
Abstract
There is evidence of early pathological changes in cholinergic neurons of patients with Alzheimer’s disease and Lewy body disease. This makes cholinergic molecular imaging one of the most important tools for studying prodromal and preclinical disease states of dementia. Various tracers have been developed for visualizing the cholinergic system. They have demonstrated profound cholinergic dysfunction in the cholinergic system of manifest dementia. Cholinergic dysfunction is also evident in prodromal stages of dementia. Evidence points to pathology first arising in the cholinergic axons and cell bodies of the basal forebrain in Alzheimer’s disease dementia and some cases of Parkinson’s disease dementia and dementia with Lewy bodies. In another group of patients with Lewy body disease, the early pathology involves peripheral cholinergic neurons of the parasympathetic nervous system. Cholinergic molecular imaging is an important tool contributing to the search of where and when the pathological processes leading to dementia begins.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- PET-CT
- Cholinergic neurons
- Dementia
- Alzheimer’s disease
- Lewy body disease
- Parkinson’s disease
- Dementia with Lewy bodies
- Parkinson’s disease dementia
-
Cholinergic molecular imaging can detect abnormalities in early disease stages of dementia and follow progression over time.
-
The cholinergic system is implicated in a myriad of functions of the central and peripheral nervous systems, several of which are affected in dementia.
-
In Alzheimer’s disease and a significant proportion of patients Lewy body disease, early changes may occur in cholinergic axons or cell bodies of the basal forebrain.
-
In another group of patients with Lewy body disease, the earliest changes most likely occur in cholinergic parasympathetic neurons innervating internal organs.
Cholinergic neurons, so called because they release the neurotransmitter acetylcholine, are found in several locations of the human body. Somatic motor neurons in the spinal cord and brainstem transmit acetylcholine to activate striated muscle tissue. Parasympathetic neurons in the brain stem and spinal cord release acetylcholine to smooth muscles in glands and organs. The interneurons of the striatum and preganglionic sympathetic neurons also transmit acetylcholine, as do the enteric neurons of the gastrointestinal tract. From a dementia research point of view, perhaps the most interesting group of cholinergic neurons are located in the poorly defined nuclei of the upper brainstem and basal forebrain. From here, the cholinergic neurons project their axons to almost all areas of the brain.
For decades, the cholinergic system has been a cornerstone in dementia research. There are at least three plausible reasons. First, important cognitive functions are heavily dependent on a well-functioning cholinergic system [1, 2]. Second, the cortical cholinergic activity is decreased in manifest dementia, particularly in Alzheimer’s disease dementia, Parkinson’s disease dementia, and dementia with Lewy bodies [3,4,5,6]. This has been demonstrated consistently in both post-mortem and in vivo studies. Third, cognitive symptoms of dementia improve when treated with inhibitors of acetylcholinesterase (AChE), the enzyme responsible for the break-down of acetylcholine.
Today, acetylcholine is acknowledged to be involved in many functions other than cognition. Correspondingly, it is increasingly acknowledged that neurodegenerative disorders affect multiple functions. For example, cholinergic dysfunction in Parkinson’s disease is implicated in falls and freezing of gait, abnormal movements during REM sleep, hyposmia, depression, visual hallucinations, autonomic dysfunction, and psychosis [7, 8].
The cholinergic system can be visualized with both single-photon emission computed tomography (SPECT) and positron emission tomography (PET) using radiotracers engaging various molecular targets involved in the synthesis, storage, reception, and hydrolysis of acetylcholine. Cholinergic molecular imaging in dementia has proved to have multiple interesting applications (Fig. 8.1).

Cholinergic imaging in dementia research. A graphic illustration of the many applications of cholinergic molecular imaging in dementia research
One example is proof of mechanism of drugs. A study used PET to measure the cerebral activity of AChE in patients with mild cognitive impairment due to Alzheimer’s disease. The authors found that the efficacy of an AChE-inhibitor depended on the activity of AChE [9]. At clinically tolerated doses, it emerged only around 25% of AChE sites were being occupied by the inhibitor donepezil.
