
Abstract
Purpose
High-dose methotrexate (HD-MTX) is widely used in the treatment of non-Hodgkin lymphoma (NHL), but the pharmacokinetic properties of HD-MTX in Chinese adult patients with NHL have not yet been established through an approach that integrates genetic covariates. The purposes of this study were to identify both physiological and pharmacogenomic covariates that can explain the inter- and intraindividual pharmacokinetic variability of MTX in Chinese adult patients with NHL and to explore a new sampling strategy for predicting delayed MTX elimination.
Methods
A total of 852 MTX concentrations from 91 adult patients with NHL were analyzed using the nonlinear mixed-effects modeling method. FPGS, GGH, SLCO1B1, ABCB1 and MTHFR were genotyped using the Sequenom MassARRAY technology platform and were screened as covariates. The ability of different sampling strategies to predict the MTX concentration at 72 h was assessed through maximum a posteriori Bayesian forecasting using a validation dataset (18 patients).
Results
A two-compartment model adequately described the data, and the estimated mean MTX clearance (CL) was 6.03 L/h (9%). Creatinine clearance (CrCL) was identified as a covariate for CL, whereas the intercompartmental clearance (Q) was significantly affected by the body surface area (BSA). However, none of the genotypes exerted a significant effect on the pharmacokinetic properties of MTX. The percentage of patients with concentrations below 0.2 µmol/L at 72 h decreased from 65.6 to 42.6% when the CrCL decreased from 90 to 60 ml/min/1.73 m2 with a scheduled dosing of 3 g/m2, and the same trend was observed with dose regimens of 1 g/m2 and 2 g/m2. Bayesian forecasting using the MTX concentrations at 24 and 42 h provided the best predictive performance for estimating the MTX concentration at 72 h after dosing.
Conclusions
The MTX population pharmacokinetic model developed in this study might provide useful information for establishing personalized therapy involving MTX for the treatment of adult patients with NHL.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
High-dose methotrexate (HD-MTX), a folic acid antagonist, serves as the cornerstone in the treatment of non-Hodgkin lymphoma (NHL) because it prevents and treats neuromeningeal localization [1].
MTX is approximately 60% protein-bound [2], competing with the enzymes of the folate cycle, including methylene tetrahydrofolate reductase (MTHFR) [3], for active transport across cell membranes through a single carrier-mediated active transport process. MTX undergoes intracellular metabolism by folylpolyglutamate synthetase (FPGS) to obtain polyglutamated MTX, and this form can be converted back to MTX by hydrolase enzymes (GGH). Small amounts of MTX polyglutamates might remain in the tissue for extended periods [4]. MTX can be pumped out of the cell by ATP-binding cassette (ABC) and transported to hepatocytes for elimination by organic anion transporting polypeptide (OATP) 1B1, which is encoded by the ABCB1 and SLCO1B1 genes, respectively [5, 6]. After intravenous infusion, 80–90% of the MTX dose is excreted unchanged in the urine within 24 h, whereas 10% or less of the dose is eliminated through biliary excretion [7].
However, treatment is often limited by severe systemic toxicity [1], which is related to exposure to MTX [8]. Delayed MTX elimination is defined as a serum MTX concentration above 50 µmol/L at 24 h, above 5 µmol/L at 48 h, or above 0.2 µmol/L at 72 h [4]. Patients that experience delayed MTX elimination are at elevated risk of toxicity, such as gastrointestinal toxicity, myelosuppression, mucositis, neurotoxicity, and liver and kidney dysfunction, which explains the importance of routinely monitoring the MTX concentration to guide leucovorin rescue and avoid potential side effects until the plasma concentrations decrease to below the threshold value of 0.2 µmol/L.
The clearance of MTX exhibits a large interindividual variability, which leads to different exposures to the drug and can thus affect the clinical outcomes of the patients [9, 10]. Therefore, the determination of individual pharmacokinetic (PK) parameters, such as through maximum posterior Bayesian (MAPB) estimation, are needed for the optimization of individual therapy.
Pharmacogenomics studies have demonstrated that the disposition, efficacy, and toxicity of MTX can be influenced by single-nucleotide polymorphisms (SNPs) in the following genes: FPGS and GGH (which encode metabolizing enzymes of MTX), SLCO1B1 and ABCB1 (which encode transporters of MTX), and MTHFR (which encodes a target of MTX) [11,12,13]. Taking SLCO1B1 as an example, a previous study showed that the plasma concentration of MTX at 48 and 72 h is increased significantly in individuals with SLCO1B1 rs4149056 variant (TC/CC genotype) compared with the wild-type individuals (TT genotype). Patients with the rs4149056 C allele exhibit a significantly higher frequency of adverse effects [14]. The significant association of SLCO1B1 rs4149056 with the serum MTX levels was also observed by Csordas et al. [15], and Lima et al. [16] and Avivi et al. [17] who also demonstrated that SLCO1B1 rs4149056 is associated with MTX-related toxicity. Lopez et al. [18] found a statistically significant association between the plasma concentration of MTX and SLCO1B1 rs11045879, and Li et al. obtained the same result [19]. Further investigations on whether these SNPs in related genes explain the interindividual variability in MTX pharmacokinetics are required prior to the inclusion of genetic information in MTX individual therapy.
