
Abstract
Purpose
Radiolabeled cyclic RGD (Arg-Gly-Asp) peptides have great potential for the early tumor detection and noninvasive monitoring of tumor metastasis and therapeutic response. 18F-labeled RGD analogs ([18F]-AH111585 and [18F]Galacto-RGD) have been investigated in clinical trials for positron emission tomography (PET) imaging of integrin expression in cancer patients. To develop new RGD radiotracers with higher tumor accumulation, improved in vivo kinetics, easy availability and low cost, we developed two new RGD peptides and labeled them with generator-eluted 68Ga (t1/2 = 68 min) for PET imaging of integrin αvβ3 expression in tumor xenograft models.
Materials and methods
The two new cyclic RGD dimers, E[PEG4-c(RGDfK)]2 (P4-RGD2, PEG4 = 15-amino-4,7,10,13-tetraoxapentadecanoic acid) and E[Gly3-c(RGDfK)]2 (G3-RGD2, G3 = Gly-Gly-Gly) were designed, synthesized and conjugated with 1,4,7-triazacyclononanetriacetic acid (NOTA) for 68Ga labeling. The microPET imaging and biodistribution of the 68Ga labeled RGD tracers were investigated in integrin αvβ3-positive tumor xenografts.
Results
The new RGD dimers with the Gly3 and PEG4 linkers showed higher integrin αvβ3 binding affinity than no-linker RGD dimer (RGD2). NOTA-G3-RGD2 and NOTA-P4-RGD2 could be labeled with 68Ga within 30 min with higher purity (>98%) and specific activity (8.88–11.84 MBq/nmol). Both 68Ga-NOTA-P4-RGD2 and 68Ga-NOTA-G3-RGD2 exhibited significantly higher tumor uptake and tumor-to-normal tissue ratios than 68Ga-NOTA-RGD2.
Conclusion
Because of their high affinity, high specificity and excellent pharmacokinetic properties, further investigation of the two novel RGD dimers for clinical PET imaging of integrin αvβ3 expression in cancer patients is warranted.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tumor angiogenesis, the sprouting of new blood vessels from preexisting vasculature, is well recognized as an essential mechanism for tumor growth and development of metastasis [1–3]. Without the formation of neovasculature to provide oxygen and nutrients, tumors cannot grow beyond about 1–2 mm in size [4, 5]. Once vascularized, previously dormant tumors begin to grow rapidly, invade surrounding tissues (invasion), and transfer to distant sites in the body (metastasis). The angiogenic process depends on vascular endothelial cell migration and invasion, and is regulated by cell adhesion receptors. Integrins are part of a family of heterodimeric transmembrane receptors involved in multiple steps of angiogenesis and metastasis. The function of integrins during tumor angiogenesis has been studied most extensively for integrin αvβ3, which is highly expressed on activated endothelial cells and some tumor cells but not on quiescent vessels and normal cells [6]. The ability to noninvasively visualize and quantify the expression level of integrin αvβ3 during tumor growth and metastasis as well as during antiangiogenesis treatment would provide new opportunities to develop individualized therapeutic approaches, select appropriate patients entering clinical trials for antiintegrin therapy, and establish optimized dosages and dose intervals for effective treatment [7–12].
Over the last decade, we and others have developed a series of Arg-Gly-Asp (RGD) radiotracers for imaging tumor integrin expression by positron emission tomography (PET) or single photon emission tomography (SPECT) [13–20]. Among the RGD radiotracers, [18F]-AH111585 and [18F]Galacto-RGD are under clinical investigation for noninvasive visualization of integrin expression in cancer patients [19, 21–23]. The 18F-labeled RGD monomer analogs can specifically bind to integrin αvβ3. However, the relatively low tumor uptake, high cost and lack of preparation modules for the 18F-labeled monomeric cyclic RGD peptides present significant challenges for their widespread clinical application.
The recent introduction of 68Ga PET imaging into clinical practice represents a landmark in the ongoing developments in functional and metabolic imaging that is independent of the availability of a cyclotron [24]. 68Ga is a positron emitter with a short half-life of 68 min, which is suitable for the pharmacokinetics of many peptides and other small molecules owing to its fast blood clearance, quick diffusion and target localization. 68Ga has suitable physical properties with a high positron yield reaching 89% of all disintegrations, and is available from an in-house 68Ge/68Ga generator (68Ge, t1/2 270.8 days) [25, 26]. In addition, the quick kinetics of 68Ga with macrocyclic chelators such as 1,4,7-triazacyclononanetriacetic acid (NOTA) makes possible kit formulation and extensive use of the corresponding imaging tracers in preclinical and clinical applications.
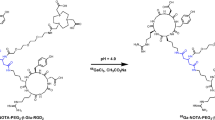
We have previously conjugated three cyclic RGD peptides, c(RGDyK) (RGD1), E[c(RGDyK)]2 (RGD2), and E{E[c(RGDyK)]2}2 (RGD4) with NOTA, which were then labeled with 68Ga for imaging integrin expression in a U87MG glioblastoma xenograft model [15]. Of the three PET tracers, the RGD monomer showed a good tumor/background ratio but rather low absolute tumor uptake, whereas the RGD tetramer had the highest tumor uptake but also high background signal especially in the kidneys. The RGD dimer, on the other hand, had higher tumor uptake than the monomer and lower background signal than the tetramer, making it a promising lead compound for further investigation. In this study we developed two new cyclic RGD dimers: E[Gly3-c(RGDfK)]2 (Gly3 = Gly-Gly-Gly) and E[PEG4-c(RGDfK)]2 (PEG4 = 15-amino-4,7,10,13-tetraoxapentadecanoic acid) [27, 28] (Fig. 1). Our goal was to investigate whether the tether linking the two RGD peptide units would affect the receptor binding affinity and pharmacokinetics of the resulting RGD dimers.

