
Abstract
Purpose
Impairment of cholinergic neurotransmission is a well-established fact in Alzheimer’s disease (AD), but there is controversy about its relevance at the early stages of the disease and in mild cognitive impairment (MCI).
Methods
In vivo positron emission tomography imaging of cortical acetylcholine esterase (AChE) activity as a marker of cholinergic innervation that is expressed by cholinergic axons and cholinoceptive neurons has demonstrated a reduction of this enzyme activity in manifest AD. The technique is also useful to measure the inhibition of cerebral AChE induced by cholinesterase inhibitors for treatment of dementia symptoms.
Results
A reduction of cortical AchE activity was found consistently in all studies of AD and in few cases of MCI who later concerted to AD.
Conclusion
The in vivo findings in MCI and very mild AD are still preliminary, and studies seem to suggest that cholinergic innervation and AChE as the main degrading enzyme are both reduced, which might result in partial compensation of their effect.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Acetylcholine (ACh) is an essential neurotransmitter in the central and the peripheral nervous system that developed very early in phylogenetical history. In the human brain, multiple areas exist where cholinergic neurons are concentrated [1]. In the context of neurodegenerative disease, cholinergic nuclei in the basal forebrain with diffuse projections into virtually all cortical areas appear to be most important. They are modulating selective attention and the processing of sensory input as well as associative thinking [2, 3]. Those cholinergic projections are severely impaired in Alzheimer’s disease [4, 5]. Another important cholinergic projection system originates in the pedunculopontine and laterodorsal tegmental nuclei of the brainstem with projections to thalamus, in particular its reticular nucleus which controls consciousness and general attention. This system is impaired in dementia with Lewy bodies which can be associated with parkinsonian symptoms or may occur without clinically manifest motor symptoms [1].
Degeneration and loss of trophic support for the cholinergic neurons of the basal forebrain and their projections is widely held as an early and pivotal event in AD, and there is evidence for interaction with amyloid deposition and plaque formation [6]. That view has been challenged by recent neuropathological studies indicating that cortical AChE immunoreactivity is well preserved in mild AD [7] or even upregulated in mild cognitive impairment (MCI) [8], and cholinergic forebrain neurons are not decreased in early AD [9]. Yet, there is a loss of calbindin in cholinergic basal forebrain neurons that corresponds with the appearance of tangles before manifestation of dementia [7], suggesting early and severe functional impairment of these neurons. Disturbance of axonal transport in cholinergic neurons has been identified as one of the earliest signs of disease in humans and in transgenic mice [8]. Thus, the current evidence from postmortem studies on the involvement of the cholinergic system in the earliest stages of AD is inconclusive.
By use of tracers that are substrates for AChE, positron emission tomography (PET) permits direct measurement of cerebral AChE activity in health and disease. AChE is being present in cholinergic axons and in some cholinoceptive pyramidal cortical neurons [10]. Loss and severe functional impairment of cholinergic axons, therefore, is associated with reduction not only of the transmitter ACh and its synthetic enzyme ChAT but also of its main degrading enzyme AChE [11]. Changes in AChE activity could also be due to alterations of the cellular environment, such as association with amyloid plaques in AD [12]. This brief review therefore concentrates on recent findings using these advanced in vivo techniques in MCI and AD.
Methods for measuring cerebral AChE activity
Labelled analogues of acetylcholine which are also substrates for AChE can be used to measure and image its activity in vivo. These are C-11-N-methyl-4-piperidyl-acetate (MP4A, also known as AMP) [13], which is 94% specific for AChE in human brain, and C-11-N-methyl-4-piperidyl-propionate (MP4P or PMP) [14]. Hydrolysis of these tracers by AChE results in tissue trapping, whereas unhydrolysed tracer is washed out from brain by blood flow.
Quantification of AChE activity is based on a standard three-compartment model comprising (1) intravascular blood plasma, (2) non-hydrolysed tracer in tissue and (3) tracer hydrolysed by AChE in tissue [13]. There also is rapid hydrolysis of the tracer in plasma, but the hydrolysis product cannot cross the blood brain barrier. Tracer hydrolysed in the brain also is trapped in brain tissue, and the hydrolysis rate in tissue, k 3, is a measure of AChE activity [15]. Due to lower affinity to AChE, hydrolysis rates of MP4P are generally lower than that of MP4A (0.02 to 0.03 min-1 for MP4P vs 0.06 to 0.09 min-1 for MP4A in normal cortex), and both tracers have been used successful in human studies (see review by [16]).