The majority of population pharmacokinetic (PPK) studies on MTX have focused on acute lymphocytic leukemia (ALL) in children, but far less information is available regarding adult patients with NHL, and few studies have considered genetic factors as covariate effects. Moreover, ethnicity is one factor that might account for the observed differences in both the pharmacokinetics (PK) and pharmacodynamics (PD) of drugs, which has resulted in variability in the responses to drug therapy [20]. Few PPK studies on MTX have provided pharmacogenetic information for non-Chinese patients, but the results have been controversial [21,22,23]. To the best of our knowledge, none of the published studies have established the PPK of HD-MTX in Chinese adult patients with NHL using a method that integrates genetic covariates.
We would like to concentrate on the genes mentioned above that have been demonstrated to affect the clinical outcome of MTX but whose impact on the PPK of MTX is inconclusive or unknown. Therefore, the following candidate genetic variants were selected for the present study: rs10106 in FPGS; rs719235, rs10464903 and rs12681874 in GGH; rs4149056 and rs11045879 in SLCO1B1; rs1045642 in ABCB1; and rs1801131 and rs1801133 in MTHFR.
In clinical practice, the routine monitoring of MTX concentrations includes several blood samplings. Patients with MTX concentrations at 72 h that remain above the rescue threshold (0.2 µmol/L) should stay longer in the hospital for additional hydration and leucovorin rescue. A new sampling strategy involves fewer samples than those used in routine practice. The early prediction of increased MTX concentrations at 72 h based on this new sampling strategy reduces the number of samples and the hospital duration.
This study aimed to identify physiological and pharmacogenomic covariates that can explain the inter- and intraindividual pharmacokinetic variability of MTX in Chinese adults with NHL and to explore a new sampling strategy for predicting delayed MTX elimination.
Materials and methods
Patients and study design
The study involved adult patients (age ≥ 18 years) who were diagnosed with NHL and treated with HD-MTX at Fujian Cancer Hospital from July 2013 to December 2018. Patients with diffuse large B-cell lymphoma were treated with MTX ± cytarabine [24, 25]. The NHL–Berlin-Frankfurt-Münster 95 protocol (NHL-BFM95) and ALL-Berlin-Frankfurt-Münster 95 protocol (ALL-BFM95) were used for the treatment of Burkitt’s and T-lymphoblastic lymphoma [26, 27], respectively, whereas the AspaMetDex protocol was used for the treatment of NK/T-cell lymphoma [28].
The administration of HD-MTX at 1–3 g/m2 was followed by leucovorin rescue, which was administered 36 h after initiation of the MTX infusion and repeated every 6 h until the MTX concentration decreased to less than 0.2 µmol/L [29].
Intravenous hydration and alkalization were achieved 12 h prior to the start of MTX therapy to provide protection against MTX-induced renal dysfunction [4]. MTX concentration monitoring was performed until the MTX plasma concentration was below 0.2 µmol/L. The study was approved by the ethics committee of Fujian Cancer Hospital. Informed consent was obtained from the patients and/or guardians.
Analytical method
The plasma was separated by centrifugation at 3500 g for 5 min, and the separated plasma samples were used for analysis. The MTX concentrations were measured by high-performance liquid chromatography (HPLC), and the chromatographic system consisted of a Beckman-C18 (150 mm × 4.6 mm, 5 μm) analytical column with a mobile phase of 40 mM sodium acetate buffer (pH 4.6):acetonitrile (88.9:11.1, v/v) and ultraviolet detection at 294 nm. The flow rate of the mobile phase was maintained at 1.0 mL/min. The injection volume was 20 μL with a run time of 6 min. The method was linear within the range of 0.09–3.52 µmol/L, and the intra- and interday precision was < 6%. The limit of quantification (LOQ) was 0.09 µmol/L.
Genotyping
Genomic DNA was isolated from whole blood using the Qiagen DNA Blood Mini Kit (Qiagen, CA, USA) following the manufacturer’s instructions. The DNA purity and concentration were checked by spectrophotometric measurement of the absorbance at 260 and 280 nm.
Genotyping for rs10106, rs719235, rs10464903, rs12681874, rs4149056, rs11045879, rs1045642, rs1801131, and rs1801133 was performed using the Sequenom MassARRAY technology platform with the complete iPLEX Gold Reagent Set (Sequenom, CA, USA) by the Beijing Genomics Institute Genomics Company (Beijing, China). Allele detection was performed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. The assay data were analyzed using Sequenom TYPER software (version 4.0). The primers were designed by ADS software 2.0 (Agena Bioscience, CA, USA).
Base model
The pharmacokinetic analysis was conducted by nonlinear mixed-effects modeling using NONMEM® Version 7.4.1 (ICON Development Solutions, Hanover, MD, USA). The first-order conditional estimate method with eta-epsilon interaction (FOCE-I) was used throughout the model-building process. The tools used to evaluate and visualize the model were R Version 3.4.1 (R Foundation for Statistical Computing, Vienna, Austria) and PsN Version 4.6.0 (https://uupharmacometrics.github.io/PsN/), which are all within the graphical interface Pirana Version 2.9.7 (Certara, St. Louis, MO, USA) [30].
Two- and three-compartment models were investigated to describe the concentration–time data based on a literature review and visual data inspection.