Chemical structures of NOTA-E[c(RGDfK)]2, NOTA-E[Gly3-c(RGDfK)]2 and NOTA-E[PEG4-c(RGDfK)]2
Materials and methods
General
All commercially obtained chemicals were of analytical grade and used without further purification. p-SCN-Bn-NOTA was purchased from Macrocyclics (Dallas, TX). RGD dimers: E[c(RGDfK)]2 (RGD2), E[Gly3-c(RGDfK)]2 (G3-RGD2) and E[PEG4-c(RGDfK)]2 (P4-RGD2), were custom-made by Peptides International (Louisville, KY). 68Ga was obtained from a 68Ge/68Ga generator (Obninsk, Russia) eluted with 0.1 N HCl. The reversed-phase high-performance liquid chromatography (HPLC) system was the same as that previously reported [18]. For NOTA peptide purification, a Vydac protein and peptide column (218TP510; 5 μm, 250 × 10 mm) was used with a flow rate of 5 ml/min. For analytical HPLC and radiolabeling purification, a Vydac 218TP54 column (5 μm, 250 × 4.6 mm) was used with a flow rate of 1 ml/min. The mobile phase was changed from 95% solvent A (0.1% trifluoroacetic acid in water) and 5% solvent B (0.1% trifluoroacetic acid in acetonitrile) (0–2 min) to 35% solvent A and 65% solvent B at 32 min. The UV absorbance was monitored at 218 nm, and the identification of the peptides was confirmed based on the UV spectrum acquired using a PDA detector. The radioactivity was detected using a model 105S single-channel radiation detector (Carroll and Ramsey Associates).
Synthesis of NOTA-conjugated RGD dimers
NOTA-RGD dimer conjugates were prepared as we have previously described [15]. In brief, a solution of 2 μmol of RGD dimer (RGD2, P4-RGD2, or G3-RGD2) was mixed with 6 μmol of p-SCN-Bn-NOTA in 0.1 N NaHCO3 solution (pH 9.0). After stirring at room temperature for 5 h, the NOTA-conjugated RGD peptides were isolated by semipreparative HPLC. The collected fractions were combined and lyophilized to yield the final product as a white powder. NOTA-E[c(RGDfK)]2 (NOTA-RGD2) was obtained in 48% yield with >95% HPLC purity. Matrix-assisted laser desorption/ionization (MALDI) time-of-light (TOF) mass spectrometry (MS): m/z 1,770.50 for [MH]+ (C79H113N23O22S, calculated molecular weight 1,768.82 Da). NOTA-E[PEG4-c(RGDfK)]2 (NOTA-P4-RGD2) was obtained in 42% yield with >95% HPLC purity. MALDI-TOF-MS was m/z 2,265.80 for [MH]+ (C101H155N25O32S, calculated molecular weight 2,263.52 Da). NOTA-E[Gly3-c(RGDyK)]2 (NOTA-G3-RGD2) was obtained in 43% yield with >95% purity. MALDI-TOF-MS was m/z 2,112.97 for [MH]+ (C91H131N29O28S, calculated molecular weight 2,110.95 Da). The retention times (Rt) of NOTA-RGD2, NOTA-P4-RGD2 and NOTA-G3-RGD2 on analytical HPLC were 17.7 min, 17.0 min, and 18.7 min, respectively.
68Ga-Labeling
NOTA-RGD dimer (10 nmol; NOTA-RGD2, NOTA-G3-RGD2, or NOTA-P4-RGD2) was dissolved in 500 μl of 0.1 M NaOAc buffer and incubated with 148 MBq (4 mCi) of 68Ga for 10 min at 42°C. 68Ga-labeled peptides were then purified by analytical HPLC. The radioactive peak containing the desired product was collected and rotary evaporated to remove the solvent. The products were then formulated in phosphate-buffered saline (PBS), and passed through a 0.22-μm Millipore filter into a sterile multidose vial for in vivo experiments. The labeling was done with 92% decay-corrected yield for NOTA-RGD2 (Rt 17.4 min), 94% for NOTA-P4-RGD2 (Rt 16.8 min), and 92% for NOTA-G3-RGD2 (Rt 18.2 min).
Cell lines and animal model
The U87MG human glioma cell line and MDA-MB-435 human breast cancer cell line were purchased from American Type Culture Collection. U87MG cells were cultured in Dulbecco’s medium (Gibco, Carlsbad, CA). MDA-MB-435 breast cancer carcinoma cells were cultured in Leibovitz’s L15 medium (Gibco). All cells were grown in medium supplemented with 10% (v/v) fetal bovine serum (Invitrogen) at 37°C in an atmosphere containing 5% CO2. Animal procedures were performed according to a protocol approved by the Stanford University Institutional Animal Care and Use Committee. The U87MG tumor model was generated by subcutaneous injection of 5 × 106 cells into the right front flank of female athymic nude mice (Harlan). The MDA-MB-435 tumor model was established by orthotopic injections of 5 × 106 cells into the left mammary fat pad of female athymic nude mice. The mice were subjected to microPET and biodistribution studies when the tumor volume had reached 100–300 mm3 (3–4 weeks after inoculation for U87MG, and 2–3 weeks for MDA-MB-435).
Cell integrin-receptor binding assay
The in vitro integrin binding affinity and specificity of cyclic RGD dimers (RGD2, G3-RGD2, and P4-RGD2) and their NOTA conjugates (NOTA-RGD2, NOTA-G3-RGD2, and NOTA-P4-RGD2) were assessed via a cellular displacement assay using 125I-echistatin (Perkin-Elmer) as the integrin-specific radioligand. Experiments were performed on U87MG glioma cells using a slight modification of a method previously described [16]. Briefly, 105 U87MG cells seeded in multiscreen DV plates were incubated with 125I-echistatin (about 30,000 cpm) in the presence of increasing concentrations of RGD dimers or their NOTA conjugates for 2 h at room temperature. After washing with PBS, hydrophilic PVDF filters were collected and the radioactivity was determined using a gamma counter (Packard, Meriden, CT). The IC50 values were calculated by fitting the data by nonlinear regression using GraphPad Prism (GraphPad Software, San Diego, CA), and reported as the averages of triplicate samples plus the standard deviation.