The first kinetic studies using these tracers involved arterial blood sampling with metabolite correction [13, 14, 17]. Kinetic measurements demonstrated that blood–tissue transfer rates K 1 and k 2 are high and correlated with cerebral blood flow (CBF), whereas k 3 is independent from CBF [18, 19]. It also became clear that hydrolysis rates in striatum are extremely high, corresponding to the very high AChE activity in this structure in humans [20]. As a consequence, virtually all tracer that enters the striatum is hydrolysed and trapped immediately, and delivery by CBF rather than hydrolysis by AChE becomes rate-limiting for tracer accumulation. In mathematical terms, this means that striatal tracer activity is the integral of plasma activity, scaled by K 1. Comparison of measured activity in arterial plasma samples with striatal activity has confirmed that relation (Fig. 1). In consequence, it is not possible with this technique to actually measure striatal k 3, but it also opens the possibility to use the striatum as a positive reference structure for non-invasive quantitation of k 3 in other brain structures. This concept for full non-invasive quantitation has been exploited in several computational implementations, either involving nonlinear least-squares fits of target-to-reference curves [21, 22] or double integration and linear least-squares fits [23, 24]. Another non-invasive technique [15] based on curve shape proved less reliable because it depends on the assumption that initially all tracer in tissue is non-hydrolysed, whereas at the end of the study, all tracer is hydrolysed.

Correspondence of measured MP4A activity in putamen with the integral of metabolite-corrected plasma activity, forming the basis for non-invasive quantitation of cortical hydrolysis rates using the putamen as a reference region to derive the time course of the input function
There are other cholinesterases besides AChE that can hydrolyse ACh in brain, most notably butyrylcholinesterase (BChE), but under physiological conditions, their quantitative contribution is small, probably less than 10% [11]. BChE is being expressed primarily in glial cells and is present in AD amyloid plaques [25]. C-11-N-methylpiperdin-4-yl-butyrate (MP4B, BMP) is hydrolysed and thus trapped with good specificity by BChE [26, 27]. Other ligands for AChE have been based on AChE inhibitors, e.g. C-11-physostigmine [28] and C-11-donepezil [29, 30], but did not provide appropriate signal quality. More recently, encouraging results were obtained using the C-11-CP-126998 [31].
Results
A study in 20 normal control subjects with a wide age range (24–89 years) did not indicate a relevant age or sex effect [19].
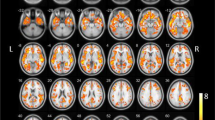
There have been several studies measuring AChE activity using MP4A or MP4P in AD, and all of them found a reduction of cortical activity [18, 32–34], most severely affecting temporal cortex (Fig. 2).

Parametric images (orthogonal slices with masking of subcortical areas) of 11C-MP4A hydrolysis rates, indicating reduced AChE activity in Alzheimer’s disease compared to normal controls
We found a reduction by 18% (in frontal cortex) to 34% (in temporal cortex) in patients with mild to moderate AD [35], while apparent AChE activity in basal forebrain was still intact. In another study, a pronounced reduction of AChE activity was found in the amygdala, whereas hippocampus was much less affected [36]. Contrary to expectations, a less severe reduction of AChE activity was found in AD patients carrying the ApoE4 allele than in those without that risk factor [37]. Correlations between reduction of AChE activity and impairment of cognitive function or dementia severity tend to be relatively weak. There appears to be a non-linear relation of reduced MP4A hydrolysis with a decline of AChE at mild stages (MMSE 15–30) and a bottom effect without further change in severe dementia (MMSE 0–15) [36], arguing for a significant decline in mild AD. On the other hand, in one study of 12 subjects with MP4P in mild AD (average MMSE score 23), an overall cortical reduction by only 9% was reported, significantly less than in patients with Parkinson’s disease with dementia of similar severity (20%) [38].
As yet, there are very few data available in patients with MCI. Subjects with MCI do not yet have dementia (which, by definition, requires cognitive impairment in multiple domains that are progressive and are severe enough to impairment of daily activities) but are at increased risk for developing AD. In a small study comparing four converters to AD within 18 months with four non-converters, we found a significant reduction of cortical AChE activity in converters at that stage already [39]. This study has limitations due to the small number of subjects studied, a relatively low age of subjects (65 ± 13 years) and a higher than expected conversion rate of 50% over 18 months. Thus, generalisation to MCI in general would be premature, and larger studies are needed.