The interindividual variability (IIV) of the PK parameters was evaluated using the exponential model shown in Equation (Eq. 1):
where \(P_{ij}\) is the parameter estimate for the ith individual on the jth occasion and \({\text{TV}}\left( P \right)\) is a typical value for the parameter. The difference between logarithms of \(P_{ij}\) and \({\text{TV}}\left( P \right)\) is described by ηi, and kij is the interoccasion variability (IOV) used to represent the random difference of occasion j from the individual i average value.
Additive (Eq. 2), proportional (Eq. 3) and combination error models (Eq. 4) were evaluated to describe the residual variability.
where Y is the observed MTX concentration, F is the model-predicted concentration, and EPS (1) and EPS (2) represent the residual error of the model with a means of 0 and variances of \(\sigma_{1}^{2}\) and \(\sigma_{2}^{2}\), respectively.
Model selection was based on the Akaike information criterion (AIC), Bayesian information criterion (BIC), objective function value (OFV), parameter precision, shrinkage values, and visual inspection of the corresponding goodness-of fit plots. A shrinkage value below 20% was considered acceptable [31].
Covariate analysis
Once the base model was selected, covariates were tested for their influence on pharmacokinetic parameters. The covariates tested were age, sex, height, body weight, body surface area (BSA), disease stage, hematocrit, serum creatinine (SCR), blood urea nitrogen (BUN), creatinine clearance (CrCL), alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin and SNPs of FPGS, GGH, SLCO1B1, ABCB1, and MTHFR. CrCL was estimated using the Chronic Kidney Disease Epidemiology Collaboration 2009Scr (CKD-EPI2009Scr) equation [32].
The influence of the continuous covariate (CO) of each patient was modeled using the following linear (Eq. 5), power (Eq. 6), and exponential (Eq. 7) equations:
where Pi is the value of P for the ith individual, TV(P) is the typical value of parameter P, COi is the covariate of the ith individual, COmedian is the median value of the covariate, and θ is the coefficient term to be estimated.
The effect of categorical covariates on parameter P was tested according to (Eq. 8), which uses sex as an example.
The influence of the SNPs on parameter P was evaluated by additive, recessive, and dominant genetic models using the following equation:
The SNPs were classified as homozygous wild-type (wt), heterozygous (het), and homozygous variant (var) genotypes. For the additive model, geno = 0, 1 or 2 for the wt, het, and var genotypes, respectively. For the recessive model, geno = 0 for both the wt and het genotypes, and geno = 1 for the var genotype. For the dominant model, geno = 0 for the wt genotype, and geno = 1 for both het and var genotypes. In addition, θ is the estimated covariate effect.
A forward inclusion and backward elimination approach was used to evaluate the statistical significance of relevant covariates. Covariates that decreased the OFV by more than 3.84 (p < 0.05) were added to the full model. Backward elimination was then performed with a stricter statistical significance of p < 0.01 (OFV > 6.64). An additional criterion for the inclusion of covariates in the final model was that the impacts of the covariates be biologically reasonable.
Model validation
The goodness of fit of the final model was evaluated by checking the plots of the observed concentrations versus the population-or individual predicted concentrations and the conditional-weighted residuals versus the population-predicted concentrations or time. Two procedures were used to evaluate the stability and predictive ability of the final model. First, a bootstrap resampling method was applied [33]. One thousand bootstrap datasets were generated by sampling randomly from the original dataset with replacement, and the medians of the parameters of the bootstrap dataset were then compared to the estimates of the original dataset. The model was further validated by the prediction-corrected visual predictive check (PC-VPC) based on 1000 simulated datasets [34]. The median and 95% confidence intervals (CI) of the simulated predictions were compared to the observed data.
Model application
The changes in the percentages of patients with renal dysfunction who had MTX concentrations below 0.2 µmol/L at 72 h were analyzed using six scenarios based on the administration of 1, 2, and 3 g/m2 to patients with the same BSA (1.65 m2) but different renal functions (CrCL = 60 or 90 ml/min/1.73 m2). Plasma MTX concentration–time profiles were simulated for 1000 hypothetical patients in each scenario based on the final population PK model.
New sampling strategy
To identify suitable sampling strategies for forecasting the concentration at 72 h, the impact of prior observed concentrations on model predictability was assessed by maximum a posteriori Bayesian forecasting using a validation dataset. The individual prediction (IPRED) of the concentration at 72 h was estimated based on one prior observation (MTX concentration at 24, 36, 42 and 48 h, respectively) and two prior observations (MTX concentrations at 24–36 h, 24–42 h, 24–48 h, 36–42 h and 36–48 h). The comparison of the observed concentration to that estimated using the Bayesian approach at 72 h was denoted by the individual prediction error [IPE%]. The median individual prediction error (MPE%), the root median squared relative individual prediction error (RMSE%), the percentage of |IPE|% within 20% (IF20) and IF30 were used to evaluate the accuracy and precision of the predictability with different prior information.
Results
Patient characteristics
A total of 852 MTX concentrations from 91 patients with NHL were available for model building. Of the patients, 70.3% were males, and 12.1% were older than 65 years. Twenty-one of these patients had renal dysfunction (CrCL < 90 mL/min/1.73 m2). Eighteen patients with 196 observations were included in the validation dataset. The demographic and clinical characteristics of the patients included in the modeling and validation datasets are presented in Table 1.