MicroPET imaging
PET scans and image analysis were performed using a microPET R4 rodent model scanner (Siemens Medical Solutions, Malvern, PA) as previously reported [16, 18]. The microPET studies were performed by tail-vein injection of about 3.7 MBq (100 μCi) of 68Ga-NOTA-RGD2, 68Ga-NOTA-G3-RGD2, or 68Ga-NOTA-P4-RGD2 into nude mice bearing U87MG or MDA-MB-435 tumor xenografts under isoflurane anesthesia. The 30-min dynamic scan (1 × 30 s, 4 × 1 min, 1 × 1.5 min, 4 × 2 min, 1 × 2.5 min, 4 × 3 min; total of 15 frames) was started 1 min after injection. The 1-h and 2-h time-point static scans were also acquired after the 30-min dynamic scan. Five-minute static PET images were also acquired separately at 30-min, 1-h, and 2-h time-points after injection (p.i.) for another set of tumor-bearing mice (n = 4 per tracer). The images were reconstructed by a two-dimensional ordered-subsets expectation maximum (OSEM) algorithm, and no correction was necessary for attenuation or scatter. For blocking experiments, each group of three mice bearing U87MG tumors were coinjected with 10 mg/kg body weight of c(RGDyK) and 3.7 MBq of 68Ga tracers. Five-minute static PET scan was then acquired at 1 h p.i. (n = 3 per group).
Biodistribution studies
Female nude mice bearing U87MG xenografts were injected with 370 kBq of 68Ga-NOTA-RGD2, 68Ga-NOTA-G3-RGD2, or 68Ga-NOTA-P4-RGD2 to evaluate the distribution of the 68Ga tracer in the tumor-bearing mice. A blocking experiment was also performed by coinjecting 68Ga-NOTA-P4-RGD2 with a saturating dose of c(RGDyK) (10 mg/kg body weight). All mice were killed 1 h after injection of the tracers. Blood, tumor, major organs, and tissues were collected and wet-weighed. The radioactivity in the tissue was measured using a γ counter (Packard, Meriden, CT). The results are presented as percentage injected dose per gram of tissue (%ID/g). Values are expressed as means±SD for a group of four animals.
Statistical analysis
Quantitative data are expressed as means±SD. Means were compared using one-way analysis of variance (ANOVA) and Student’s t test. P values <0.05 were considered statistically significant.
Results
Chemistry and radiochemistry
NOTA-RGD2, NOTA-G3-RGD2, and NOTA-P4-RGD2 were prepared by direct conjugation of RGD2, G3-RGD2, and P4-RGD2 with p-SCN-Bn-NOTA in the yields of 48%, 42% and 43%, respectively. The products were purified by HPLC and characterized by MALDI-TOF MS. Their HPLC purity was >95% before being used for the integrin αvβ3 binding assay and 68Ga labeling. The labeling procedure was done within 30 min, including the radioisotope incorporation, HPLC purification, rotary evaporation and formulation in PBS, with a decay-corrected yield ranging from 92% to 94% and a radiochemical purity of more than 98%. The specific activity of purified 68Ga-NOTA-RGD dimers was about 8.9–11.8 MBq/nmol.
Integrin αvβ3 binding affinity
The integrin αvβ3-positive U87MG human glioma cells were used for the integrin αvβ3 binding studies. We determined the integrin αvβ3 binding affinity of RGD2, G3-RGD2, P4-RGD2, and their NOTA conjugates NOTA-RGD2, NOTA-G3-RGD2, and NOTA-P4-RGD2 by competitive displacement of 125I-echistatin bound to U87MG cells. The IC50 values for RGD2, G3-RGD2, P4-RGD2, NOTA-RGD2, NOTA-G3-RGD2, and NOTA-P4-RGD2 were obtained by curve fitting from Fig. 2, and were calculated to be 88.83 ± 5.42 nM, 61.64 ± 3.28 nM, 41.83 ± 5.81 nM, 100.04 ± 2.85 nM, 66.38 ± 3.75 nM and 33.96 ± 2.17 nM, respectively (n = 3).

