II secretory phospholipase A2. J Immunol 159(1):439–446
Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM (1995) A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 83(5):813–819
Herlong JL, Scott TR (2006) Positioning prostanoids of the D and J series in the immunopathogenic scheme. Immunol lett 102(2):121–131. doi:10.1016/j.imlet.2005.10.004
Sinha D, Addya S, Murer E, Boden G (1999) 15-Deoxy-delta(12,14) prostaglandin J2: a putative endogenous promoter of adipogenesis suppresses the ob gene. Metab: Clin Exp 48(6):786–791
Zhang J, Shi GP (2012) Mast cells and metabolic syndrome. Biochim Biophys Acta 1822(1):14–20. doi:10.1016/j.bbadis.2010.12.012
Caspar-Bauguil S, Cousin B, Galinier A, Segafredo C, Nibbelink M, Andre M, Casteilla L, Penicaud L (2005) Adipose tissues as an ancestral immune organ: site-specific change in obesity. FEBS lett 579(17):3487–3492. doi:10.1016/j.febslet.2005.05.031
Barra NG, Chew MV, Reid S, Ashkar AA (2012) Interleukin-15 treatment induces weight loss independent of lymphocytes. PloS One 7(6):e39553. doi:10.1371/journal.pone.0039553
Lynch LA, O'Connell JM, Kwasnik AK, Cawood TJ, O'Farrelly C, O'Shea DB (2009) Are natural killer cells protecting the metabolically healthy obese patient? Obesity (Silver Spring) 17(3):601–605. doi:10.1038/oby.2008.565
Duffaut C, Galitzky J, Lafontan M, Bouloumie A (2009) Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun 384(4):482–485. doi:10.1016/j.bbrc.2009.05.002
O'Rourke RW, White AE, Metcalf MD, Olivas AS, Mitra P, Larison WG, Cheang EC, Varlamov O, Corless CL, Roberts CT Jr, Marks DL (2011) Hypoxia-induced inflammatory cytokine secretion in human adipose tissue stromovascular cells. Diabetologia 54(6):1480–1490. doi:10.1007/s00125-011-2103-y
Barra NG, Reid S, MacKenzie R, Werstuck G, Trigatti BL, Richards C, Holloway AC, Ashkar AA (2010) Interleukin-15 contributes to the regulation of murine adipose tissue and human adipocytes. Obesity (Silver Spring) 18(8):1601–1607. doi:10.1038/oby.2009.445
Ahima RS, Flier JS (2000) Leptin. Annu Rev Physiol 62:413–437. doi:10.1146/annurev.physiol.62.1.413
La Cava A, Matarese G (2004) The weight of leptin in immunity. Nat Rev Immunol 4(5):371–379. doi:10.1038/nri1350
Faggioni R, Feingold KR, Grunfeld C (2001) Leptin regulation of the immune response and the immunodeficiency of malnutrition. FASEB J : Off Publ Fed Am Soc Exp Biol 15(14):2565–2571. doi:10.1096/fj.01-0431rev
Gainsford T, Willson TA, Metcalf D, Handman E, McFarlane C, Ng A, Nicola NA, Alexander WS, Hilton DJ (1996) Leptin can induce proliferation, differentiation, and functional activation of hemapoietic cells. Proc Natl Acad Sci U S A 93(25):14564–14568
Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, Klein AS, Bulkley GB, Bao C, Noble PW, Lane MD, Diehl AM (1998) Leptin regulates proinflammatory immune responses. FASEB J : Off Publ Fed Am Soc Exp Biol 12(1):57–65
Lee FY, Li Y, Yang EK, Yang SQ, Lin HZ, Trush MA, Dannenberg AJ, Diehl AM (1999) Phenotypic abnormalities in macrophages from leptin-deficient, obese mice. Am J Physiol 276(2 Pt 1):C386–394
Mattioli B, Giordani L, Quaranta MG, Viora M (2009) Leptin exerts an anti-apoptotic effect on human dendritic cells via the PI3K-Akt signaling pathway. FEBS lett 583(7):1102–1106. doi:10.1016/j.febslet.2009.02.029
Smith AG, Sheridan PA, Harp JB, Beck MA (2007) Diet-induced obese mice have increased mortality and altered immune responses when infected with influenza virus. J Nutr 137(5):1236–1243
