
Abstract
Purpose
The objectives of this study were to characterize the population pharmacokinetics of MTX in patients with acute lymphoblastic leukemia (ALL) with ages ranging from 2 to 16 years and to propose a limited sampling strategy to estimate individual pharmacokinetic parameters.
Methods
Seventy-nine children were enrolled in this study; they received 1–4 courses of chemotherapy. MTX was administered at a dose of 5 g/m². MTX population parameters were estimated from 61 patients (231 courses; age range: 2–16 years). The data were analyzed by nonlinear mixed-effect modeling with use of a two-compartment structural model. The interoccasion variability was taken into account in the model. Eighteen additional patients (70 courses) were used to evaluate the predictive performances of the Bayesian approach and to devise a limited sampling strategy.
Results
The following population parameters were obtained: total clearance (CL) = 8.8 l/h (inter-individual variability: 43%), initial volume of distribution (V 1) = 17.3 l (48%), k 12 = 0.0225 h−1 (41%), and k 21 = 0.0629 h−1 (24%). The inter-individual variability in the initial volume of distribution was partially explained by the fact that this parameter was weight-dependent. Intercourse variability was limited, with a mean variation of 13.2%. The protocol involving two sampling times, 24 and 48 h after the beginning of infusion, allows precise and accurate determination of individual pharmacokinetic parameters and consequently, it was possible to predict the time at which the MTX concentration reached the predicted threshold (0.2 μM) below which the administration of folinic acid could be stopped.
Conclusion
The results of this study combine the relationships between the pharmacokinetic parameters of MTX and patient covariates that may be useful for dose adjustment, with a convenient sampling procedure that may aid in optimizing pediatric patient care.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute lymphoblastic leukemia (ALL) in pediatrics is a largely curable disease, with approximately 80% of children achieving 5-year survival on current intensive protocols [1–3]. The common treatment of ALL is based on induction–remission therapy, followed by intensification and maintenance therapy [2]. High dose intravenous methotrexate (HD-MTX) is an important component of many chemotherapeutic protocols [4] although the superiority of high versus low dose MTX is still a matter of debate [5, 6]. Hospitalization is required for hydration and urinary alkalinization. Following HD-MTX, variability in MTX elimination requires routine monitoring of drug concentrations as both the plasma concentration of MTX and the duration of MTX concentration above a certain threshold are important in the occurrence of toxicity [7–9]. Patients with high concentrations are at elevated risk of toxicity and should receive adjusted doses of folinic acid until the concentrations fall below the threshold, allowing the termination of the folinic acid rescue [9, 10].
Several studies on MTX pharmacokinetics in children have been performed [9, 11–24]. Conventional compartmental or noncompartmental approach [9, 11–17] and population approach [23, 24] have been used to compute individual pharmacokinetic parameters. In some studies, pharmacokinetic parameters were estimated by parametric and nonparametric methods using the software package USC*PACK [19, 20] or by using a Bayesian algorithm implemented in the software package ADAPT [21, 22]. But only few studies reported Bayesian adaptative strategies after HD-MTX infusion in patients with ALL or osteosarcoma [18, 21, 23, 24]. Dose adjustment by Bayesian forecasting [25] could play an important role in the optimization of drug dosage regimens in this vulnerable population of patients with large inter-patient variability.
The present study was carried out to provide data on HD-MTX pharmacokinetics in order to optimize care in pediatric patients. It was performed in 79 pediatric patients aged from 2 to 16 years. The purpose of this study was (1) to determine accurate population pharmacokinetic parameters of MTX by using a two-compartment open model in a population of patients covering a wide range of age; (2) to accurately estimate both inter- and intra-individual variability in pharmacokinetic parameters; (3) to estimate the inter-occasion (inter-course) variability; (4) to examine which of the patient physio-pathological parameters could have influenced drug disposition; and (5) to propose a limited sampling strategy to estimate individual pharmacokinetic parameters. The predictive performances of the Bayesian procedure were evaluated using an independent group of patients, and thus, predicted MTX concentrations were compared to measured concentrations.
Materials and methods
Patients and treatment
A total of 79 children with ALL were studied. They were enrolled at the time of diagnosis in the European Organization for Research and Treatment of Cancer (EORTC) protocol 58951 approved by an institutional review committee for clinical trials or in the French protocol 95020 approved by the Ethics committee of Paris Bichat-Claude Bernard. Written informed consent was signed by the parents of the patients. Children were treated in the department of Pediatric Immuno Hematology of the Robert Debré hospital (Paris, France) from Mars 1999 to April 2004. Nine patients were classified as being at very low risk (VLR), 60 as being at average risk (AR), and 10 as being at very high risk (VHR) on the basis of blast count, immunophenotype of leukemic cells, and hepatic and splenic dimensions.