Genotyping
Six SNPs in three genes encoding enzymes of the folate metabolic pathway (FPGS, GGH, and MTHFR) and three SNPs in two MTX transporter genes (SLCO1B1 and ABCB1) were analyzed. All genotype groups were in Hardy–Weinberg equilibrium. The genotypes of the targeted SNPs and the associated Hardy–Weinberg equilibrium results are shown in Table 2.
Base model
The data were best described by a two-compartment model according to the OFV, AIC, BIC, shrinkage values, and diagnostic plots. The inclusion of the IIV on the clearance (CL), the intercompartmental clearance (Q), and the volume of the central compartment (V1) significantly improved the model, and the OFV decreased further after the introduction of the IOV on the CL. The residual variability was described by a proportional error model.
Covariate analysis
Covariate analysis performed by forward inclusion into the base model indicated that the CL was affected by CrCL (ΔOFV = 17.1, p < 0.001). After inclusion of the CrCL on the CL, only the BSA could be added on the Q. After backward exclusion, the final model that included the effect of the CrCL on the CL and that of the BSA on the Q was obtained (ΔOFV = 24.766, p < 0.001).
None of the genotypes exerted a significant effect on MTX CL or V1, and the relationship among the CL, V1 and the target SNPs is shown in Fig. 1.






Relationship among CL, V1 and the target SNPs
The final PK model for MTX in Chinese adult patients with NHL was as follows:
Model validation
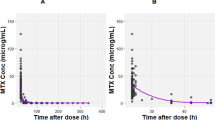
The goodness-of-fit plots of the observations versus population and individual predictions obtained with the final model were densely distributed around the line of identity, and the conditional-weighted residuals were normally distributed, which indicated that the model adequately described the observed concentrations (Fig. 2).

Goodness-of-fit plots of the final model. a Observed concentration vs. individual-predicted concentration, b observed concentration vs. population-predicted concentration (PRED), c conditional-weighted residuals (CWRES) vs. PRED, and d CWRES vs. time after the last dose. The solid lines in a and b are identity lines, and the solid lines in c and d are zero lines. The Y-axis is on a log scale
The bootstrap analysis was successful in 86% of the 1000 runs, and the median parameter values as well as the 2.5% and 97.5% percentiles agreed with the final model estimations and errors, which indicated the stability of the final model (Table 3).
The PC-VPC, which exhibited an acceptable overlap of the 5th, 50th, and 95th percentiles of the observed data and the 95% CI of the corresponding simulations indicated the good predictive performance of the final model (Fig. 3).

Prediction-corrected visual predictive check of the final model. The dots indicate the observed concentrations. The lines represent the 5th, 50th, and 95th percentiles of the observed data. The shaded areas are the 95% confidence intervals for the same percentiles of the simulations
Model application
The population medians of the simulated plasma concentration-versus-time profiles are shown in Fig. 4. The percentage of patients with concentrations below 0.2 µmol/L at 72 h was dependent on the CrCL and related to the dose.

Simulated plasma profiles of MTX under conditions involving different renal functions and dosages
The percentage decreased from 65.6 to 42.6% when the CrCL decreased from 90 to 60 ml/min/1.73 m2 with the scheduled dosing of 3 g/m2, and the same trend was obtained with dose regimens of 1 and 2 g/m2.
New sampling strategy
Bayesian forecasting demonstrated that prior information could improve the prediction accuracy for the concentration at 72 h. Among the prior information groups, the one that included the MTX concentration at 36 h provided the best estimation of the concentration at 72 h, whereas prior information of the MTX concentration at 24 and 42 h provided the best estimation among all the tested groups (MPE% − 3.47%, RMSE% 12.76%, IF20 = 66.67%, IF30 = 80.56%). Boxplots of the individual prediction errors obtained with different prior information are presented in Fig. 5.

Box plots of the individual prediction errors in different scenarios
Discussion
This study provides the first reported PPK model of MTX in Chinese adult patients with NHL that involves genetic factors as covariate effects. Our results show that the CrCL significantly influenced the CL and that the BSA was associated with the Q, but none of the target SNPs exerted a significant effect on the CL or V1.
Growing evidence suggests that polymorphisms in genes encoding transporters or enzymes might partly account for the MTX-PK variability. A study of European children and adolescents with lymphoblastic leukemia and malignant lymphoma screened ABCB1 rs1045642, MTHFR rs1801131, and MTHFR rs1801133 and found that only MTHFR rs1801133 exhibited a significant effect on the MTX pharmacokinetics. The MTX CL in patients homozygous for the variant allele of MTHFR rs1801133 was 26% lower than that in homozygous wild-type individuals [21]. However, Kim et al. [22] found a significant association between ABCB1 rs1045642, but not MTHFR rs1801133 and the MTX CL. Their study included 20 Korean patients treated with low-dose MTX (10–15 mg/kg) after hematopoietic stem cell transplantation.