Another application of cholinergic imaging focuses on understanding the neuropathological basis of symptoms and signs. For example, studies have looked at the role of cholinergic dysfunction in visual hallucinations, hyposmia, gait disturbances, and of course; cognition [7]. The contribution of cholinergic dysfunction to symptoms and signs, however, can be difficult to disentangle, as the processes occur on a background of pathology involving multiple transmitter systems and cell types.
A third example on how cholinergic imaging is used in research is to understand cholinergic changes in relation to other pathological markers such as metabolic activity [10], inflammation [11], amyloid [12], and structural atrophy [13]. Unfortunately, there is not yet a tracer that specifically binds to aggregated alpha-synuclein, the pathological substrate of Lewy body diseases. So far, cholinergic imaging is not routinely used in the clinic to diagnose or differentiate disorders. This chapter will focus on cholinergic imaging in dementia research [14].
In the first part of this chapter, we will introduce the cholinergic neuron. To understand PET-images, it is crucial to know where the different PET-tracers bind and the function of these target-molecules. Then we present the organization of the cholinergic system on a macroscopic level. The second part of the chapter will focus on the role of cholinergic imaging in identifying preclinical or prodromal disease stages of Alzheimer’s disease dementia, Parkinson’s disease dementia, and dementia with Lewy bodies [15]. From a clinical perspective, patients at very early stages represent optimal candidates for evaluating disease modifying therapies and possibly curative treatments while the disease remains rather localized. From a basic scientific perspective, studying early disease stages can shed light on fundamental questions about when and where the pathology begins and how it spreads. Cholinergic imaging may be a very important tool for exploring these questions.
Cholinergic Neurons and Tracer Molecules
Acetylcholine is synthesized in the nerve terminal by choline acetyltransferase (ChAT) and loaded into vesicles by the vesicular acetylcholine transporter (VAChT) (Fig. 8.2). Even small decreases of VAChT may have large effects on the release of acetylcholine by reducing the amount of acetylcholine release by vesicles [16]. [18F]FEOBV is a PET-tracer for VAChT and a very specific marker of cholinergic terminals (Fig. 8.3) [12, 17, 18]. [123I]IBVM is the corresponding tracer for SPECT.

Acetylcholine metabolism in cholinergic nerve terminals and relevant positron emission tomography (PET) tracers. Acetylcholine is synthesized from acetyl coenzyme A and choline by choline acetyl transferase in the cholinergic terminal. The vesicular acetylcholine transporter loads acetylcholine into pre-synaptic vesicles. Upon release, acetylcholine can engage its receptors. Acetylcholine esterase hydrolyses acetylcholine into acetic acid and choline. Tracers marked by an asterisk are used in single photon emission computed tomography (SPECT)

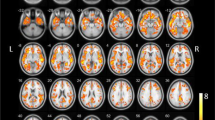
[18F]fluroetoxybenzovesamicol (FEOBV) positron emission tomography (PET) of healthy control and patient with dementia with Lewy bodies. A 75-year-old man with DLB (right side) and lower uptake of [18F]FEOBV, a PET-ligand for the vesicular acetylcholine transporter, compared to a non-demented age- and sex-matched control (left). The images are sliced axially on the AC-PC line and present an [18F]FEOBV-PET superimposed on a T1-MRI scan. The colorbar is scaled to a standard uptake value ratio (SUVR) of 0 to 4 where red colors indicate high uptake of tracer, and blue colors low uptake. SUVR standard uptake value ratio
In the synaptic cleft, acetylcholine is hydrolyzed by acetylcholinesterase (AChE). PET-tracers targeting AChE are among the most used in dementia research. The activity of AChE can be measured by substrate-tracers that are metabolized by AChE, such as [11C]MP4A and [11C]MP4P ([11C]PMP). These are lipophilic and pass the blood–brain barrier but become hydrophilic when metabolized and trapped inside the brain [19]. Another group of radioligands for AChE act as ligands that bind to AChE, such as [11C]donepezil. In the CNS, AChE is expressed by cholinergic as well as cholinoceptive non-cholinergic neurons.