Inhibition of 125I-echistatin binding to integrin αvβ3 on U87MG cells by RGD2, G3-RGD2, P4-RGD2 and their NOTA conjugates: closed squares RGD2 (IC50 88.83 ± 5.42 nM), open circles G3-RGD2 (IC50 61.64 ± 3.28 nM), closed triangles P4-RGD2 (IC50 41.83 ± 5.81 nM), closed circles NOTA-RGD2 (IC50 100.04 ± 2.85 nM), open triangles NOTA-G3-RGD2 (IC50 66.38 ± 3.75 nM), open squares NOTA-P4-RGD2 (IC50 33.96 ± 2.17 nM) (n = 3, means±SD)
MicroPET imaging study
Static microPET scans were performed on a U87MG tumor model and representative decay-corrected coronal images at 30, 60, and 120 min after tail vein injection of 68Ga-NOTA-G3-RGD2, 68Ga-NOTA-P4-RGD2, and 68Ga-NOTA-RGD2 are shown in Fig. 3a (n = 4 per group). The U87MG tumors were clearly visualized with good tumor-to-background contrast for all three tracers. The uptake in the tumor or other organs was measured from the region of interest (ROI) analysis and shown in Fig. 3b. For 68Ga-NOTA-G3-RGD2, the tumor uptake was 9.04 ± 2.05, 7.36 ± 2.08, and 6.38 ± 1.31%ID/g, at 30, 60, and 120 min p.i., respectively. For 68Ga-NOTA-P4-RGD2, the tumor uptake was 10.13 ± 1.81, 7.40 ± 0.39, and 7.24 ± 0.45%ID/g, at 30, 60, and 120 min p.i., respectively. For 68Ga-NOTA-RGD2, the tumor uptake was 5.28 ± 1.03, 4.36 ± 0.85, and 3.78 ± 0.85%ID/g, at 30, 60, and 120 min p.i., respectively. The tumor uptake of both 68Ga-NOTA-P4-RGD2 and 68Ga-NOTA-G3-RGD2 was significantly higher than that of 68Ga-NOTA-RGD2 (p < 0.01, Fig. 3b) at all three time points examined. All three tracers cleared rapidly from the blood, and were excreted mainly through the kidneys. The blood activity of 68Ga-NOTA-RGD2 was slightly lower than that of 68Ga-NOTA-P4-RGD2 and 68Ga-NOTA-G3-RGD2. The renal uptakes of the three tracers were similar at early time-points. However, at 2 h p.i., the kidney uptake of 68Ga-NOTA-RGD2 (4.23 ± 0.65%ID/g) was lower than that of 68Ga-NOTA-G3-RGD2 (5.90 ± 0.59%ID/g) and 68Ga-NOTA-P4-RGD2 (5.99 ± 0.39%ID/g) (p < 0.05, Fig. 3b). The liver uptakes of 68Ga-NOTA-G3-RGD2, 68Ga-NOTA-P4-RGD2 and 68Ga-NOTA-P4-RGD2 were almost identical at all time-points except for the much higher uptake of 68Ga-NOTA-G3-RGD2 at 0.5 h p.i. The nonspecific uptake in the muscle was at a very low level for all three tracers. The tumor-to-nontumor ratios of the three tracers at 1 h and 2 h were calculated and are compared in Fig. 3c. Other than the lack of a significant difference of the tumor/kidney ratios between 68Ga-NOTA-G3-RGD2 and 68Ga-NOTA-RGD2 at 2 h p.i (p > 0.05), the tumor/liver, tumor/muscle, tumor/blood and tumor/kidney ratios of 68Ga-NOTA-G3-RGD2 and 68Ga-NOTA-P4-RGD2 were all significantly higher than those of 68Ga-NOTA-RGD2 (p < 0.01) at 0.5 h, 1 h, and 2 h. Taken together, the two novel 68Ga-labeled RGD dimers provide better image quality than 68Ga-NOTA-RGD2.

a, b Coronal microPET images and radioactivity accumulation quantification in tumor, kidney, liver and muscle of the U87MG tumor-bearing mice at 30 min, 60 min and 120 min after injection of 3.7 MBq (100 μCi) of 68Ga-NOTA-RGD2, 68Ga-NOTA-G3-RGD2, or 68Ga-NOTA-P4-RGD2 (arrows U87MG tumors). All microPET images were decay-corrected to the injection time. c Tumor/nontumor (T/NT) ratios at 1 h and 2 h after injection were calculated from b
The integrin αvβ3 targeting specificity of the novel 68Ga-labeled RGD dimers was demonstrated by coinjection of excess c(RGDyK) as the blocking agent. Figure 4a shows the microPET images of 68Ga-NOTA-P4-RGD2 and 68Ga-NOTA-G3-RGD2 at 60 min p.i. in the absence/presence of c(RGDyK). Coinjection of excess dose of c(RGDyK) resulted in almost complete blockage of tumor uptake for 68Ga-NOTA-G3-RGD2 (1.71 ± 0.39%ID/g with c(RGDyK) vs. 7.36 ± 2.08%ID/g without c(RGDyK)) and 68Ga-NOTA-P4-RGD2 (1.40 ± 0.31%ID/g with c(RGDyK) vs. 7.40 ± 0.39%ID/g without c(RGDyK)) (Fig. 4c).

Decay-corrected whole-body coronal microPET images of U87MG tumor-bearing mice at 1 h after injection of 3.7 MBq (100 μCi) 68Ga-NOTA-G3-RGD2 (a 1) and 68Ga-NOTA-P4-RGD2 (a 2) without/with a blocking dose of c(RGDyK) (10 mg/kg body weight), respectively (n = 3 or 4 per group). b Decay-corrected coronal microPET images of the MDA-MB-435 tumor-bearing mice at 30 min, 60 min and 120 min after injection of 3.7 MBq (100 μCi) of 68Ga-NOTA-P4-RGD2 (arrows tumors). c Quantification of uptake of 68Ga-NOTA-G3-RGD2 and 68Ga-NOTA-P4-RGD2 in U87MG tumors without/with cold RGD blocking from a 1 and a 2 (n = 3 or 4 per group, means±SD). d Comparison of the uptake of 68Ga-NOTA-P4-RGD2 in U87MG tumors, MDA-MB-435 tumors and U87MG tumors (cold RGD blocking) at 30 min, 60 min and 120 min after injection (n = 3 or 4 per group, means±SD)
MicroPET imaging of 68Ga-NOTA-P4-RGD2 was also tested in the orthotopic MDA-MB-435 breast tumor model. As shown in Fig. 4b, the tumors could be visualized at 0.5 h p.i., and also gave good contrast with the clearance of the tracer in normal organs with time. The tumor uptake of 68Ga-NOTA-P4-RGD2 in U87MG and MDA-MB-435 tumor models is compared in Fig. 4d. The tumor uptake of 68Ga-NOTA-P4-RGD2 with coinjection of excess c(RGDyK) in U87MG tumor-bearing nude mice is also shown in Fig. 4d as a control. The tumor uptakes of 68Ga-NOTA-P4-RGD2 in U87MG tumors (10.13 ± 1.81%ID/g at 30 min p.i. and 7.40 ± 0.39%ID/g at 60 min p.i.) were significantly higher (p < 0.01) than those in MDA-MB-435 breast tumors (6.42 ± 1.06%ID/g at 30 min p.i. and 5.78 ± 0.95%ID/g at 60 min p.i.). This finding is consistent with the fact that the U87MG glioma cells have a higher level of integrin αvβ3 expression than MDA-MB-435 breast tumor cells [18, 29]. At 120 min, the tumor uptake of 68Ga-NOTA-P4-RGD2 in U87MG tumor model was also higher than that in MDA-MB-435 breast tumor models, but the difference was not significant (p > 0.05). The tumor uptakes of 68Ga-NOTA-P4-RGD2 in U87MG tumors and MDA-MB-435 tumor were all significantly higher than those in U87MG tumors with excess c(RGDyK) blocking (p < 0.001), demonstrating the specific targeting of the tracer in the two different integrin αvβ3-positive tumor models.