Tian Z, Sun R, Wei H, Gao B (2002) Impaired natural killer (NK) cell activity in leptin receptor deficient mice: leptin as a critical regulator in NK cell development and activation. Biochem Biophys Res Commun 298(3):297–302
Wrann CD, Laue T, Hubner L, Kuhlmann S, Jacobs R, Goudeva L, Nave H (2012) Short-term and long-term leptin exposure differentially affect human natural killer cell immune functions. Am J Physiol Endocrinol Metab 302(1):E108–116. doi:10.1152/ajpendo.00057.2011
Beckman EM, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB (1994) Recognition of a lipid antigen by CD1-restricted alpha beta + T cells. Nature 372(6507):691–694. doi:10.1038/372691a0
Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, Balk SP, O'Shea D, O'Farrelly C, Exley MA (2012) Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity 37(3):574–587. doi:10.1016/j.immuni.2012.06.016
Ohmura K, Ishimori N, Ohmura Y, Tokuhara S, Nozawa A, Horii S, Andoh Y, Fujii S, Iwabuchi K, Onoe K, Tsutsui H (2010) Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler, Thromb, Vasc Biol 30(2):193–199. doi:10.1161/ATVBAHA.109.198614
Mantell BS, Stefanovic-Racic M, Yang X, Dedousis N, Sipula IJ, O'Doherty RM (2011) Mice lacking NKT cells but with a complete complement of CD8+ T-cells are not protected against the metabolic abnormalities of diet-induced obesity. PloS One 6(6):e19831. doi:10.1371/journal.pone.0019831
Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, Prop S, Meerding J, Hamers N, Besra G, Boon L, Nieuwenhuis EE, Elewaut D, Prakken B, Kersten S, Boes M, Kalkhoven E (2012) Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Investig 122(9):3343–3354. doi:10.1172/JCI62739
Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, Gao B, Lee CH, Kersten S, Qi L (2012) Activation of natural killer T cells promotes M2 macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem 287(17):13561–13571. doi:10.1074/jbc.M112.350066
Ji Y, Sun S, Xia S, Yang L, Li X, Qi L (2012) Short-term high-fat-diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer T cells and interleukin-4. J Biol Chem. doi:10.1074/jbc.M112.371807
Wu L, Parekh VV, Gabriel CL, Bracy DP, Marks-Shulman PA, Tamboli RA, Kim S, Mendez-Fernandez YV, Besra GS, Lomenick JP, Williams B, Wasserman DH, Van Kaer L (2012) Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice. Proc Natl Acad Sci U S A 109(19):E1143–1152. doi:10.1073/pnas.1200498109
Lynch L, O'Shea D, Winter DC, Geoghegan J, Doherty DG, O'Farrelly C (2009) Invariant NKT cells and CD1d(+) cells amass in human omentum and are depleted in patients with cancer and obesity. Eur J Immunol 39(7):1893–1901. doi:10.1002/eji.200939349
Kenna T, Golden-Mason L, Porcelli SA, Koezuka Y, Hegarty JE, O'Farrelly C, Doherty DG (2003) NKT cells from normal and tumor-bearing human livers are phenotypically and functionally distinct from murine NKT cells. J Immunol 171(4):1775–1779
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392(6673):245–252. doi:10.1038/32588
Dominguez PM, Ardavin C (2010) Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol Rev 234(1):90–104. doi:10.1111/j.0105-2896.2009.00876.x
Joffre O, Nolte MA, Sporri R, Reis e Sousa C (2009) Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev 227(1):234–247. doi:10.1111/j.1600-065×.2008.00718.x
Bedford PA, Todorovic V, Westcott ED, Windsor AC, English NR, Al-Hassi HO, Raju KS, Mills S, Knight SC (2006) Adipose tissue of human omentum is a major source of dendritic cells, which lose MHC Class II and stimulatory function in Crohn's disease. J Leukoc Biol 80(3):546–554. doi:10.1189/jlb.0905501