Upon release from the cholinergic neuron, acetylcholine can bind to metabotropic muscarinic receptors or ionotropic nicotinic receptors [20, 21]. These receptors comprise several subtypes that are located on dendrites, cell bodies, and axons, on pre-terminal and post-terminal membranes of cholinergic and non-cholinergic cells. In general, presynaptic and preterminal nicotinic receptors enhance release of neurotransmitter, whereas post-synaptic and non-synaptic nicotinic receptors mediate excitation. The PET-tracers [18F]flubatine and [18F]FA have been implemented in clinical research and bind the alpha 4 beta 2 nicotinic acetylcholine receptor [22, 23]. The PET-tracer [11C]nicotine is a non-selective agonist to nicotinic receptor subtypes [24]. [11C]NMBP binds to all muscarinic receptor subtypes [25].
In summary, the cholinergic molecules and their corresponding tracers are markers of different aspects of the cholinergic system.
Cholinergic Neurons in the Human Organism
Cholinergic neurons in the basal forebrain provide the principal source of acetylcholine to the cortex and limbic structures (Fig. 8.4). They can be divided into four overlapping groups of cell bodies [26]. The cholinergic neurons located on the medial septum and horizontal band project to the hippocampus and hypothalamus. The neurons associated with the diagonal band project to the olfactory tubercle. The largest group of cholinergic cell bodies is associated with the nucleus basalis of Meynert (NBM) and project to the cortex and amygdala [27]. From the NBM the fiber tracts bundle in a lateral and a medial pathway before they fan out to the cortex [28]. The long and unmyelinated axons pass close to the lateral ventricles and reach the cortex through the underlying white matter. Interestingly, recent studies have documented that periventricular white matter lesions correlate with decreased cognition and decreased cortical cholinergic activity. Thus, periventricular white matter lesions may disrupt the cholinergic projections from the NBM to the cortex [29].

Main cholinergic projecting neurons of the central nervous system. The basal forebrain is composed of four overlapping cell groups and provide the principal source of acetylcholine to the cortex and limbic structures. The pedunculopontine nucleus and laterodorsal tegmental nucleus provide the main cholinergic innervation of the thalamus. The cholinergic projecting neurons of the medial habenula and parabigeminal nucleus are not presented on this figure. Neither are the cholinergic interneurons of the striatum or the motor neurons of the brainstem
When reaching the cortex, the projecting neurons from the NBM arborize extensively and each cover an area of about 1–1.5 mm2. Along the thin axons there are multiple varicosities. These represent sites for transmitter release, and some make direct contact with other neurons to form a synapse. Other varicosities release their transmitter molecules into the extracellular space to signal multiple cells simultaneously. The NBM innervates the entire cortex, but only limbic areas project back to the NBM. These limbic areas assign relevance to sensory stimuli and thereby modulate the response by the NBM [27]. If stimuli have high salience, acetylcholine is released to augment or amplify the signal in relevant cortical areas. For example, acetylcholine increases the responsiveness of neurons in the visual cortex to inputs from the lateral geniculate nucleus [2].
There is another significant group of cholinergic neurons on the junction of the pons and mesencephalon. The neurons are dispersed around the pedunculopontine and laterodorsal tegmental nuclei (PPN/LDT), and project their axons mainly to the thalamus.
Alzheimer’s Disease and Lewy Body Disease
Alzheimer’s disease (AD) is characterized by the accumulation of beta amyloid and tau tangles. The aggregation of these misfolded proteins is closely linked with neuronal dysfunction and degeneration. Over years, AD pathology can lead to Alzheimer’s disease dementia (ADD), the most common cause of dementia worldwide. The second most common cause of neurodegenerative dementia is Lewy body disease. Lewy bodies are pathological aggregates that are formed in neurons and defined by their content of phosphorylated alpha synuclein. Lewy body disease can lead to several clinical syndromes including Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), dementia with Lewy bodies (DLB), pure autonomic failure, and REM sleep behavior disorder (RBD), a sleep disorder characterized by abnormal movements during REM sleep. These are collectively referred to as Lewy body disorders (LBD). The related disorder multiple system atrophy is also caused by aggregated alpha synuclein, but these aggregates do not form Lewy bodies and the patients rarely become demented, so this disease will not be dealt with in this chapter. Up to 95% of patients with isolated RBD (iRBD) will eventually develop either PD or DLB, and more than 80% of patients with PD will eventually develop PDD [30, 31]. Thus, the end-stages of Lewy body diseases converge on dementia. Therefore, in this chapter, we will consider LBD in general, and not only the manifest dementia stages.