Briefly, the EORTC protocol consisted of four phases: induction (including a prephase), consolidation, interval, and maintenance therapies. During the interval therapy, children with a low or a standard risk received four courses of HD-MTX at days 8, 22, 36, and 50 while children with a high risk only received the three first courses. MTX was administered as a 24-h infusion of 5 g/m2. One-tenth of the dose (500 mg/m²) was given over the first hour and the remaining dose (4,500 mg/m²) was given over the following 23 h at a constant rate. Urine pH must be over seven before starting MTX. Alkalinization was achieved through administration of bicarbonate 1 mEq/kg in 20–50 ml (according to age) of 5% glucose solute over 15 min. On the first day, hydratation was implemented intravenously with the following mixture: 5% glucose (2/3 of the total volume), 1.4% sodium bicarbonate (1/3 of the total volume), and potassium chloride (30 mEq/l). Leucovorin (5-formyl-tetrahydrofolic acid), 12 mg/m² (preferentially orally) was given 36 h after the start of the MTX infusion, Doses were repeated every 6 h and MTX levels were measured, according to the protocol at 48 and 72 h after the start of MTX infusion and adjusted until MTX concentrations were below 0.2 μM. Patients were required to have no biochemical signs of hepatic dysfunction, a leucocyte count ≥1.5 × 109/l and a platelet count >80 × 109/l as well as normal levels of s-carbamide and s-creatinine before the start of the MTX infusion. The following patient’s characteristics were collected: age, gender, weight, height, alanine aminotransferase (ALAT), and serum creatinine. A nomogram derived from Du Bois’s [26] formula was used to estimate body surface area (BSA) from height and weight. Creatinine clearance was estimated using Schwartz and Haycock’s [27] formula for children less than 12 years old and Cockcroft and Gault’s [28] formula for the older ones.
Sample collection and processing
Blood samples were collected from all patients with the purpose of MTX monitoring at the following times: 24 (or 36), 48, and 72 h after the beginning of the infusion. If the concentrations of MTX 72 h following the start of infusion were higher than 0.2 μM, additional blood samples were drawn (Table 1). Additional blood samples were also obtained at 26 (16 children), 28 (7 children), 30 (6 children), and 34 h (5 children) after the beginning of the infusion. These samples represent about 16% of total number of samples colleted during this study. All samples were drawn from a central venous catheter. The blood was kept cold in the dark until centrifugation at 4°C (2,000g for 15 min) and immediately analyzed in order to adjust leucovorin administration.
Measurement of MTX concentrations
The plasma MTX concentrations were measured by an enzyme multiplied immunoassay technique Emit (Dade Behring Limited, Milton Keynes, UK). Calibration concentrations ranged from 0.02 to 2 μM. Quality controls (QC, 0.33, 1.33 and 7.1 μM) were from Bio-Rad (Marnes la Coquette, France). The precision values were 13, 11, and 12%, respectively. The accuracy ranged from 90 to 110%. The lower limit of quantitation was 0.02 μM; at this concentration accuracy was 115% and precision was 19%.
Population pharmacokinetic analysis
The database consisted of one to four courses per patient. The subjects included were randomly allocated into a model building group (population group: 61 patients) and a test group (validation group: 18 patients). Potential explanatory covariables such as patients’ age, weight, gender, height, body surface area (BSA), serum creatinine, creatinine clearance, ALAT, corticoid administration (one for dexamethasone, two for prednisolone), administration of either Ca–folic acid and folinic acid (one for yes, two for no) and the risk group of ALL were included in the original data files.
Pharmacostatistical model (base model)
Pharmacokinetic analyses were performed using the nonlinear mixed-effect modeling approach as implemented in the NONMEM computer program (version 5.1) [29] through the visual-NM graphical interface [30]. The population characteristics of the pharmacokinetic parameters (fixed and random effects) were estimated using the subroutines ADVAN-4 and TRANS-1 from the library of programs provided with the NONMEM-PREDPP package. Both, first-order (FO) and first-order conditional estimation (FOCE) methods were used to estimate population pharmacokinetic parameters. The FO method was used to fit the model because it markedly decreases the objective function and the residuals between observed concentrations (DV) and individual predicted concentrations (IPRED).
A two-compartment structural model was selected based on previous knowledge of MTX pharmacokinetics [18, 20–24]. The four-dimensional vector θ of kinetic parameters considered in the population analysis consists of total clearance (CL), transfer rate constants (k 12 and k 21), and initial volume of distribution (V 1).
Deviations of CL, V 1, k 12, and k 21 of the jth individual from the estimated population average values were modeled with the use of an exponential interindividual variability (IIV) error model:
in which P j is the required pharmacokinetic parameter in the jth individual and η j is a random variable distributed with mean zero and variance of ω 2 η about the average value (P mean) in the population.
Various error models were also tested (additive, exponential, or combined additive and exponential). The smallest −2 log likelihood function value was associated to the better model. The error on the concentration measurements of the individual j was best described by a combined additive and exponential model given below:
in which P j refers to the parameter vector of the subject j; t ij is the time of the ith measurement; D j is the dosing history of the subject j; f is the pharmacokinetic model; ε 1ij and ε 2ij represent the residual departure of the model from the observations and contain contributions from intra-individual variability, assay error, and model misspecification for the dependent variable. ε 1 and ε 2 are assumed to be random Gaussian variables with mean zero and variances of \( \sigma ^{2}_{{\varepsilon _{1} }} , \) and \( \sigma ^{2}_{{\varepsilon _{2} }} . \) .