Bertola A, Ciucci T, Rousseau D, Bourlier V, Duffaut C, Bonnafous S, Blin-Wakkach C, Anty R, Iannelli A, Gugenheim J, Tran A, Bouloumie A, Gual P, Wakkach A (2012) Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 61(9):2238–2247. doi:10.2337/db11-1274
Stefanovic-Racic M, Yang X, Turner MS, Mantell BS, Stolz DB, Sumpter TL, Sipula IJ, Dedousis N, Scott DK, Morel PA, Thomson AW, O'Doherty RM (2012) Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c + cells in adipose tissue and liver. Diabetes 61(9):2330–2339. doi:10.2337/db11-1523
Winer S, Paltser G, Chan Y, Tsui H, Engleman E, Winer D, Dosch HM (2009) Obesity predisposes to Th17 bias. Eur J Immunol 39(9):2629–2635. doi:10.1002/eji.200838893
Gao B, Jeong WI, Tian Z (2008) Liver: an organ with predominant innate immunity. Hepatology 47(2):729–736. doi:10.1002/hep.22034
Bjorkstrom NK, Kekalainen E, Mjosberg J (2013) Tissue-specific effector functions of innate lymphoid cells. Immunology. doi:10.1111/imm.12098
Sakaguchi S, Takahashi S, Sasaki T, Kumagai T, Nagata K (2011) Progression of alcoholic and non-alcoholic steatohepatitis: common metabolic aspects of innate immune system and oxidative stress. Drug Metab Pharmacokinet 26(1):30–46
Williams KH, Shackel NA, Gorrell MD, McLennan SV, Twigg SM (2013) Diabetes and nonalcoholic fatty liver disease: a pathogenic duo. Endocr Rev 34(1):84–129. doi:10.1210/er.2012-1009
Larrain S, Rinella ME (2012) A myriad of pathways to NASH. Clin liver Dis 16(3):525–548. doi:10.1016/j.cld.2012.05.009
Bohinc BN, Diehl AM (2012) Mechanisms of disease progression in NASH: new paradigms. Clin liver Dis 16(3):549–565. doi:10.1016/j.cld.2012.05.002
Zhan YT, An W (2010) Roles of liver innate immune cells in nonalcoholic fatty liver disease. World J Gastroenterol : WJG 16(37):4652–4660
Bilzer M, Roggel F, Gerbes AL (2006) Role of Kupffer cells in host defense and liver disease. Liver Int : Off J Int Assoc Study of the Liver 26(10):1175–1186. doi:10.1111/j.1478-3231.2006.01342.x
Szabo G, Petrasek J, Bala S (2012) Innate immunity and alcoholic liver disease. Dig Dis 30(Suppl 1):55–60. doi:10.1159/000341126
Cubero FJ, Nieto N (2012) Arachidonic acid stimulates TNFalpha production in Kupffer cells via a reactive oxygen species-pERK1/2-Egr1-dependent mechanism. Am J Physiol Gastrointest Liver Physiol 303(2):G228–239. doi:10.1152/ajpgi.00465.2011
Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D (2011) An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology 54(6):2185–2197. doi:10.1002/hep.24599
Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E (2010) Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 139(1):323–334. doi:10.1053/j.gastro.2010.03.052, e327
Yang YY, Huang YT, Tsai TH, Hou MC, Lee FY, Lee SD, Lin HC (2012) Kupffer cell depletion attenuates leptin-mediated methoxamine-stimulated portal perfusion pressure and thromboxane A2 release in a rodent model of NASH-cirrhosis. Clin Sci 123(12):669–680. doi:10.1042/CS20110572
Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS (2012) Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. J Biol Chem 287(48):40161–40172. doi:10.1074/jbc.M112.417014
Seki E, Schnabl B (2012) Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J Physiol 590(Pt 3):447–458. doi:10.1113/jphysiol.2011.219691
Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, Naveau S, Prevot S, Makhzami S, Perlemuter G, Cassard-Doulcier AM (2012) Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol 57(1):141–149. doi:10.1016/j.jhep.2012.02.028