The natural history of AD and LBD can be illustrated on a timeline (Fig. 8.5). In the beginning, there is no pathology. An individual’s risk of dementia is defined by genetic predisposition and environmental exposure. Some genetic traits and environmental factors increase the risk of disease, others protect against disease. This stage can be referred to as the risk phase. Then follows the preclinical phase where pathology is present, at least at a cellular level, but there are not yet any symptoms or detectable signs. These incidental cases are asymptomatic with signs of pathology on post-mortem examination or PET scans. During the prodromal stage the pathological processes evolve to a degree where symptoms and signs develop. An example is mild cognitive impairment (MCI) where subjects have cognitive difficulties that are not yet severe enough to cause loss of independence. Another example is iRBD which is considered a prodromal stage of PD and DLB. The final step to manifest dementia occurs when the cognitive difficulties are severe enough to interfere with daily life functions.

Timeline of early disease stages of Alzheimer’s disease dementia and Lewy body disorders. The stages in Alzheimer’s disease and Lewy body disorders depicted on a timeline. The figure should be read from left to right. In the beginning (left), there is no pathology. The balance of genetic traits and environmental factors determine the likelihood of developing neurodegenerative disease (risk phase). In the preclinical phase pathology has developed, but there are not yet any signs or symptoms. Then follows the prodromal phase with emergence of signs and symptoms, and finally manifest dementia when cognitive difficulties begin to interfere with daily life functions
MCI can be diagnosed when there is concern about a decline in cognition and evidence of impairment in one or more cognitive domains in combination with preserved independence in activities of daily living [32]. MCI with affected memory increases the likelihood that the cause of MCI is AD (MCI-AD). Biomarkers showing brain beta-amyloid aggregation and neuronal injury can further increase the likelihood of MCI due to AD. These markers include PET amyloid imaging and low β-amyloid 42 in the cerebrospinal fluid. Markers of neuronal injury include high tau in the cerebrospinal fluid, atrophy on structural imaging, and decreased perfusion or glucose metabolism on PET.
Cholinergic Molecular Imaging in Alzheimer’s Disease and Lewy Body Disorders
The first molecular imaging studies using cholinergic markers in vivo with ADD, PD, and PDD were performed in the mid 1990s. Studies on DLB appeared a few years later, as formal diagnostic criteria for DLB were defined in 1996. These in vivo studies confirmed decades of post-mortem studies showing cortical cholinergic depletion in manifest dementia [10, 33]. They also confirmed the general tendency of a severe and indistinguishable cortical depletion in PDD and DLB, milder cholinergic involvement in cases at early stages, and more severe depletion in PDD and DLB compared to ADD [23, 34,35,36]. Overall, the cortical cholinergic integrity correlates with disease severity and dementia. We will focus initially on studies that investigated early disease stages using cholinergic imaging.
A study used the AChE substrate-tracer [11C]MP4A to investigate the cholinergic system of patients with MCI-AD. Apart from MCI, the patients were characterized by memory impairment, atrophy of the medial temporal lobe, and lowered amyloid in the CSF. The authors found decreased cortical activity of AChE, particularly in the temporal, parietal, and occipital lobes [29]. A similar pattern of reduced AChE activity was reported in a group of MCI characterized by memory decline [37]. Again, the activity of AChE was reduced in several cortical regions, but mostly in the temporal cortex. A third study using the same tracer on a comparable MCI population came to similar results, except that they found the hippocampus to be the structure most severely affected [38]. These findings suggest that cholinergic dysfunction in the temporal lobe may be a sign of early AD.
Another study followed a group of patients with MCI and affected memory and decreased performance in at least one other cognitive domain. They found that individuals with MCI who progressed to ADD had widespread reductions of cortical AChE activity, particularly in the parietotemporal regions. Activity in the hippocampus and thalamus was preserved among MCI who did not progress [39]. The cholinergic projections to the hippocampus come from the rostral sectors of the basal forebrain, and the cholinergic projections to the thalamus come from the PPN/LDT complex in the brainstem. This suggests that more rostral sectors of the basal forebrain are affected later and marks the transition to dementia. A similar sequential involvement could be true for the PPN/LDT, although evidence of thalamic reductions in cholinergic signal in ADD is less convincing.