A large cohort and genome-wide association study showed that the SLCO1B1 genotypes were significant predictors of the AUC0–48 h of MTX and significantly associated with the C24 h and clearance of MTX [11]. Although OATP1B1 is not expressed in the kidney, SNPs of SLCO1B1 might interfere with renal transporters responsible for MTX elimination in urine by affecting the accumulation of endogenous metabolites in blood, which are subsequently excreted in urine [35]. Gabrielle et al. [23] evaluated the influence of 54 SNPs located in the SLCO1B1, ABCB1, ABCC1, ABCC2, ABCC3, ABCC4, ABCG2, SLC19A1, and UGT1A1 genes on the MTX PPK model for 187 osteosarcoma patients; in the patient cohort, SLCO1B1the. Woillard et al. [36] developed a three-parameter time-dependent MTX PPK model that accounted for the nonlinearity in the clearance using Pmetrics and found that the time-dependent model might be better than a previously published two-compartment model developed using NONMEM.
In the present study, we analyzed nine SNPs in FPGS, GGH, SLCO1B1, ABCB1, and MTHFR as covariates, and none of the genotypes exerted a significant effect on the MTX pharmacokinetic properties. The main MTX elimination route might vary among treatment protocols used for different types of cancer [23], which might partly account for the above-mentioned inconformity. Furthermore, the present study might not have sufficient power to detect a significant association between the targeted SNPs and the MTX pharmacokinetic properties.
In our final PPK model, the typical values of CL (6.03 L/h) and V1 (20.7 L) were comparable to the values reported by Faltaos et al. [37] (CL, 7.1 L/h; V1, 25.1 L), Kotnik et al. [21] (CL, 7.43 L/h; V1, 16.7 L), and Mei et al. [38] (CL, 6.67 L/h; V1, 24.5 L).
Some studies have reported that the MTX CL is influenced by the CrCL [29, 39, 40]. Our study showed that incorporation of the CrCL for prediction of the MTX CL led to a decrease in the OFV by 17.1 points (p < 0.001). We also found that a decrease in CrCL corresponded to a lower body weight-normalized CL in both men and women (Fig. 6). The significant influence of CrCL on MTX CL could be explained by the fact that renal excretion is the predominant MTX elimination route.

Relationship between body weight-normalized clearance and creatinine clearance
We found an IOV on CL, which suggested that elimination parameters might vary across MTX courses. The IOV on CL was estimated to equal 15.4%, in agreement with previous PPK reports (14.8% and 16.6%, respectively) [21, 23].
We also found that BSA was a significant covariate in Q, which was similar to the results obtained in two previous studies [38, 39]. Although both body weight and hematocrit exerted a significant effect on methotrexate CL and V1 in some published models [41,42,43], no significant results were observed in the present study, which was similar to the results reported by Mei et al. [38].
Patients with high doses and renal dysfunction are at higher risk of delayed elimination. The time it took for the MTX concentration to decrease below the rescue threshold (0.2 µmol/L) exhibited a wide variability between groups with different renal functions or different doses, as shown in Fig. 4, which suggests that MTX monitoring should remain an important strategy for providing information regarding the elimination of MTX.
A study performed in children with ALL showed that Bayesian estimation using a one-sample (48 h) schedule yielded better results for predicting delayed elimination to reach a concentration less than 0.2 µmol/L compared with a two-sample schedule (24 and 48 h) [44]. However, Christine et al. [45] tested a one-sample schedule (each of the available blood samples) and a two-sample schedule (five combinations) and demonstrated that two sampling times (24 and 48 h) allowed more precise and accurate determination of individual pharmacokinetic parameters. In the present study, the two-sample schedule (24 and 42 h) showed the most accurate and precise prediction of the concentration at 72 h.
This study has some limitations. First, the sample size was relatively small (852 observations from 91 patients). Second, some important SNPs, such as SLC19A1 rs1051266, ABCB1 rs1128503, ABCC1 rs28364006, ABCC2 rs3740065, ABCG2 rs2231142, and ABCG2 rs13120400, were not identified in this study but will be taken into consideration in our future studies. Third, the present study relied on routine MTX monitoring data, which did not include concentrations of MTX in urine or concentrations of MTX metabolites.
Conclusion
An MTX population pharmacokinetic model for Chinese adult patients with NHL was developed and validated. Lower CrCL resulted in lower MTX CL, and the BSA was positively correlated with Q. It is important to assess the renal function in each patient prior to the initiation of chemotherapy, and to pay attention to the MTX accumulations in patients with a high BSA. None of the targeted SNPs in FPGS, GGH SLCO1B1 ABCB1 or MTHFR exerted a significant effect on the MTX pharmacokinetic properties. Model simulation quantified the differences in reaching the threshold at 72 h between patients with different renal functions and doses. A schedule involving two samples, at 24 and 42 h, might be able to forecast delayed elimination, but this finding needs to be validated in a prospective study.