As for most of the patients, four pharmacokinetic evaluations were performed; the intercourse variability in CL and V 1 was evaluated by using the interoccasion variability (IOV) as described by Karlsson and Sheiner [31]. This model takes account of random variability in subject’s parameters between study occasions and allows one to obtain specific values of CL and V 1 for each occasion. IOV can be modeled using NONMEM at the same level of random effects as IIV by assigning a different type of η to parameter P on each occasion studied, and assuming that the variance–covariance of these ηs is \( \pi ^{2}_{P} I \) (where I is the identity matrix of dimension equal to the maximum number of occasions studied in any individual in the data set).
In the first step, the population parameters, fixed and random effects together with the individual posterior estimates were computed assuming that no dependency existed between the pharmacokinetic parameters and the covariates.
Several secondary pharmacokinetic parameters were calculated from the individual (Bayesian estimates) primary pharmacokinetic parameters: the volume of distribution at steady-state (V dss), the elimination half-life (t 1/2 elim), and the area under plasma concentration–time curves (AUC).
The uncertainty (coefficient of variation) in estimating fixed and random parameter values was determined by expressing the standard error of estimation (calculated in NONMEM) as a percentage of the estimated value.
The individual predicted plasma concentrations (C IPRED) were calculated for each individual by means of the empirical Bayes estimate of pharmacokinetic parameters using the POSTHOC option in the NONMEM program.
Covariate analysis
Following selection of the basic structural and statistical models, a preliminary assessment of covariate influence was conducted by plotting individual Bayesian pharmacokinetic estimates against all the preselected potential covariates. On the basis of these results, models were built with the use of a stepwise forward addition process followed by a backward elimination process. When a significant relationship was observed, the selected covariates were included individually in the model and tested for statistical significance. The change in the NONMEM objective function produced by the inclusion of a covariate term (asymptotically distributed as χ 2 with degrees of freedom equal to the number of parameters added to the model) was used to compare alternative models. A change in objective function of at least 3.8 (P < 0.05 with one degree of freedom) was required for statistical significance at the initial covariate screening stage. Finally, accepted covariates were added to the model and the population pharmacokinetic parameters were estimated. To demonstrate that retained covariates contributed to an improvement of the fit of the population pharmacokinetic model, each covariate was deleted sequentially from the proposed final model (backward elimination) in order to confirm statistical significance (χ 2 test). If the objective function did not vary significantly, the relationship between the covariate and the pharmacokinetic parameter was ignored.
Final model
Only the covariates providing a significant change in the objective function when introduced in the model were retained in the analysis. The final population parameters were estimated considering the relationship with the covariates.
Structural model validation
Individual pharmacokinetic parameters of the patients in the validation group (18 children not included in the calculation of population parameters) were calculated on the basis of the Bayesian approach. This approach combines the prior knowledge of mean and dispersion of pharmacokinetic parameters in the population to which the selected individual belongs and the individual samples. From the resulting individualized parameter values, t 1/2 elim, V dss, and plasma MTX concentrations at each sampling time (IPRED) were calculated for each patient.
Validation of a limited sampling strategy
Individual pharmacokinetic parameters of the 18 patients in the validation group were estimated based on Bayesian estimation using the NONMEM program from a limited number of samples [32–35]. Moreover, it is very interesting for practical purposes to limit the number of samples to one. Thus, one (each of the available plasma samples) or two samples’ (five combinations) schedules were tested. The database consisted of the data of the validation group.
Statistical analysis
Model acceptance (population group)
At each step of the model building, diagnostic plots were analyzed for closeness to and randomness along the line of identity on DV versus predicted (PRED) concentration plot, as well as randomness along the residual (DV-PRED) and weighted residual zero line on the predicted concentrations or time versus residual or weighted residual plot. Moreover, IPRED were plotted versus observed concentrations (DV) and the results were compared with the reference line of slope = 1 and intercept = 0. PRED concentrations were computed based on population parameter estimates; IPRED concentrations were computed based on individual parameter estimates. Descriptive statistics were used to compare mean residual values to 0 and to calculate 95% confidence intervals. The model was accepted when (1) plots showed no systematic pattern and (2) descriptive statistics did not show any systematic deviation from the initial hypothesis (mean supposed to be 0).
Performance of Bayesian individual parameter estimates (validation group)
The performance of Bayesian estimation was assessed in the validation group (18 patients), by comparing the observed concentrations (DV) to the ones estimated using the Bayesian approach (IPRED). Given the wide range of concentrations, the error has been defined as relative error, both bias and precision have been calculated as follows [36, 37]:
-
1.
bias or mean relative predictor error:
$$ {\text{Bias}}\,{\text{ = }}\,\frac{{\text{1}}} {N}{\sum\limits_{i\,{\text{ = }}\,{\text{1}}}^{i\,{\text{ = }}\,N} {{\text{[DV - IPRED]/DV}}} } $$ -
2.
precision or root mean relative square error:
$$ {\text{Precision = }}{\sqrt {\frac{1} {N}{\sum\limits_{i = 1}^{i = N} {[{\text{DV - IPRED/DV}}]^{2} } }} } $$
In these expressions the index i refers to the concentration number and N is the sample size. The 95% confidence interval for bias was computed and the t test was used to compare the bias to 0.