The 30-min decay-corrected dynamic microPET imaging followed by 1-h and 2-h time-point static scans of the three 68Ga-labeled RGD dimers was performed in mice bearing U87MG glioma. The tumor and major organ uptake levels were quantitated by measuring ROIs encompassing the entire tissue or organ in the coronal orientation of the microPET images. The time–activity curves of the tracers are shown in Fig. 5. While the tumor uptakes of 68Ga-NOTA-P4-RGD2 and 68Ga-NOTA-G3-RGD2 were comparable, they were both higher than that of 68Ga-NOTA-RGD2, which is consistent with the static scans at different time points. The uptake of the three tracers showed similar temporal trends in the kidneys, liver and muscle. All three tracers revealed dominant renal clearance. Some hepatic clearance was also observed.

Time–activity curves of U87MG tumor (a), kidney (b), liver (c) and muscle (d) in U87MG tumor-bearing nude mice after intravenous injection of 3.7 MBq (100 μCi) of 68Ga-NOTA-RGD2, 68Ga-NOTA-G3-RGD2, and 68Ga-NOTA-P4-RGD2, respectively
Biodistribution studies
To validate the microPET studies, we also performed biodistribution and blocking studies. Female nude mice bearing U87MG xenografts were injected intravenously with 370 kBq of 68Ga-labeled NOTA-G3-RGD2, NOTA-P4-RGD2, or NOTA-RGD2, and then killed at 1 h after injection of the tracer. The data are expressed as the percentage injected dose per gram of tissue (%ID/g) in Fig. 6 and supplementary Table S1. As shown in Fig. 6a, the tumor uptakes were 7.05 ± 1.06%ID/g for 68Ga-NOTA-G3-RGD2, 7.98 ± 0.94%ID/g for 68Ga-NOTA-P4-RGD2, and 4.17 ± 1.10%ID/g for 68Ga-NOTA-RGD2, respectively. The tumor uptakes of 68Ga-NOTA-G3-RGD2 and 68Ga-NOTA-P4-RGD2 were both significantly higher than that of 68Ga-NOTA-RGD2 (p < 0.01), which is consistent with the microPET data. All the tracers exhibited higher kidney uptakes with 10.86 ± 1.28%ID/g for 68Ga-NOTA-G3-RGD2, 11.61 ± 0.63%ID/g for 68Ga-NOTA-P4-RGD2, and 11.07 ± 0.32%ID/g for 68Ga-NOTA-RGD2, respectively (n = 4). The tumor-to-nontumor ratios of 68Ga-NOTA-G3-RGD2 and 68Ga-NOTA-P4-RGD2 were all higher than that of 68Ga-NOTA-RGD2 as calculated from the biodistribution data, the only exception being that the tumor/blood ratio of 68Ga-NOTA-Gly3-RGD2 (5.83 ± 3.03) was lower than that of 68Ga-NOTA-RGD2 (6.96 ± 4.10) (supplementary Table S2)

a Biodistribution of 68Ga-NOTA-RGD2, 68Ga-NOTA-G3-RGD2, and 68Ga-NOTA-P4-RGD2 in U87MG tumor-bearing nude mice at 1 h after injection. b Biodistribution of 68Ga-NOTA-P4-RGD2 in U87MG tumor-bearing nude mice with and without coinjection of 10 mg/kg of c(RGDyK) as a blocking agent. Data are expressed as %ID/g±SD (n = 4 per group)
The in vivo receptor binding specificity of 68Ga-NOTA-P4-RGD2 was also confirmed by coinjection with a blocking dose of c(RGDyK) (10 mg/kg). A decrease in radioactivity was seen in all dissected tissues and organs (Fig. 6b), which is similar to the findings of studies with other radiolabeled RGD peptides. The tumor uptake was reduced markedly from 7.98 ± 0.94 to 1.58 ± 0.30%ID/g at the 1-h time-point, indicating the tumor targeting specificity of 68Ga-NOTA-P4-RGD2 in the U87MG tumor model.
Discussion
Previously, we and others have proposed that the binding affinity of dimeric and multimeric RGD peptides would be better than that of monomeric RGD peptides based upon the polyvalency effect [15, 16, 30–32]. Given the short distance between the two cyclic RGD peptides in the RGD dimer molecule, it is unlikely that they would bind simultaneously to the two adjacent integrin αvβ3 receptors. However, the receptor binding of the one RGD peptide may significantly enhance the “local concentration” of the other RGD peptide in the vicinity of the receptor, which may lead to a faster rate of receptor binding and/or a slower rate of dissociation of the radiolabeled multimeric RGD peptide [11, 12]. The high “local RGD peptide concentration” may explain the higher tumor uptake and longer tumor retention times of the radiolabeled (99mTc, 111In, 90Y, 18F, and 64Cu, 68Ga) [14, 32–36] cyclic RGD dimers as compared to their monomeric analogues. Although the RGD tetramer and octamer have higher integrin affinity in vitro and higher tumor uptake in vivo than the RGD dimer and monomer, they have lower tumor/kidney ratios than the RGD dimer and monomer. In order to further improve the dimeric RGD peptide’s tumor uptake without compromising the tumor/background contrast, we designed two novel RGD dimers with the Gly3 and PEG4 linkers (G3-RGD2, and P4-RGD2), and labeled them with the positron-emitting isotope 68Ga. We then investigated the potential advantage of using 68Ga-labeled novel RGD dimer tracers for integrin imaging.