[18F]FA binds to the alpha 4 beta 2 nicotinic receptor and has been used to image MCI with PET. On follow up, those MCI cases who progressed to dementia had significant baseline reductions in [18F]FA binding in several cortical areas, most pronounced in the temporal cortex and caudate [40]. Another study used the same tracer to study amnestic MCI and reported significant decreases in signal in all investigated cortical regions in addition to the hippocampus and caudate. The most severe reductions were found in those patients who progressed to ADD. Overall, the reductions among patients with MCI who progressed were almost as severe as those seen in fully developed ADD. This suggests that marked cholinergic dysfunction occurs at an early disease stage of AD [41].
Another more recent study used [18F]FA to investigate patients with MCI and impairment in the memory domain. They found that the level of change in nicotinic cholinergic receptor binding was between that of healthy controls and manifest ADD [23]. This supports the view that AD-MCI represents a stage on the spectrum to fulminant dementia. Furthermore, it seems that the density of nicotinic cholinergic receptors declines initially in the entorhinal cortex and other limbic structures suggesting that the cholinergic projections from the NBM to the entorhinal cortex are affected at a very early stage. Interestingly, the study found [18F]FA uptake in the hippocampus to be almost normal in AD-MCI, but severely decreased in ADD. This implies that the cholinergic cell bodies in the medial septum and horizontal limb are affected later than those in the NBM, in line with AD-pathology spreading through the basal forebrain in a rostral direction [42]. Another possible explanation for preserved hippocampal uptake in AD-MCI could be a compensatory upregulation in the density of receptors.
Overall, PET imaging has shown that there is impaired cholinergic integrity in MCI-AD that is more severe in limbic and medial temporal structures. Also, impairment is more severe in MCI cases who are close to converting to dementia [43]. While early-stage MCI patients also have impaired cholinergic integrity, their reductions often fall short of statistical significance, probably due to small study sample sizes. It is likely that the cholinergic system is affected from the beginning of the MCI-stage and possibly before the onset of MCI. This is supported by longitudinal data showing that the cognitive decline in MCI can be detected 4–6 years before MCI is diagnosed, implying a long-term temporal window of Alzheimer progression before MCI diagnosis [44]. Molecular cholinergic imaging in preclinical stages of AD has not yet been performed.
Magnetic resonance imaging (MRI) can be used to measure the size of the basal forebrain [45]. Decreases in grey matter volume detected with MRI-based volumetry can be used to measure rates of neurodegeneration. A study found an association between atrophy in the basal forebrain measured using MRI and amyloid deposition measured using PET in preclinical and prodromal stages of AD [46]. Moreover, among MCI cases, lower volumes of the basal forebrain were found to be associated with impaired cognition and cortical hypometabolism assessed using PET [47]. A study using MRI to investigate patients with MCI-AD found that atrophy of the NBM precedes atrophy of the entorhinal cortex, which is then followed by memory impairment. Interestingly, pathology of the NBM alone does not lead to memory impairment. Rather, the memory impairments of early AD arise when there is degeneration of the projections from the NBM to the entorhinal cortex. Thus, a subcortical to cortical spread of pathology may be a very early stage in AD pathology [48].
iRBD
Isolated RBD (iRBD) is characterized by accumulation of Lewy bodies in neurons and abnormal movement and behavior during REM sleep. The vast majority of patients with iRBD will develop either PD or DLB, and the majority of patients with PD progress to dementia (PDD) [30, 31]. This makes iRBD an excellent disorder for studying prodromal stages of DLB and PDD. Surprisingly, very little research has yet been carried out on the cholinergic system in iRBD. A study investigated patients with iRBD using PET and [11C]donepezil, a tracer that binds to AChE. Although the patients did not have any symptoms or signs of cognitive decline or motor disturbances, the authors found decreased uptake of tracer in the cortex of patients with iRBD compared to controls [49]. The cortical levels of [11C]donepezil were lower in the superior temporal cortex, the cingulum, dorsolateral prefrontal, and occipital cortices. Interestingly, this pattern of dysfunction resembles that seen in manifest Lewy body disorders. This observation has two important implications. First, it supports that the cholinergic system in prodromal PD and DLB is impaired and dysfunctional. Second, it suggests that investigations of patients with iRBD may be a key to understand early changes in the cholinergic system related to Lewy body disease.