Odegaard JI, Chawla A (2008) Mechanisms of macrophage activation in obesity-induced insulin resistance. Nat Clin Pract Endocrinol Metab 4(11):619–626. doi:10.1038/ncpendmet0976
Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW, Chawla A (2008) Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab 7(6):496–507. doi:10.1016/j.cmet.2008.04.003
de las Casas-Engel M, Dominguez-Soto A, E S-F, Bragado R, Nieto C, Puig-Kroger A, Samaniego R, Loza M, Corcuera MT, Gomez-Aguado F, Bustos M, Sanchez-Mateos P, Corbi AL (2013) Serotonin skews human macrophage polarization through HTR2B and HTR7. J Immunol 190(5):2301–2310. doi:10.4049/jimmunol.1201133
Muldoon MF, Mackey RH, Korytkowski MT, Flory JD, Pollock BG, Manuck SB (2006) The metabolic syndrome is associated with reduced central serotonergic responsivity in healthy community volunteers. J Clin Endocrinol Metab 91(2):718–721. doi:10.1210/jc.2005-1654
Gao B, Radaeva S (2012) Natural killer and natural killer T cells in liver fibrosis. Biochim Biophys Acta. doi:10.1016/j.bbadis.2012.09.008
Kahraman A, Fingas CD, Syn WK, Gerken G, Canbay A (2012) Role of stress-induced NKG2D ligands in liver diseases. Liver Int : Off J Int Assoc Study Liver 32(3):370–382. doi:10.1111/j.1478-3231.2011.02608.x
Mondelli MU (2012) NKG2D and its ligands: key to immunotherapy of liver cancer? J Hepatol 56(2):308–310. doi:10.1016/j.jhep.2011.07.008
Zou Y, Chen T, Han M, Wang H, Yan W, Song G, Wu Z, Wang X, Zhu C, Luo X, Ning Q (2010) Increased killing of liver NK cells by Fas/Fas ligand and NKG2D/NKG2D ligand contributes to hepatocyte necrosis in virus-induced liver failure. J Immunol 184(1):466–475. doi:10.4049/jimmunol.0900687
Tian Z, Chen Y, Gao B (2012) Natural killer cells in liver disease. Hepatology. doi:10.1002/hep.26115
Kahraman A, Schlattjan M, Kocabayoglu P, Yildiz-Meziletoglu S, Schlensak M, Fingas CD, Wedemeyer I, Marquitan G, Gieseler RK, Baba HA, Gerken G, Canbay A (2010) Major histocompatibility complex class I-related chains A and B (MIC A/B): a novel role in nonalcoholic steatohepatitis. Hepatology 51(1):92–102. doi:10.1002/hep.23253
O'Shea D, Cawood TJ, O'Farrelly C, Lynch L (2010) Natural killer cells in obesity: impaired function and increased susceptibility to the effects of cigarette smoke. PloS One 5(1):e8660. doi:10.1371/journal.pone.0008660
Glassner A, Eisenhardt M, Kramer B, Korner C, Coenen M, Sauerbruch T, Spengler U, Nattermann J (2012) NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab Investig; J Tech Methods Pathol 92(7):967–977. doi:10.1038/labinvest.2012.54
Gomez-Santos L, Luka Z, Wagner C, Fernandez-Alvarez S, Lu SC, Mato JM, Martinez-Chantar ML, Beraza N (2012) Inhibition of natural killer cells protects the liver against acute injury in the absence of glycine N-methyltransferase. Hepatology 56(2):747–759. doi:10.1002/hep.25694
Liaskou E, Wilson DV, Oo YH (2012) Innate immune cells in liver inflammation. Mediat Inflamm 2012:949157. doi:10.1155/2012/949157
Rahman AH, Aloman C (2013) Dendritic cells and liver fibrosis. Biochim Biophys Acta. doi:10.1016/j.bbadis.2013.01.005
Ibrahim J, Nguyen AH, Rehman A, Ochi A, Jamal M, Graffeo CS, Henning JR, Zambirinis CP, Fallon NC, Barilla R, Badar S, Mitchell A, Rao RS, Acehan D, Frey AB, Miller G (2012) Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology 143(4):1061–1072. doi:10.1053/j.gastro.2012.06.003