The in vitro competition binding assay showed that both P4-RGD2 and G3-RGD2 had a higher integrin αvβ3 binding affinity than RGD2. After conjugation with NOTA, the respective binding affinity of the NOTA conjugates also followed the order of NOTA-P4-RGD2 > NOTA-G3-RGD2 > NOTA-RGD2. Apparently, the addition of PEG4 and G3 linkers between two cyclic RGD motifs improves integrin αvβ3 binding affinity of the cyclic RGD peptide dimers.
The property of the linker between the two RGD motifs in cyclic RGD dimer peptides is important for its bivalent binding and enhanced affinity (avidity) to integrin αvβ3 receptors. First, the linker must be long enough to present the two RGD motifs simultaneously to the receptors with minimal entropic penalty. Second, the linker must be flexible enough to present a high conformational entropic cost. The distance between two cyclic RGD motifs is 6 bonds in RGD2, 26 bonds in G3-RGD2 and 38 bonds in P4-RGD2 (excluding side-arms of K-residues). Although we did not find direct evidence that the Gly3 or PEG4 linkers can induce the simultaneous binding of the two RGD motifs with the integrin receptors, after inserting the linkers, the integrin binding affinity of the RGD dimers with Gly3 and PEG4 linkers (IC50 values were 66.38 ± 3.75 nM for NOTA-G3-RGD2 and 33.96 ± 2.17 nM for NOTA-P4-RGD2) did increase as compared with NOTA-RGD2 (IC50 100.04 ± 2.85 nM). The enhanced receptor binding affinity is well supported by the significantly higher tumor uptake seen in 68Ga-NOTA-G3-RGD2 (9.04 ± 2.05%ID/g, 7.35 ± 2.08%ID/g, and 7.35 ± 2.08%ID/g at 30, 60 and 120 min p.i., respectively) and 68Ga-NOTA-P4-RGD2 (10.13 ± 1.81%ID/g, 7.40 ± 0.39%ID/g, and 7.24 ± 0.45%ID/g at 30, 60 and 120 min p.i., respectively) compared to that of 68Ga-NOTA-RGD2 (5.28 ± 1.03%ID/g, 4.36 ± 0.85%ID/g, and 3.78 ± 0.85%ID/g at 30 min, 60 min and 120 min p.i, respectively) in the same tumor xenograft model (Fig. 3).
It is possible that the enhanced tumor uptake of 68Ga-NOTA-G3-RGD2, and 68Ga-NOTA-P4-RGD2 could also be due in part to the slightly increased molecular weights over 68Ga-NOTA-RGD2, resulting in a prolonged circulation (higher blood uptake) and sustained tumor retention. However, the uptakes of 68Ga-NOTA-G3-RGD2 and 68Ga-NOTA-P4-RGD2 was almost identical to that of 68Ga-NOTA-RGD2 in almost all the normal organs (Figs. 3 and 6a). More importantly, the tumor/blood, tumor/muscle, tumor/liver, and tumor/kidney ratios of 68Ga-NOTA-G3-RGD2 and 68Ga-NOTA-P4-RGD2 were all significantly higher than those of 68Ga-NOTA-RGD2 at 30 min, 60 min and 120 min p.i. (p < 0.01), the only exception being that the tumor/kidney ratio of 68Ga-NOTA-Gly3-RGD2 (1.08 ± 0.11) at 120 min was not quite significantly higher than that of 68Ga-NOTA-RGD2 (0.89 ± 0.14) (p < 0.05; Fig. 3c). Taken together, our studies demonstrate that G3-RGD2 and P4-RGD2 are better targeting biomolecules than RGD2.
Our finding that the tumor uptake of 68Ga-NOTA-G3-RGD2 and 68Ga-NOTA-P4-RGD2 was almost completely blocked by coinjection of excess c(RGDyK) (Fig. 4a, c) is clear evidence that their tumor localization is indeed integrin receptor-mediated. 68Ga-NOTA-P4-RGD2 also showed good tumor targeting in MDA-MB-435 tumors, but the tumor uptake was much lower than that in U87MG tumors. MDA-MB-435 tumors are known to express lower integrin αvβ3 levels than U87MG [18, 29]. The fact that the higher integrin αvβ3-expressing tumors also have higher 68Ga-NOTA-P4-RGD2 uptake further confirms the specific integrin αvβ3-targeting ability of 68Ga-NOTA-P4-RGD2. Note that we did not consider the different physiological properties (e.g. blood flow, vascular permeability) of U87MG and MDA-MB-435 tumors, because we performed the microPET studies using similar sizes of tumors to minimize the effects of the nonspecific targeting of the tracers. Future studies should investigate whether the tumor uptake (%ID/g) or the tumor-to-background ratios of 68Ga-NOTA-G3-RGD2 or 68Ga-NOTA-P4-RGD2 derived from the PET scans of various tumors are positively correlated with the integrin expression level as measured by ex vivo Western blot.