References
Reiter A, Schrappe M, Tiemann M, Ludwig WD, Yakisan E, Zimmermann M, Mann G, Chott A, Ebell W, Klingebiel T, Graf N, Kremens B, Muller-Weihrich S, Pluss HJ, Zintl F, Henze G, Riehm H (1999) Improved treatment results in childhood B-cell neoplasms with tailored intensification of therapy: a report of the Berlin-Frankfurt-Munster Group Trial NHL-BFM 90. Blood 94(10):3294–3306
Maia MB, Saivin S, Chatelut E, Malmary MF, Houin G (1996) In vitro and in vivo protein binding of methotrexate assessed by microdialysis. Int J Clin Pharmacol Ther 34(8):335–341
Fotoohi AK, Albertioni F (2008) Mechanisms of antifolate resistance and methotrexate efficacy in leukemia cells. Leuk Lymphoma 49(3):410–426. https://doi.org/10.1080/10428190701824569
Hospira (2017) Label for methotrexate injection. Lake Forest, IL; Hospira; 2011. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/011719s117lbl.pdf. Accessed 29 Dec 2019
Takatori R, Takahashi KA, Tokunaga D, Hojo T, Fujioka M, Asano T, Hirata T, Kawahito Y, Satomi Y, Nishino H, Tanaka T, Hirota Y, Kubo T (2006) ABCB1 C3435T polymorphism influences methotrexate sensitivity in rheumatoid arthritis patients. Clin Exp Rheumatol 24(5):546–554
Trevino LR, Shimasaki N, Yang W, Panetta JC, Cheng C, Pei D, Chan D, Sparreboom A, Giacomini KM, Pui CH, Evans WE, Relling MV (2009) Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J Clin Oncol 27(35):5972–5978. https://doi.org/10.1200/JCO.2008.20.4156
Bleyer WA (1978) The clinical pharmacology of methotrexate: new applications of an old drug. Cancer 41(1):36–51
Sterba J, Valik D, Bajciova V, Kadlecova V, Gregorova V, Mendelova D (2005) High-dose methotrexate and/or leucovorin rescue for the treatment of children with lymphoblastic malignancies: do we really know why, when and how? Neoplasma 52(6):456–463
Evans WE, Crom WR, Abromowitch M, Dodge R, Look AT, Bowman WP, George SL, Pui CH (1986) Clinical pharmacodynamics of high-dose methotrexate in acute lymphocytic leukemia. Identification of a relation between concentration and effect. N Engl J Med 314(8):471–477. https://doi.org/10.1056/NEJM198602203140803
Suthandiram S, Gan GG, Zain SM, Bee PC, Lian LH, Chang KM, Ong TC, Mohamed Z (2014) Effect of polymorphisms within methotrexate pathway genes on methotrexate toxicity and plasma levels in adults with hematological malignancies. Pharmacogenomics 15(11):1479–1494. https://doi.org/10.2217/pgs.14.97
Radtke S, Zolk O, Renner B, Paulides M, Zimmermann M, Moricke A, Stanulla M, Schrappe M, Langer T (2013) Germline genetic variations in methotrexate candidate genes are associated with pharmacokinetics, toxicity, and outcome in childhood acute lymphoblastic leukemia. Blood 121(26):5145–5153. https://doi.org/10.1182/blood-2013-01-480335
Chen Y, Shen Z (2015) Gene polymorphisms in the folate metabolism and their association with MTX-related adverse events in the treatment of ALL. Tumour Biol 36(7):4913–4921. https://doi.org/10.1007/s13277-015-3602-0
Lopez-Lopez E, Martin-Guerrero I, Ballesteros J, Garcia-Orad A (2013) A systematic review and meta-analysis of MTHFR polymorphisms in methotrexate toxicity prediction in pediatric acute lymphoblastic leukemia. Pharmacogenomics J 13(6):498–506. https://doi.org/10.1038/tpj.2012.44
Zhang HN, He XL, Wang C, Wang Y, Chen YJ, Li JX, Niu CH, Gao P (2014) Impact of SLCO1B1 521T %3e C variant on leucovorin rescue and risk of relapse in childhood acute lymphoblastic leukemia treated with high-dose methotrexate. Pediatr Blood Cancer 61(12):2203–2207. https://doi.org/10.1002/pbc.25191
Csordas K, Lautner-Csorba O, Semsei AF, Harnos A, Hegyi M, Erdelyi DJ, Eipel OT, Szalai C, Kovacs GT (2014) Associations of novel genetic variations in the folate-related and ARID5B genes with the pharmacokinetics and toxicity of high-dose methotrexate in paediatric acute lymphoblastic leukaemia. Br J Haematol 166(3):410–420. https://doi.org/10.1111/bjh.12886
Lima A, Bernardes M, Azevedo R, Monteiro J, Sousa H, Medeiros R, Seabra V (2014) SLC19A1, SLC46A1 and SLCO1B1 polymorphisms as predictors of methotrexate-related toxicity in Portuguese rheumatoid arthritis patients. Toxicol Sci 142(1):196–209. https://doi.org/10.1093/toxsci/kfu162
Avivi I, Zuckerman T, Krivoy N, Efrati E (2014) Genetic polymorphisms predicting methotrexate blood levels and toxicity in adult non-Hodgkin lymphoma. Leuk Lymphoma 55(3):565–570. https://doi.org/10.3109/10428194.2013.789506