Computing of a limited sampling strategy (validation group)
To evaluate the reliability of the parameter estimates using a limited sampling strategy (LSS), for each combination, pharmacokinetic parameters (only CL, V 1, and t 1/2elim were considered for this purpose due to their clinical interest) estimated using a Bayesian methodology and a limited sampling strategy were compared to the ones estimated using a Bayesian methodology and the entire set of data. These comparisons were performed by computing the bias and the precision.
Results
Patients characteristics
Seventy-nine children (46 boys and 33 girls) received repeated high dose MTX according to the protocol. The baseline demographic characteristics of the patients enrolled in this study are summarized in Table 2 and the age distribution frequency is presented in Fig. 1. Nine patients were classified as being VLR, 60 as being AR and 10 as being VHR. Except for ALAT, there was no statistically significant difference (t test) in the demographic and biological data between the patients included in the population group and in the validation group, Table 2.

Age frequency of distribution in the studied population
Therapeutic drug monitoring of MTX
For most of the patients (83.5%), four pharmacokinetic evaluations were carried out; 15% of patients had three pharmacokinetic evaluations and one patient had only one pharmacokinetic evaluation. So, a total of 301 pharmacokinetic data from 79 patients were analyzed. The therapeutic drug monitoring data evidenced a wide interpatient variability in MTX concentrations at each sampling time (Fig. 2). Based on our data, 264 pharmacokinetic evaluations (87.7%) obtained during MTX monitoring followed the exact sampling protocol with MTX concentrations measured at 24 (or 36), 48, and 72 h post-infusion, with additional samples if required. Results are presented in Table 1. Among the 296 and 294 MTX concentrations available at H48 and H72 h, 5 and 165 concentrations were ≤0.2 μM.

MTX concentrations from 301 courses in 79 children. Concentrations below the LLOQ are not presented
Population modeling
Base model
The population database consisted of 943 MTX concentrations (231 courses) from 61 children. The basic pharmacokinetic parameters (before inclusion of covariates, step 1) are shown in Table 3.
Covariate inclusion
In step 2, there were significant relationships between V 1 and weight (r = 0.57, P < 0.0001), V 1 and BSA (r = 0.40, P = 0.002), V 1 and age (r = 0.30, P = 0.0188), CL and age (r = 0.40, P = 0.0019), CL and weight (r = 0.47, P = 0.0002), and CL and BSA (r = 0.49, P = 0.0001). No differences in estimated parameters were found between boys and girls. The other covariates did not influence pharmacokinetic parameters. In the model-building phase, after inclusion of each covariate in the model, only the relationships between V 1 and weight (V 1 = a × weightb [38]) and between CL and age and BSA [CL = a × age (or BSA) + b] decreased significantly the objective function by 3.8 or more when tested against the baseline model. At this stage, weight appeared to be the most important of these factors. In a forward modeling building step, the cumulative inclusion of weight, age, and BSA reduced the objective function value at each addition. Finally, in the backward elimination step, age and BSA did not modify significantly the objective function when they were omitted individually from the model. Thus, only the relationship between V 1 and weight was retained in the final model. The inclusion of this second stage model significantly improved both the relationship between model-predicted and observed concentrations (the objective function decreased from the baseline model value of 998.8–990.6) and the plot of weighted residuals versus model-predicted concentrations. Moreover, the inclusion of this covariate in the model substantially reduced interindividual variability in V 1 (from 63.1 to 51.6%) and residual error as compared to the baseline model.
Final model
Population pharmacokinetic parameters are presented in Table 3. Intercourse variability was limited, with a mean variation of 13.2%. From the individual (Bayesian estimates) primary pharmacokinetic parameters, the following secondary pharmacokinetic parameters were calculated: the elimination half-life (t 1/2 elim = 11.9 h; interindividual variability: 17%), the volume of distribution at steady-state (V dss = 155 l; interindividual variability: 41.2%) and the area under plasma concentration versus time curve (AUC normalized to a total administered dose of 11 mM = 1,316 μM × h; interindividual variability: 49%).
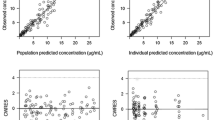
Plots of model-predicted versus observed concentrations obtained from the final model based on individual and population parameter estimates are shown in Fig. 3a and b. Various statistical tests were carried out which showed that (1) there was no significant difference when the regression line of individual predicted concentrations versus observed concentrations (slope = 0.9997, SE = 0.0082; intercept = −0.19 μM, SE = 0.096) was compared to the reference line (slope = 1 and intercept = 0); and (2) the frequency of distribution histogram of the normalized residuals was as expected (normal with zero mean and unitary variance). The vast majority of the weighted residuals lay within two units of perfect agreement and were symmetrically distributed around the zero ordinate (Fig. 3c). The mean relative error was low, −0.0654%; however, a small bias was observed, the 95% confidence interval did not included the zero value (−0.87, −0.02%). Such bias was attributable to five IPRED concentrations (i.e., 0.5% of MTX concentrations) found 2.5-times higher than observed MTX concentrations that are close to the LLOQ of the analytical method (0.02–0.04 μM,). By removing these five values, no bias occurred.