Inhibition of AChE by cholinesterase inhibitors which are used as therapeutic agents in AD and dementia with Lewy bodies has been measured using MP4A and MP4P. For all currently available cholinesterase inhibitors at standard clinical dose, the reduction of cerebral AChE activity is in the range of 30 to 40% [40–42], and slightly stronger inhibition by 50% has been observed using intravenous infusion of 1.5 mg physostigmine salicylate [32].
Comments
It is controversial whether there is significant impairment of the cholinergic system very early in AD, but there are some in vivo studies indicating such impairment. In addition to the non-linear relation between dementia severity and reduction of MP4A which is steeper in mild than in severe disease, and the preliminary findings in MCI, a study in 27 patients with mild AD found a relation between attentional tests (digit symbol and trail making test A) and using C-11-nicotine binding [43]. These studies seem to suggest that presynaptic receptors and AChE as the main degrading enzyme are both reduced in AD, which might result in partial compensation. In conclusion, PET has demonstrated a reduction of cortical AChE activity in manifest AD in vivo, while the findings in MCI are still preliminary.
References
Perry E, Walker M, Grace J, Perry R. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 1999;22:273–80.
Mesulam M. The cholinergic lesion of Alzheimer’s disease: pivotal factor or side show? Learn. Mem. 2004;11:43–9.
Sarter M, Hasselmo ME, Bruno JP, Givens B. Unraveling the attentional functions of cortical cholinergic inputs: interactions between signal-driven and cognitive modulation of signal detection. Brains Res Rev. 2005;48:98–111.
Bierer LM, Haroutunian V, Gabriel S, Knott PJ, Carlin LS, Purohit DP, Perl DP, Schmeidler J, Kanof P, Davis KL. Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. J Neurochem. 1995;64:749–60.
Reinikainen KJ, Soininen H, Riekkinen PJ. Neurotransmitter changes in Alzheimer’s disease: implications to diagnostics and therapy. J Neurosci Res. 1990;27:576–86.
Auld DS, Kornecook TJ, Bastianetto S, Quirion R. Alzheimer’s disease and the basal forebrain cholinergic system: relations to [beta]-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol. 2002;68:209–45.
Davis KL, Mohs RC, Marin D, Purohit DP, Perl DP, Lantz M, Austin G, Haroutunian V. Cholinergic markers in elderly patients with early signs of Alzheimer disease. JAMA. 1999;281:1401–6.
DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, Cochran EJ, Kordower JH, Mufson EJ. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002;51:145–55.
Gilmor ML, Erickson JD, Varoqui H, Hersh LB, Bennett DA, Cochran EJ, Mufson EJ, Levey AI. Preservation of nucleus basalis neurons containing choline acetyltransferase and the vesicular acetylcholine transporter in the elderly with mild cognitive impairment and early Alzheimer’s disease. J Comp Neurol. 1999;411:693–704.
Mesulam MM, Geula C. Acetylcholinesterase-rich neurons of the human cerebral cortex: cytoarchitectonic and ontogenetic patterns of distribution. J Comp Neurol. 1991;306:193–220.
Perry EK, Perry RH, Blessed G, Tomlinson BE. Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol Appl Neurobiol. 1978;4:273–7.
Inestrosa NC, Alarcon R. Molecular interactions of acetylcholinesterase with senile plaques. J Physiol Paris. 1998;92:341–4.
Namba H, Irie T, Fukushi K, Iyo M. In vivo measurement of acetylcholinesterase activity in the brain with a radioactive acetylcholine analog. Brain Res. 1994;667:278–82.
Kilbourn MR, Snyder SE, Sherman PS, Kuhl DE. In vivo studies of acetylcholinesterase activity using a labeled substrate, n-[C-11]methylpiperdin-4-yl propionate ([C-11]PMP). Synapse. 1996;22:123–31.
Koeppe RA, Frey KA, Snyder SE, Meyer P, Kilbourn MR, Kuhl DE. Kinetic modeling of N-[11C]methylpiperidin-4-yl propionate: alternatives for analysis of an irreversible positron emission tomography trace for measurement of acetylcholinesterase activity in human brain. J Cereb Blood Flow Metab. 1999;19:1150–63.