Lopez-Lopez E, Martin-Guerrero I, Ballesteros J, Pinan MA, Garcia-Miguel P, Navajas A, Garcia-Orad A (2011) Polymorphisms of the SLCO1B1 gene predict methotrexate-related toxicity in childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 57(4):612–619. https://doi.org/10.1002/pbc.23074
Li J, Wang XR, Zhai XW, Wang HS, Qian XW, Miao H, Zhu XH (2015) Association of SLCO1B1 gene polymorphisms with toxicity response of high dose methotrexate chemotherapy in childhood acute lymphoblastic leukemia. Int J Clin Exp Med 8(4):6109–6113
Yasuda SU, Zhang L, Huang SM (2008) The role of ethnicity in variability in response to drugs: focus on clinical pharmacology studies. Clin Pharmacol Ther 84(3):417–423. https://doi.org/10.1038/clpt.2008.141
Faganel Kotnik B, Grabnar I, Bohanec Grabar P, Dolzan V, Jazbec J (2011) Association of genetic polymorphism in the folate metabolic pathway with methotrexate pharmacokinetics and toxicity in childhood acute lymphoblastic leukaemia and malignant lymphoma. Eur J Clin Pharmacol 67(10):993–1006. https://doi.org/10.1007/s00228-011-1046-z
Kim IW, Yun HY, Choi B, Han N, Park SY, Lee ES, Oh JM (2012) ABCB1 C3435T genetic polymorphism on population pharmacokinetics of methotrexate after hematopoietic stem cell transplantation in Korean patients: a prospective analysis. Clin Ther 34(8):1816–1826. https://doi.org/10.1016/j.clinthera.2012.06.022
Lui G, Treluyer JM, Fresneau B, Piperno-Neumann S, Gaspar N, Corradini N, Gentet JC, Marec Berard P, Laurence V, Schneider P, Entz-Werle N, Pacquement H, Millot F, Taque S, Freycon C, Lervat C, Le Deley MC, Mahier Ait Oukhatar C, Brugieres L, Le Teuff G, Bouazza N, Sarcoma Group of U (2018) A pharmacokinetic and pharmacogenetic analysis of osteosarcoma patients treated with high-dose methotrexate: data from the OS2006/sarcoma-09 trial. J Clin Pharmacol 58(12):1541–1549. https://doi.org/10.1002/jcph.1252
Cheah CY, Herbert KE, O'Rourke K, Kennedy GA, George A, Fedele PL, Gilbertson M, Tan SY, Ritchie DS, Opat SS, Prince HM, Dickinson M, Burbury K, Wolf M, Januszewicz EH, Tam CS, Westerman DA, Carney DA, Harrison SJ, Seymour JF (2014) A multicentre retrospective comparison of central nervous system prophylaxis strategies among patients with high-risk diffuse large B-cell lymphoma. Br J Cancer 111(6):1072–1079. https://doi.org/10.1038/bjc.2014.405
Hoang-Xuan K, Bessell E, Bromberg J, Hottinger AF, Preusser M, Ruda R, Schlegel U, Siegal T, Soussain C, Abacioglu U, Cassoux N, Deckert M, Dirven CM, Ferreri AJ, Graus F, Henriksson R, Herrlinger U, Taphoorn M, Soffietti R, Weller M, European Association for Neuro-Oncology Task Force on Primary CNSL (2015) Diagnosis and treatment of primary CNS lymphoma in immunocompetent patients: guidelines from the European Association for Neuro-Oncology. Lancet Oncol 16(7):e322–e332. https://doi.org/10.1016/S1470-2045(15)00076-5
Woessmann W, Seidemann K, Mann G, Zimmermann M, Burkhardt B, Oschlies I, Ludwig WD, Klingebiel T, Graf N, Gruhn B, Juergens H, Niggli F, Parwaresch R, Gadner H, Riehm H, Schrappe M, Reiter A, Group BFM (2005) The impact of the methotrexate administration schedule and dose in the treatment of children and adolescents with B-cell neoplasms: a report of the BFM Group Study NHL-BFM95. Blood 105(3):948–958. https://doi.org/10.1182/blood-2004-03-0973
Moricke A, Reiter A, Zimmermann M, Gadner H, Stanulla M, Dordelmann M, Loning L, Beier R, Ludwig WD, Ratei R, Harbott J, Boos J, Mann G, Niggli F, Feldges A, Henze G, Welte K, Beck JD, Klingebiel T, Niemeyer C, Zintl F, Bode U, Urban C, Wehinger H, Niethammer D, Riehm H, Schrappe M, German-Austrian-Swiss ALLBFMSG (2008) Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111(9):4477–4489. https://doi.org/10.1182/blood-2007-09-112920
Jaccard A, Gachard N, Marin B, Rogez S, Audrain M, Suarez F, Tilly H, Morschhauser F, Thieblemont C, Ysebaert L, Devidas A, Petit B, de Leval L, Gaulard P, Feuillard J, Bordessoule D, Hermine O, Gela IG (2011) Efficacy of L-asparaginase with methotrexate and dexamethasone (AspaMetDex regimen) in patients with refractory or relapsing extranodal NK/T-cell lymphoma, a phase 2 study. Blood 117(6):1834–1839. https://doi.org/10.1182/blood-2010-09-307454
Dupuis C, Mercier C, Yang C, Monjanel-Mouterde S, Ciccolini J, Fanciullino R, Pourroy B, Deville JL, Duffaud F, Bagarry-Liegey D, Durand A, Iliadis A, Favre R (2008) High-dose methotrexate in adults with osteosarcoma: a population pharmacokinetics study and validation of a new limited sampling strategy. Anticancer Drugs 19(3):267–273
Keizer RJ, Karlsson MO, Hooker A (2013) Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacomet Syst Pharmacol 2:e50. https://doi.org/10.1038/psp.2013.24