Model performance and diagnostic plots (n = 61 patients; 231 courses, 943 MTX concentrations). a Model-predicted versus observed concentrations obtained for the final model based on estimated individual parameters. b Model-predicted versus observed concentrations obtained with the final model based on population parameter estimates. c Weighted residuals versus predicted concentrations. Dotted line represents the line of identity
Evaluation of the Bayesian pharmacokinetic parameter prediction
Individual pharmacokinetic parameters for the 18 patients in the validation group (398 MTX concentrations, 70 courses) were determined using the population characteristics and all the available concentration–time points. Mean pharmacokinetic parameters were CL = 9.4 l/h (CV = 25.5%), V 1 = 27.7 l (CV = 23.7%), k 12 = 0.0200 h−1 (CV = 27.7%), and k 21 = 0.0608 h−1 (CV = 14.4%). The regression line of empirical Bayes predicted values and individual observed MTX concentrations did not differ significantly from the reference line of slope = 1 and intercept = 0. The bias (−0.0349%) was not statistically different from zero (t test) and the 95% confidence interval included the zero value (−0.07/1.67 × 10−4). The precision was equal to 0.358% (95% confidence interval, 0.307/0.41). The corresponding predicted curves are shown in Fig. 4 for two representative patients.

Concentration time curves for MTX in two representative children after four courses. a Age: 2 years; weight: 12 kg. b Age: 10 years; weight: 30.4 kg. Individual predicted concentrations (filled symbols); observed concentrations (open symbols). Lines are obtained from individual predicted values (IPRED) connected point by point
Validation of a limited-sampling strategy
Validation consisted of comparing CL, V 1 and t ½ elim determined by Bayesian estimation and all data concentrations (considered as the reference), to CL, V 1, and t ½ elim determined by Bayesian estimation and limited-sampling strategies. Bias and precision values corresponding to the best of each schedule (i.e., one- and two-sample schedules) are given in Table 4. The two-sample schedule (end of infusion and 48 h after the end of infusion) combines accurate prediction and convenience. The comparison between observed and individual predicted concentrations was performed by computing the mean relative error. Bias was equal to −0.042% (95% confidence interval, 0.084/0.0012) and precision was 0.258 (95% confidence interval, 0.21/0.30). When only one sample (48 h after the end of infusion) was considered, CL, V 1 and t ½ elim were correctly estimated but the interindividual variability of these parameters decreased. Figure 5 compares CL determined by considering all data concentrations and that obtained from these two samples.

Comparison of CL determined by considering all data concentrations (reference CL) and that obtained from two samples: 24 and 48 h (estimated CL)
Population pharmacokinetic parameters from all chemotherapy courses (79 patients)
In the final step, population pharmacokinetic parameters of MTX were determined from all patients (n = 79, 301 courses). The calculated population parameters (Table 3) were similar to those calculated from the patients in the population group. From the individual (Bayesian estimates) primary pharmacokinetic parameters, the following secondary pharmacokinetic parameters were calculated: the elimination half-life (t 1/2 elim = 12.2 h; interindividual variability: 15%), the volume of distribution at steady-state (V dss = 159 l; interindividual variability: 39.4%), and the area under plasma concentration versus time curve (AUC normalized to a total administered dose of 11 mM = 1,295 μM × h; interindividual variability: 46%).
Discussion
The association of the antifolate MTX and LV prevents MTX toxicity, including major neurotoxicity, without reducing the antitumor activity of the drug. As adequate MTX concentrations have been shown to be critical to avoid a risk of toxicity, routine MTX monitoring after HD-MTX infusions is performed to identify the children who require adjusted LV, associated with prolonged hospitalization. The current monitoring of MTX concentrations requires a follow-up period up to 96 or even 108 h after the end of infusion with numerous samplings. However, for both ethical and practical reasons [39], it is essential to select a strategy that allows to reduce the number of samples and the time spent in the hospital for sampling. Bayesian estimation of individual pharmacokinetic parameters during MTX monitoring would be helpful in predicting the concentration–time curve of this drug in each patient. Thus, the present study was undertaken to estimate population pharmacokinetic parameters of MTX in children with ALL. In addition to the population approach used for analyzing the MTX data, the originality of the present work consists in the careful evaluation of the intercourse pharmacokinetic variability. Moreover, a limited sampling strategy was developed for determining individual pharmacokinetic parameters and plasma MTX concentrations. In this study, MTX population characteristics were estimated using NONMEM. This population modeling method described MTX data well.