Shinotoh H, Fukushi K, Nagatsuka S, Irie T. Acetylcholinesterase imaging: its use in therapy evaluation and drug design. Curr Pharm Des. 2004;10:1505–17.
Shinotoh H, Namba H, Fukushi K, Nagatsuka S, Tanaka N, Aotsuka A, Tanada S, Irie T. Brain acetylcholinesterase activity in Alzheimer disease measured by positron emission tomography. Alzheimer Dis Assoc Disord. 2000;14(Suppl 1):S114–8.
Herholz K, Bauer B, Wienhard K, Kracht L, Mielke R, Lenz O, Strotmann T, Heiss WD. In-vivo measurements of regional acetylcholine esterase activity in degenerative dementia: comparison with blood flow and glucose metabolism. J Neural Transm. 2000;12:1457–68.
Namba H, Iyo M, Fukushi K, Shinotoh H, Nagatsuka S, Suhara T, Sudo Y, Suzuki K, Irie T. Human cerebral acetylcholinesterase activity measured with positron emission tomography: procedure, normal values and effect of age. Eur J Nucl Med. 1999;26:135–43.
Atack JR, Perry EK, Bonham JR, Candy JM, Perry RH. Molecular forms of acetylcholinesterase and butyrylcholinesterase in the aged human central nervous system. J Neurochem. 1986;47:263–77.
Herholz K, Lercher M, Wienhard K, Bauer B, Lenz O, Heiss WD. PET measurement of cerebral acetylcholine esterase activity without blood sampling. Eur J Nucl Med. 2001;28:472–7.
Zundorf G, Herholz K, Lercher M, Wienhard K, Bauer B, Weisenbach S, Heiss WD. In: Senda M, Kimura Y, Herscovitch P, editors. PET functional parametric images of acetylcholine esterase activity without blood sampling Brain imaging using PET. San Diego, CA.: Academic; 2002. p. 41–6.
Nagatsuka S, Fukushi K, Shinotoh H, Namba H, Iyo M, Tanaka N, Aotsuka A, Ota T, Tanada S, Irie T. Kinetic analysis of [(11)C]MP4A using a high-radioactivity brain region that represents an integrated input function for measurement of cerebral acetylcholinesterase activity without arterial blood sampling. J Cereb Blood Flow Metab. 2001;21:1354–66.
Sato K, Fukushi K, Shinotoh H, Nagatsuka S, Tanaka N, Aotsuka A, Ota T, Shiraishi T, Tanada S, Iyo M, Irie T. Evaluation of simplified kinetic analyses for measurement of brain acetylcholinesterase activity using N-[11C]Methylpiperidin-4-yl propionate and positron emission tomography. J Cereb Blood Flow Metab. 2004;24:600–11.
Huff FJ, Reiter CT, Rand JB. The ratio of acetylcholinesterase to butyrylcholinesterase influences the specificity of assays for each enzyme in human brain. J Neural Transm. 1989;75:129–34.
Roivainen A, Rinne J, Virta J, Jarvenpaa T, Salomaki S, Yu M, Nagren K. Biodistribution and blood metabolism of 1-11C-methyl-4-piperidinyl n-butyrate in humans: an imaging agent for in vivo assessment of butyrylcholinesterase activity with PET. The J Nucl Med. 2004;45:2032–9.
Snyder SE, Gunupudi N, Sherman PS, Butch ER, Skaddan MB, Kilbourn MR, Koeppe RA, Kuhl DE. Radiolabeled cholinesterase substrates: in vitro methods for determining structure-activity relationships and identification of a positron emission tomography radiopharmaceutical for in vivo measurement of butyrylcholinesterase activity. J Cereb Blood Flow Metab. 2001;21:132–43.
Traykov L, Tavitian B, Jobert A, Boller F, Forette F, Crouzel C, Di Giamberardino L, Pappata S. In vivo PET study of cerebral [11C] methyl-tetrahydroaminoacridine distribution and kinetics in healthy human subjects. Eur J Neurol. 1999;6:273–8.
Funaki Y, Kato M, Iwata R, Sakurai E, Sakurai E, Tashiro M, Ido T, Yanai K. Evaluation of the binding characteristics of [5-(11)C-methoxy]donepezil in the rat brain for in vivo visualization of acetylcholinesterase. J Pharmacol Sci. 2003;91:105–12.