PET-based molecular imaging provides a sensitive way to identify and characterize the nature of disease early. For a new integrin αvβ3-targeted PET imaging tracer to succeed clinically, it must show high tumor uptake with diagnostically useful tumor/background ratios in a short period of time (preferably <2 h p.i.). The radiotracer must be prepared in high yield and radiochemical purity with very high specific activity. In addition, the cost of the radiotracer must be sufficiently low to achieve widespread clinical utility and availability. 18F is one of the most important positron-emitting candidates for the labeling of RGD derivatives, and various RGD derivatives have been labeled with 18F for PET [13, 14, 17–19, 21–23, 33, 37–39]. 18F-labeled RGD analogs ([18F]-AH111585 [22] and [18F]Galacto-RGD [23]) have also been successful in clinical trials. Nevertheless, at present 18F has some drawbacks, namely its relatively low tumor uptake and tumor/background ratio, need for an in-house cyclotron system, a long irradiation time for production, and a complicated and time-consuming multistep procedure, that may limit its clinical utility for noninvasive imaging of integrin αvβ3 expression in cancer patients. Until these issues are resolved satisfactorily, the clinical utility of 18F for noninvasive imaging of integrin αvβ3 expression in cancer patients will be limited. There is therefore a continuing need to develop a more suitable integrin αvβ3-targeted radiotracer. For example, the 68Ga-labeled novel cyclic RGD peptide dimers described here meet these essential needs because of their superior nuclear properties, easy availability and relatively low cost. Moreover, the desirable properties of the 68Ga-labeled RGD dimers in conjugation with Gly3 and PEG4 linkers, such as rapid labeling procedure, high specific activity, high radiochemical purity, outstanding tumor targeting ability, and in vivo kinetics, make them attractive alternatives to cyclotron-based radiopharmaceuticals for integrin αvβ3 imaging.
Conclusion
In this study, two novel cyclic RGD peptide dimers with Gly3 and PEG4 linkers (G3-RGD2 and P4-RGD2) were designed, labeled with generator-eluted 68Ga, and then investigated in vitro and in vivo. The Gly3 and PEG4 linkers were inserted between the two RGD motifs in NOTA-G3-RGD2 and NOTA-P4-RGD2, in order to induce their enhanced binding affinity (avidity) to integrin αvβ3. The in vitro and in vivo characteristics of the new RGD dimers were improved as evidenced by their higher integrin αvβ3 binding affinity compared to that of NOTA-RGD2, and the significantly higher tumor uptake of 68Ga-NOTA-G3-RGD2 and 68Ga-NOTA-P4-RGD2 relative to that of 68Ga-NOTA-RGD2. The high affinity, high specificity and excellent pharmacokinetic properties of the two novel RGD dimers make them promising agents for PET imaging of integrin αvβ3 expression for clinical application.
References
Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995;1:27–31.
Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol 2002;29:15–8.
Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature 2000;407:249–57.
Sharma RA, Harris AL, Dalgleish AG, Steward WP, O’Byrne KJ. Angiogenesis as a biomarker and target in cancer chemoprevention. Lancet Oncol 2001;2:726–32.
Folkman J. Seminars in medicine of the Beth Israel hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med 1995;333:1757–63.
Eliceiri BP, Cheresh DA. The role of alphav integrins during angiogenesis: insights into potential mechanisms of action and clinical development. J Clin Invest 1999;103:1227–30.
Cai W, Chen X. Multimodality molecular imaging of tumor angiogenesis. J Nucl Med 2008;49(Suppl 2):113S–28S.
Cai W, Niu G, Chen X. Imaging of integrins as biomarkers for tumor angiogenesis. Curr Pharm Des 2008;14:2943–73.
Hsu AR, Chen X. Advances in anatomic, functional, and molecular imaging of angiogenesis. J Nucl Med 2008;49:511–4.
Cai W, Rao J, Gambhir SS, Chen X. How molecular imaging is speeding up antiangiogenic drug development. Mol Cancer Ther 2006;5:2624–33.
Chen X. Multimodality imaging of tumor integrin αvβ3 expression. Mini Rev Med Chem 2006;6:227–34.
Liu S. Radiolabeled multimeric cyclic RGD peptides as integrin αvβ3 targeted radiotracers for tumor imaging. Mol Pharm 2006;3:472–87.
Cai W, Zhang X, Wu Y, Chen XA. Thiol-reactive 18F-labeling agent, N-[2-(4-18F-fluorobenzamido)ethyl]maleimide, and synthesis of RGD peptide-based tracer for PET imaging of αvβ3 integrin expression. J Nucl Med 2006;47:1172–80.
Chen X, Park R, Shahinian AH, Tohme M, Khankaldyyan V, Bozorgzadeh MH, et al. 18F-labeled RGD peptide: initial evaluation for imaging brain tumor angiogenesis. Nucl Med Biol 2004;31:179–89.
Li ZB, Chen K, Chen X. 68Ga-labeled multimeric RGD peptides for microPET imaging of integrin αvβ3 expression. Eur J Nucl Med Mol Imaging 2008;35:1100–8.
Wu Y, Zhang X, Xiong Z, Cheng Z, Fisher DR, Liu S, et al. microPET imaging of glioma integrin αvβ3 expression using 64Cu-labeled tetrameric RGD peptide. J Nucl Med 2005;46:1707–18.
Wu Z, Li ZB, Chen K, Cai W, He L, Chin FT, et al. MicroPET of tumor integrin αvβ3 expression using 18F-labeled PEGylated tetrameric RGD peptide (18F-FPRGD4). J Nucl Med 2007;48:1536–44.
Zhang X, Xiong Z, Wu Y, Cai W, Tseng JR, Gambhir SS, et al. Quantitative PET imaging of tumor integrin αvβ3 expression with 18F-FRGD2. J Nucl Med 2006;47:113–21.
Haubner R, Weber WA, Beer AJ, Vabuliene E, Reim D, Sarbia M, et al. Noninvasive visualization of the activated αvβ3 integrin in cancer patients by positron emission tomography and [18F]Galacto-RGD. PLoS Med 2005;2:e70.
Liu S, Hsieh WY, Jiang Y, Kim YS, Sreerama SG, Chen X, et al. Evaluation of a 99mTc-labeled cyclic RGD tetramer for noninvasive imaging integrin αvβ3-positive breast cancer. Bioconjug Chem 2007;18:438–46.
Beer AJ, Grosu AL, Carlsen J, Kolk A, Sarbia M, Stangier I, et al. [18F]galacto-RGD positron emission tomography for imaging of αvβ3 expression on the neovasculature in patients with squamous cell carcinoma of the head and neck. Clin Cancer Res 2007;13:6610–6.