Population pharmacokinetic analyses after HD-MTX infusion in children have been previously published [18–24]. In theses studies, MTX was administered as 4–6 h [19, 20, 22, 23] in patients with osteosarcoma or as 24-h infusion [18, 21, 24] in patients with ALL. In the study published by Evans et al. [18] and Wall et al. [21], infusion rates were adjusted at 8 h based on clearance estimates using the 1 and 6 h plasma concentrations using a Bayesian algorithm as implemented in ADAPTII. In the study published by Odoul et al. [24], 23 children (9 months to 15 years) with ALL received HD-MTX administered as 24-h infusion. The authors reported that a sampling schedule involving only one sample at H48 and Bayesian parameter estimation allowed to predict the delay required to reach 0.2 μM. In this study, patients received 1–4 courses of chemotherapy and each course was considered to be an independent administration to a different subject. Such an approach did not allow accurate estimations of either inter- and intraindividual variability in pharmacokinetic parameters. In a recent study performed in 70 patients (4.5–23 years) with osteosarcoma receiving MTX administered as 6-h infusion, Rousseau et al. [23] reported that two samples (6 and 24 h) after the beginning of the infusion allowed an accurate estimation of individual pharmacokinetic parameters.
Both nonlinear mixed effects modeling (NONMEM) [18, 21, 22, 24] and nonparametric expectation maximization (NPEM) [19, 20, 23] methods have been used for population pharmacokinetic modeling. But, to date, there are only a few studies comparing these methods in the same age group. The study conducted by Patoux et al. [40] was carried out after carboplatin administration. The authors concluded that there are no differences in bias or precision between both methods. While, de Hoog et al. [41] after administration of tobramycin in neonates showed that the NONMEM model gave a significant smaller bias; but there was no significant difference in precision.
In the present study, the population data modeling was performed using a NONMEM method. The computed population pharmacokinetic parameters (k el = 0.36 h−1, CV = 37.5%; V 1 = 26.8 l, CV = 48.1%; k 12 = 0.0225 h−1, CV = 40.7%; k 21 = 0.063 h−1, CV = 24.3%, and CL = 8.8 l/h, CV = 42.8%) were close to those reported by Rousseau et al. [23] (k el = 0.41 h−1, CV = 42.3%; V 1 = 18.2 l, CV = 54.1%; k 12 = 0.0168 h−1, CV = 68.7%; k 21 = 0.107 h−1, CV = 61.3%, and CL = 7.1 l/h, CV = 45.1%). However, the elimination half-life calculated from these parameters was lower in the study of Rousseau et al. [23], 7 h than in our study, 12.2 h. Our analysis of the plasma pharmacokinetics of MTX confirms the large interpatient variations in pharmacokinetic parameters, as previously reported by Odoul et al. [24] and Rousseau et al. [23] We observed a ratio of almost six in CL between the extreme values (4.2 and 21 l/h/m²). However, variability between courses was limited, lower than 14%.
In the last step of the population analysis, only the relationship between V 1 and weight was retained in the final model. Other authors have previously reported relationships between MTX clearance and the markers of renal function and age [11, 12, 42]. The results of the covariates analysis performed in the present study show a weak relationship between CL or CL/BSA and age. Children less than 4 years have a higher clearance than in the older ones (12.7 ± 4.7 vs. 10.9 ± 3.7 l/h/m²). However, this relationship was not retained in the final model. As previously reported, no differences in estimated parameters were found between boys and girls [11, 12, 24].
The validity of population parameters estimated (population group) was confirmed with an independent group of patients (validation group). Moreover, in this paper, from the data of patients included in the validation group, different sampling strategies were investigated for the estimation of MTX CL, V 1, and t ½ elim, among them that published by Odoul et al. [24]. These pharmacokinetic parameters, particularly CL and t 1/2 elim, were selected due to their importance in the occurrence of toxicity. For determining these three parameters in clinical routine with minimal constraints for patients, we propose a limited sampling strategy based on Bayesian estimation using the NONMEM program. The two-sample schedules based on times 24 and 48 h after the beginning of infusion combines good precision and convenience. For both ethical and practical reasons it is essential to reduce the number of samples and the time spent in the hospital for sampling. However, by using only one sample at 48 h following the beginning of infusion, the estimations of CL, V 1, and t ½ elim were not biased but the interindividual variability in these parameters decreased, particularly the interindividual variability on V 1 and t ½ elim (Table 4). Indeed, these two parameters were overestimated for low V 1 and t ½ elim values and underestimated for high V 1 and t ½ elim values. The decrease of interindividual variability is due to lack of information about the parameters, resulting in a shift toward the population mean as a result of the Bayesian principle.
The high quality (as indicated by the small residual variability and error of estimate in percent obtained by NONMEM analysis) of the pharmacokinetic data collected during this trial shows the feasibility of MTX drug monitoring at a large scale. Using two sampling times (24 and 48 h), it was possible to predict the time at which the MTX concentration reached a predicted threshold below which the administration of folinic acid could be stopped (i.e., 0.2 μM). According to our data, 93.7 and 87.3% of patients have MTX concentrations higher than the threshold of 0.2 μM, 48 and 72 h after the end of the infusion, respectively. Therefore, more than half of the HDMTX courses will benefit for the LSS model, allowing hospitalization in the corresponding patients to be reduced by 24 h or more. An additional retrospective study, based on the proposed LSS will be performed to further validate this model before introducing it in the strategy of MTX monitoring for children with ALL.