Henning JR, Graffeo CS, Rehman A, Fallon NC, Zambirinis CP, Ochi A, Barilla R, Jamal M, Deutsch M, Greco S, Ego-Osuala M, Saeed UB, Rao RS, Badar S, Quesada JP, Acehan D, Miller G (2013) Dendritic cells limit fibro-inflammatory injury in NASH. Hepatology. doi:10.1002/hep.26267
Satoh M, Andoh Y, Clingan CS, Ogura H, Fujii S, Eshima K, Nakayama T, Taniguchi M, Hirata N, Ishimori N, Tsutsui H, Onoe K, Iwabuchi K (2012) Type II NKT cells stimulate diet-induced obesity by mediating adipose tissue inflammation, steatohepatitis and insulin resistance. PloS One 7(2):e30568. doi:10.1371/journal.pone.0030568
Tajiri K, Shimizu Y, Tsuneyama K, Sugiyama T (2009) Role of liver-infiltrating CD3 + CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol 21(6):673–680. doi:10.1097/MEG.0b013e32831bc3d6
Xu CF, Yu CH, Li YM, Xu L, Du J, Shen Z (2007) Association of the frequency of peripheral natural killer T cells with nonalcoholic fatty liver disease. World J Gastroenterol : WJG 13(33):4504–4508
Johnson AR, Milner JJ, Makowski L (2012) The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev 249(1):218–238. doi:10.1111/j.1600-065×.2012.01151.x
Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, Xie G, Philips G, Chan IS, Karaca GF, Pereira Tde A, Chen Y, Mi Z, Kuo PC, Choi SS, Guy CD, Abdelmalek MF, Diehl AM (2012) NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut 61(9):1323–1329. doi:10.1136/gutjnl-2011-301857
Locatelli I, Sutti S, Vacchiano M, Bozzola C, Albano E (2013) NF-kappaB1 deficiency stimulates the progression of non-alcoholic steatohepatitis (NASH) in mice by promoting NKT-cell-mediated responses. Clin Sci 124(4):279–287. doi:10.1042/CS20120289
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259(5091):87–91
Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356(15):1517–1526. doi:10.1056/NEJMoa065213
Sauter NS, Schulthess FT, Galasso R, Castellani LW, Maedler K (2008) The antiinflammatory cytokine interleukin-1 receptor antagonist protects from high-fat diet-induced hyperglycemia. Endocrinology 149(5):2208–2218. doi:10.1210/en.2007-1059
Rizos CV, Elisaf MS, Mikhailidis DP, Liberopoulos EN (2009) How safe is the use of thiazolidinediones in clinical practice? Expert Opin Drug Saf 8(1):15–32. doi:10.1517/14740330802597821
De Vito R, Alisi A, Masotti A, Ceccarelli S, Panera N, Citti A, Salata M, Valenti L, Feldstein AE, Nobili V (2012) Markers of activated inflammatory cells correlate with severity of liver damage in children with nonalcoholic fatty liver disease. Int J Mol Med 30(1):49–56. doi:10.3892/ijmm.2012.965
Inzaugarat ME, Ferreyra Solari NE, Billordo LA, Abecasis R, Gadano AC, Chernavsky AC (2011) Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J Clin Immunol 31(6):1120–1130. doi:10.1007/s10875-011-9571-1
Acknowledgments
We apologize to our colleagues whose contributions could not be cited due to space limitations. We thank Tatiana Golea for the support with the figure design. Our work is supported by grants from the Deutsche Forschungsgemeinschaft, the FP7 EU Project “DIABAT”, the German Cancer Aid, the Helmholtz cross program topic “Metabolic Dysfunction”, and the Helmholtz Alliance “ICEMED.”
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Metabolic Syndrome - Guest Editor: T. Miyazaki
Ashley Eheim and Dasa Medrikova contributed equally to the manuscript preparation.
Rights and permissions
About this article
Cite this article
Eheim, A., Medrikova, D. & Herzig, S. Immune cells and metabolic dysfunction. Semin Immunopathol 36, 13–25 (2014). https://doi.org/10.1007/s00281-013-0403-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-013-0403-7