Kenny LM, Coombes RC, Oulie I, Contractor KB, Miller M, Spinks TJ, et al. Phase I trial of the positron-emitting Arg-Gly-Asp (RGD) peptide radioligand 18F-AH111585 in breast cancer patients. J Nucl Med 2008;49:879–86.
Beer AJ, Haubner R, Sarbia M, Goebel M, Luderschmidt S, Grosu AL, et al. Positron emission tomography using [18F]Galacto-RGD identifies the level of integrin αvβ3 expression in man. Clin Cancer Res 2006;12:3942–9.
Al-Nahhas A, Win Z, Szyszko T, Singh A, Khan S, Rubello D. What can gallium-68 PET add to receptor and molecular imaging? Eur J Nucl Med Mol Imaging 2007;34:1897–901.
Fani M, Andre JP, Maecke HR. 68Ga-PET: a powerful generator-based alternative to cyclotron-based PET radiopharmaceuticals. Contrast Media Mol Imaging 2008;3:67–77.
Maecke HR, Andre JP. 68Ga-PET radiopharmacy: a generator-based alternative to 18F-radiopharmacy. In: Schubiger PA, Lehmann L, Friebe M, editors. PET chemistry: the driving force in molecular imaging. Ernst Schering Research Foundation Workshop. Berlin: Springer; 2007. p. 215–42.
Shi J, Kim Y-S, Zhai S, Liu Z, Chen X, Liu S. Improving tumor uptake and pharmacokinetics of 64Cu-labeled cyclic RGD peptide dimers with Gly3 and PEG4 Linkers. Bioconjug Chem. in press.
Shi J, Wang L, Kim Y-S, Zhai S, Liu Z, Chen X, et al. Improving tumor uptake and excretion kinetics of 99mTc-labeled cyclic arginine-glycine-aspartic (RGD) dimers with triglycine linkers. J Med Chem 2008;51:7980–90.
Cai W, Wu Y, Chen K, Cao Q, Tice DA, Chen X. In vitro and in vivo characterization of 64Cu-labeled Abegrin, a humanized monoclonal antibody against integrin αvβ3. Cancer Res 2006;66:9673–81.
Li ZB, Cai W, Cao Q, Chen K, Wu Z, He L, et al. 64Cu-labeled tetrameric and octameric RGD peptides for small-animal PET of tumor αvβ3 integrin expression. J Nucl Med 2007;48:1162–71.
Dijkgraaf I, Kruijtzer JA, Liu S, Soede AC, Oyen WJ, Corstens FH, et al. Improved targeting of the αvβ3 integrin by multimerisation of RGD peptides. Eur J Nucl Med Mol Imaging 2007;34:267–73.
Janssen M, Oyen WJ, Massuger LF, Frielink C, Dijkgraaf I, Edwards DS, et al. Comparison of a monomeric and dimeric radiolabeled RGD-peptide for tumor targeting. Cancer Biother Radiopharm 2002;17:641–6.
Chen X, Tohme M, Park R, Hou Y, Bading JR, Conti PS. Micro-PET imaging of αvβ3-integrin expression with 18F-labeled dimeric RGD peptide. Mol Imaging 2004;3:96–104.
Janssen ML, Oyen WJ, Dijkgraaf I, Massuger LF, Frielink C, Edwards DS, et al. Tumor targeting with radiolabeled αvβ3 integrin binding peptides in a nude mouse model. Cancer Res 2002;62:6146–51.
Jia B, Liu Z, Shi J, Yu Z, Yang Z, Zhao H, et al. Linker effects on biological properties of 111In-labeled DTPA conjugates of a cyclic RGDfK dimer. Bioconjug Chem 2008;19:201–10.
Chen X, Liu S, Hou Y, Tohme M, Park R, Bading JR, et al. MicroPET imaging of breast cancer alphav-integrin expression with 64Cu-labeled dimeric RGD peptides. Mol Imaging Biol 2004;6:350–9.
Beer AJ, Niemeyer M, Carlsen J, Sarbia M, Nahrig J, Watzlowik P, et al. Patterns of αvβ3 expression in primary and metastatic human breast cancer as shown by 18F-Galacto-RGD PET. J Nucl Med 2008;49:255–9.
Wu Z, Li ZB, Cai W, He L, Chin FT, Li F, et al. 18F-labeled mini-PEG spacered RGD dimer (18F-FPRGD2): synthesis and microPET imaging of alphavbeta3 integrin expression. Eur J Nucl Med Mol Imaging 2007;34:1823–31.
Glaser M, Morrison M, Solbakken M, Arukwe J, Karlsen H, Wiggen U, et al. Radiosynthesis and biodistribution of cyclic RGD peptides conjugated with novel [18F]fluorinated aldehyde-containing prosthetic groups. Bioconjug Chem 2008;19:951–7.
Acknowledgments
This work was supported, in part, by the National Cancer Institute (NCI R01 CA119053, R21CA121842, P50 CA114747 and U54 CA119367). We thank Dr. Kai Chen for excellent technical support. Z. Liu would like to thank Dr. Zibo Li for training in the use of the 68Ge/68Ga generator and also acknowledges the China Scholarship Council (CSC) for partial financial support during his visit to Stanford University.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table S1
Biodistribution of 68Ga labeled RGD dimers in U87MG tumor xenografts at 1 h p.i (n = 4, Mean±SD) (DOC 33.5 KB)
Supplementary Table S2
Tumor-to-nontumor (T/NT) Ratios (n = 4, Means±SD) (DOC 30.0 KB)
Rights and permissions
About this article
Cite this article
Liu, Z., Niu, G., Shi, J. et al. 68Ga-labeled cyclic RGD dimers with Gly3 and PEG4 linkers: promising agents for tumor integrin αvβ3 PET imaging. Eur J Nucl Med Mol Imaging 36, 947–957 (2009). https://doi.org/10.1007/s00259-008-1045-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-008-1045-1