De Vos F, Santens P, Vermeirsch H, Dewolf I, Dumont F, Slegers G, Dierckx RA, De Reuck J. Pharmacological evaluation of [11C]donepezil as a tracer for visualization of acetylcholinesterase by PET. Nucl Med Biol. 2000;27:745–7.
Bencherif B, Endres CJ, Musachio JL, Villalobos A, Hilton J, Scheffel U, Dannals RF, Williams S, Frost JJ. PET imaging of brain acetylcholinesterase using [11C]CP-126,998, a brain selective enzyme inhibitor. Synapse. 2002;45:1–9.
Kuhl DE, Koeppe RA, Minoshima S, Snyder SE, Ficaro EP, Foster NL, Frey KA, Kilbourn MR. In vivo mapping of cerebral acetylcholinesterase activity in aging and Alzheimer’s disease. Neurology. 1999;52:691–9.
Iyo M, Namba H, Fukushi K, Shinotoh H, Nagatsuka S, Suhara T, Sudo Y, Suzuki K, Irie T. Measurement of acetylcholinesterase by positron emission tomography in the brains of healthy controls and patients with Alzheimers disease. Lancet. 1997;349:1805–9.
Rinne JO, Kaasinen V, Jarvenpaa T, Nagren K, Roivainen A, Yu M, Oikonen V, Kurki T. Brain acetylcholinesterase activity in mild cognitive impairment and early Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74:113–5.
Herholz K, Weisenbach S, Zundorf G, Lenz O, Schroder H, Bauer B, Kalbe E, Heiss WD. In-vivo study of acetylcholine esterase in basal forebrain, amygdala, and cortex in mild to moderate Alzheimer disease. Neuroimage. 2004;21:136–43.
Shinotoh H, Fukushi K, Nagatsuka S, Tanaka N, Aotsuka A, Ota T, Namba H, Tanada S, Irie T. The amygdala and Alzheimer’s disease: positron emission tomographic study of the cholinergic system. Ann NY Acad Sci. 2003;985:411–9.
Eggers C, Herholz K, Kalbe E, Heiss WD. Cortical acetylcholine esterase activity and ApoE4-genotype in Alzheimer disease. Neurosci Lett. 2006;408:46–50.
Bohnen NI, Kaufer DI, Ivanco LS, Lopresti B, Koeppe RA, Davis JG, Mathis CA, Moore RY, DeKosky ST. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol. 2003;60:1745–8.
Herholz K, Weisenbach S, Kalbe E, Diederich NJ, Heiss WD. Cerebral acetylcholine esterase activity in mild cognitive impairment. Neuroreport. 2005;16:1431–4.
Bohnen NI, Kaufer DI, Hendrickson R, Ivanco LS, Lopresti BJ, Koeppe RA, Meltzer CC, Constantine G, Davis JG, Mathis CA, DeKosky ST, Moore RY. Degree of inhibition of cortical acetylcholinesterase activity and cognitive effects by donepezil treatment in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2005;76:315–9.
Kaasinen V, Nagren K, Jarvenpaa T, Roivainen A, Yu M, Oikonen V, Kurki T, Rinne JO. Regional effects of donepezil and rivastigmine on cortical acetylcholinesterase activity in Alzheimer’s disease. J Clin Psychopharmacol. 2002;22:615–20.
Kadir A, Darreh-Shori T, Almkvist O, Wall A, Grut M, Strandberg B, Ringheim A, Eriksson B, Blomquist G, Langstrom B, Nordberg A. PET imaging of the in vivo brain acetylcholinesterase activity and nicotine binding in galantamine-treated patients with AD. Neurobiol Aging. 2007 (in press). http://dx.doi.org/10.1016/j.neurobiolaging.2007.02.020
Kadir A, Almkvist O, Wall A, Langstrom B, Nordberg A. PET imaging of cortical 11C-nicotine binding correlates with the cognitive function of attention in Alzheimer's disease. Psychopharmacology (Berl). 2006;188:509–20.
Conflict of interest statement
The author declares that he has no relevant financial or any other interests in this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Herholz, K. Acetylcholine esterase activity in mild cognitive impairment and Alzheimer’s disease. Eur J Nucl Med Mol Imaging 35 (Suppl 1), 25–29 (2008). https://doi.org/10.1007/s00259-007-0699-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-007-0699-4