Karlsson MO, Savic RM (2007) Diagnosing model diagnostics. Clin Pharmacol Ther 82(1):17–20. https://doi.org/10.1038/sj.clpt.6100241
Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J, Ckd EPI (2009) A new equation to estimate glomerular filtration rate. Ann Intern Med 150(9):604–612. https://doi.org/10.7326/0003-4819-150-9-200905050-00006
Yafune A, Ishiguro M (1999) Bootstrap approach for constructing confidence intervals for population pharmacokinetic parameters. I: a use of bootstrap standard error. Stat Med 18(5):581–599
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO (2011) Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J 13(2):143–151. https://doi.org/10.1208/s12248-011-9255-z
Martinez D, Muhrez K, Woillard JB, Berthelot A, Gyan E, Choquet S, Andres CR, Marquet P, Barin-Le Guellec C (2018) Endogenous metabolites-mediated communication between OAT1/OAT3 and OATP1B1 may explain the association between SLCO1B1 SNPs and methotrexate toxicity. Clin Pharmacol Ther 104(4):687–698. https://doi.org/10.1002/cpt.1008
Woillard JB, Debord J, Benz-de-Bretagne I, Saint-Marcoux F, Turlure P, Girault S, Abraham J, Choquet S, Marquet P, Barin-Le Guellec C (2017) A time-dependent model describes methotrexate elimination and supports dynamic modification of MRP2/ABCC2 activity. Ther Drug Monit 39(2):145–156. https://doi.org/10.1097/FTD.0000000000000381
Faltaos DW, Hulot JS, Urien S, Morel V, Kaloshi G, Fernandez C, Xuan KH, Leblond V, Lechat P (2006) Population pharmacokinetic study of methotrexate in patients with lymphoid malignancy. Cancer Chemother Pharmacol 58(5):626–633. https://doi.org/10.1007/s00280-006-0202-0
Mei S, Li X, Jiang X, Yu K, Lin S, Zhao Z (2018) Population pharmacokinetics of high-dose methotrexate in patients with primary central nervous system lymphoma. J Pharm Sci 107(5):1454–1460. https://doi.org/10.1016/j.xphs.2018.01.004
Zhang W, Zhang Q, Tian X, Zhao H, Lu W, Zhen J, Niu X (2015) Population pharmacokinetics of high-dose methotrexate after intravenous administration in Chinese osteosarcoma patients from a single institution. Chin Med J (Engl) 128(1):111–118. https://doi.org/10.4103/0366-6999.147829
Simon N, Marsot A, Villard E, Choquet S, Khe HX, Zahr N, Lechat P, Leblond V, Hulot JS (2013) Impact of ABCC2 polymorphisms on high-dose methotrexate pharmacokinetics in patients with lymphoid malignancy. Pharmacogenomics J 13(6):507–513. https://doi.org/10.1038/tpj.2012.37
Aumente D, Buelga DS, Lukas JC, Gomez P, Torres A, Garcia MJ (2006) Population pharmacokinetics of high-dose methotrexate in children with acute lymphoblastic leukaemia. Clin Pharmacokinet 45(12):1227–1238. https://doi.org/10.2165/00003088-200645120-00007
Colom H, Farre R, Soy D, Peraire C, Cendros JM, Pardo N, Torrent M, Domenech J, Mangues MA (2009) Population pharmacokinetics of high-dose methotrexate after intravenous administration in pediatric patients with osteosarcoma. Ther Drug Monit 31(1):76–85. https://doi.org/10.1097/FTD.0b013e3181945624
Min Y, Qiang F, Peng L, Zhu Z (2009) High dose methotrexate population pharmacokinetics and Bayesian estimation in patients with lymphoid malignancy. Biopharm Drug Dispos 30(8):437–447. https://doi.org/10.1002/bdd.678
Odoul F, Le Guellec C, Lamagnere JP, Breilh D, Saux MC, Paintaud G, Autret-Leca E (1999) Prediction of methotrexate elimination after high dose infusion in children with acute lymphoblastic leukaemia using a population pharmacokinetic approach. Fundam Clin Pharmacol 13(5):595–604
Plard C, Bressolle F, Fakhoury M, Zhang D, Yacouben K, Rieutord A, Jacqz-Aigrain E (2007) A limited sampling strategy to estimate individual pharmacokinetic parameters of methotrexate in children with acute lymphoblastic leukemia. Cancer Chemother Pharmacol 60(4):609–620. https://doi.org/10.1007/s00280-006-0394-3
Funding
This study was supported by the Fujian Provincial Health Technology Project (No: 2018-ZQN-18), the Natural Science Foundation of Fujian Province (No: 2016J01509), and the Science and Technology Program of Fuiian Province, China (No: 2018Y2003).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, L., Wu, H., de Winter, B.C.M. et al. Pharmacokinetics and pharmacogenetics of high-dose methotrexate in Chinese adult patients with non-Hodgkin lymphoma: a population analysis. Cancer Chemother Pharmacol 85, 881–897 (2020). https://doi.org/10.1007/s00280-020-04058-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-020-04058-4