References
Pui CH, Sandlund JT, Pei D, Rivera GK, Howard SC, Ribeiro RC, Rubnitz JE, Razzouk BI, Hudson MM, Cheng C, Raimondi SC, Behm FG, Downing JR, Relling MV, Evans WE (2003) Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA 290:2001–2007
Pui CH, Reilling MV, Downing JR (2004) Acute lymphoblastic leukemia. N Engl J Med 350:1535–1548
Entz-Werle N, Suciu S, van der Werff ten Bosch J, Vilmer E, Bertrand Y, Benoit Y, Margueritte G, Plouvier E, Boutard P, Vandecruys E, Ferster A, Lutz P, Uyttebroeck A, Hoyoux C, Thyss A, Rialland X, Norton L, Pages MP, Philippe N, Otten J, Behar C, EORTC Children Leukemia Group (2005) Results of 58872 and 58921 trials in acute myeloblastic leukemia and relative value of chemotherapy vs allergenic bone marrow transplantation in first complete remission: the EORTC Children Leukemia Group report. Leukemia 19:2072–2081
Moe PJ, Holen A (2000) High-dose methotrexate in childhood ALL. Paediatr Hematol Oncol 17:615–622
Cohen IJ (2004) Defining the appropriate dosage of folinic acid after high-dose methotrexate for childhood acute lymphatic leukemia that will prevent neurotoxicity without rescuing malignant cells in the central nervous system. J Pediatr Hematol Oncol 26:156–163
Mantadakis E, Cole PD, Kamen BA (2005) High-dose methotrexate in acute lymphoblastic leukemia: where is the evidence for its continued use? Pharmacotherapy 25:748–755
Bleyer WA (1977) Methotrexate: clinical pharmacology. Current status and therapeutic guidelines. Cancer Treat Rev 4:87–101
Sirotnak FM, Moccio DM (1980) Pharmacokinetic basis for differences in methotrexate sensitivity of normal proliferative tissues in the mouse. Cancer Res 40:1230–1234
Relling MV, Fairclough D, Ayers D, Crom WR, Rodman JH, Pui CH, Evans WE (1994) Patient characteristics associated with high-risk methotrexate concentrations and toxicity. J Clin Oncol 12:1667–1672
Treon SP, Chabner BA (1996) Concepts in use of high-dose methotrexate. Clin Chem 42:1322–1329
Donelli MG, Zuccheti M, Robatto A, Perlangeli V, D’Incalci M, Masera G, Rossi MR (1995) Pharmacokinetics of HD-MTX in infants, children and adolescents with non-B acute lymphoblastic leukaemia. Med Pediatr Oncol 24:154–159
Crom WR, Glynn AM, Abromowitch M, Pui CH, Dodge R, Evans WE (1986) Use of the automatic interaction detector method to identify patient characteristics related to methotrexate clearance. Clin Pharmacol Ther 39:592–597
Rodman JH, Sunderland M, Kavanagh RL, Ochs J, Yalowich J, Evans WE, Rivera GK (1990) Pharmacokinetics of contineuous infusion of methotrexate and teniposide in paediatric cancer patients. Cancer Res 50:4267–4271
Garre ML, Relling MV, Kalwinski D, Dodge R, Crom WR, Abromowitch M, Pui CH, Evans WE (1987) Pharmacokinetics and toxicity of methotrexate in children with Down syndrome and acute lymphocytic leukaemia. J Pediatr 111:606–612
Borsi JD, Moe PJ (1987) A comparative study of t he pharmacokinetic of methotrexate in a dose range of 0.5 g to 33.6 g/m² in children with acute lymphoblastic leukaemia. Cancer 60:5–13
Lawrence JR, Steele WH, Stuart JF, McNeill CA, McVie JG, Whiting B (1980) Dose dependent methotrexate elimination following bolus intravenous injection. Eur J Clin Pharmacol 17:371–374
Rask C, Albertioni F, Bentzen SM, Schroeder H, Peterson C (1998) Clinical and pharmacokinetic risk factors for high-dose methotrexate-induced toxicity in children with acute lymphoblastic leukaemia. Acta Oncol 37:277–284
Evans WE, Relling MV, Rodman JH, Crom WR, Boyett JM, Pui CH (1998) Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med 338:499–505
Aquerreta I, Aldaz A, Giraldez J, Sierrasesumaga L (2002) Pharmacodynamics of high-dose methotrexate in pediatric patients. Oncology 36:1344–1349
Aquerreta I, Aldaz A, Giraldez J, Sierrasesumaga L (2004) Methotrexate pharmacokinetics and survival in osteosarcoma. Pediatr Blood Cancer 42:52–58
Wall AM, Gajjar A, Link A, Mahmoud H, Pui CH, Relling MV (2000) Individualized methotrexate dosing in children with relapsed acute lymphoblastic leukemia. Leukemia 14:221–225
Crews KR, Liu T, Rodriguez-Galindo C, Tan M, Meyer WH, Panetta JC, Link MP, Daw NC (2004) High-dose methotrexate pharmacokinetics and outcome of children and young adults with osteosarcoma. Cancer 100:1724–1733
Rousseau A, Sabot C, Delepine N, Delepine G, Debord J, Lachatre G, Marquet P (2002) Baysian estimation of methotrexate pharmacokinetic parameters and area under the curve in children and young adults with localised osteosarcoma. Clin Pharmacokinet 41:1095–1104
Odoul F, Le Guellec C, Lamagnere JP, Breilh D, Saux MC, Paintaud G, Autret-Leca E (1999) Prediction of MTX elimination after high dose infusion in children with acute lymphoblastic leukaemia using a population pharmacokinetic approach. Fundam Clin Pharmacol 13:595–604
Vinks AA (2002) The application of population pharmacokinetic modeling to individualized antibiotic therapy. Int J Antimicrob Agents 19:313–322
Du Bois D, Du Bois EF (1989) A formula to estimate the approximate surface area if height and weight be known. 1916. Nutrition 5:303–311
Schwartz GJ, Haycock GB, Edelmann CM Jr, Spitzer A (1976) A simple estimate of glomerular filtration rate in children derived from body length and plasma creatinine. Pediatrics 58:259–263
Cockcroft DW, Gault MH (1976) Prediction of creatinine clearance from serum creatinine. Nephron 16:31–41
Beal SL, Sheiner LB (1994) NONMEM user’s guide, University of California at San Francisco, San Francisco, USA
Visual-NM program (1988) Visual-NM user’s manual, version 5.1. Research Development Population Pharmacokinetics, Montpellier, France
Karlsson MO, Scheiner LB (1993) The importance of modelling interoccasion variability in population pharmacokinetic analyses. J Pharmacokinet Biopharm 21:735–750
Al-Banna MK, Kelman AW, Whitng B (1990) Experimental design and efficient parameter estimation in population pharmacokinetics. J Pharmacokinet Biopharm 18:347–360
Drusano GL (1991) Optimal sampling theory and population modelling: application to determination of the influence of the microgravity environment on drug distribution and elimination. J Clin Pharmacol 31:962–967
Endrenyi L (1981) Design of experiments for estimating enzyme and pharmacokinetic experiments. In: Endrenyi L (eds) Kinetic data analysis, design and analysis of enzyme and pharmacokinetic experiments. Plenum Press, New York, 137–167
Hurst AK, Yoshinaga MA, Mitani GH, Foo KA, Jelliffe RW, Harrison EC (1990) Application of a Bayesian method to monitor and adjust vancomycin dosage regimens. Antimicrob Agents Chemother 34:1165–1171
Sheiner LB, Beal SL (1981) Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm 9:503–512
Sheiner LB, Beal SL (1981) Evaluation of methods for estimating population pharmacokinetic parameters. II. Biexponential model and experimental pharmacokinetic data. J Pharmacokinet Biopharm 9:635–651
Meibohm B, Läer S, Panetta JC, Barrett J (2005) Population pharmacokinetic studies in pediatrics: issues in design and analysis. AAPS J 7:E475–E487
Note for guidance on clinical investigation of medicinal products in the paediatric population (CPMP/ICH/2711/99). http://www.emea.eu.int/pdfs/human/ich/271199EN.pdf
Patoux A, Bleyzac N, Boddy AV, Doz F, Rubie H, Bastian G, Maire P, Canal P, Chatelut E (2001) Comparison of nonlinear mixed-effect and non-parametric expectation maximisation modelling for Bayesian estimation of carboplatin clearance in children. Eur J Clin Pharmacol 57:297–303
de Hoog M, Schoemaker RC, van den Anker JN, Vinks A (2002) NONMEM and NPEM2 population modeling: a comparison using tobramycin data in neonates. Ther Drug Monit 24:359–365
Skarby T, Jonsson P, Hjorth L, Behrentz M, Bjork O, Forestier E, Jarfelt M, Lonnerholm G, Hoglund P (2003) High-dose methotrexate: on the relationship of methotrexate elimination time vs renal function and serum methotrexate levels in 1164 courses in 264 Swedish children with acute lymphoblastic leukaemia (ALL). Cancer Chemother Pharmacol 51:311–320
Acknowledgments
To the children and their parents, to Maria-Claude Linus-Sorbet for secretary assistance in preparing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00280-007-0550-4
Rights and permissions
About this article
Cite this article
Piard, C., Bressolle, F., Fakhoury, M. et al. A limited sampling strategy to estimate individual pharmacokinetic parameters of methotrexate in children with acute lymphoblastic leukemia. Cancer Chemother Pharmacol 60, 609–620 (2007). https://doi.org/10.1007/s00280-006-0394-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-006-0394-3