Another finding of the study was that the affected cortical structures in iRBD are known to receive dense cholinergic projections from the NBM. Furthermore, the cortical tracer uptake was lower in those patients who performed worse on tests of cognitive function. This underlines that early preclinical or prodromal cognitive decline is associated with dysfunction of the cholinergic system in Lewy body disease, and that this dysfunction can be visualized using PET. Furthermore, there is evidence that the cholinergic dysfunction occurs in parallel with nigrostriatal dopaminergic dysfunction, implying that the pathological processes in the NBM and substantia nigra are linked [50, 51].
PET using cholinergic tracers has been used to visualize the peripheral autonomous nervous system. Patients with iRBD show decreased uptake of [11C]donepezil in their colon and small intestine. This signifies a dysfunction of the enteric and parasympathetic nervous system [52]. It may well be that the Lewy pathology was initiated in the peripheral nervous system in those patients with Lewy body dementia that were RBD-positive during their prodromal stage [53]. In iRBD there is pathology in nuclei of the upper brainstem. This means that at the time point of diagnosing iRBD, pathology is already relatively widespread in the brainstem and limbic system. Supposing that pathology in iRBD arose in the peripheral nervous system, and spread to the CNS, it should be theoretically possible to identify cases with pathology confined to the periphery. Pure autonomic failure, another Lewy body disorder, may represent such a population [54].
As such, cholinergic molecular markers are successfully being used to further characterize the prodromal disease stages of RBD-first Lewy body disorders. However, about two-thirds of patients with PD and one-quarter with DLB do not have iRBD in their prodromal stage [55]. The question is then how to identify this group in their prodromal stage? Part of the answer may be MCI-LB—see the next section.
Table 8.1 presents a list of molecular cholinergic human in vivo studies in preclinical and prodromal AD and LBD.
MCI-LB
MCI plus core features of DLB (MCI-LB) is a prodromal stage of DLB [56]. A recent publication describes the recommendations for identifying prodromal DLB. In short, the prodromal phase may include MCI, delirium-onset, and psychiatric-onset manifestations [56]. So far, no studies using cholinergic in vivo imaging have been published on patients who presented with one of these three phenotypes.
A few recent structural MRI-studies have been published on MCI-LB. A follow-up study investigated gray matter atrophy in MCI-LB. Comparing patients who remained MCI-LB to those who progressed to DLB, both groups had atrophy in the NBM at baseline [57]. Those who progressed to DLB had more longitudinal atrophy in entorhinal and parahippocampal cortices, temporoparietal association cortices, thalamus, and basal ganglia. Thus, atrophy of the NBM is a feature of prodromal DLB regardless of proximity to dementia and there is gradual atrophy of cortical regions with significant cholinergic innervation. The findings suggest that atrophy of the NBM occurs very early in the pathogenesis of DLB and reaches a plateau when the first cognitive symptoms appear. It seems that the initial phase of MCI-LB is associated with little cortical grey matter atrophy and that the later accelerated cortical grey matter atrophy in MCI-LB is a marker of impending dementia. In the study, RBD was assessed using a questionnaire. Overall, 89% had probable RBD. So far, no studies have focused on RBD-negative MCI-LB, which may represent a separate prodromal phenotype.
In PD without dementia, basal forebrain volume is correlated with cognition and reduced volume predicts future dementia [58,59,60].
In summary, early patients with MCI-LB show significant atrophy in the NBM [61]. This is followed by atrophy of the entorhinal cortex [57]. Later, on conversion to dementia, there is widespread gray matter atrophy in multiple cortical and subcortical regions, in particular those areas receiving input from the NBM. Both MCI-AD and MCI-LB exhibit atrophy of the NBM, although this is to a lesser degree in MCI-AD [61, 62]. Interestingly, both prodromal groups show evidence of degeneration in the axonal projections from the NBM to the cortex, and the integrity of cholinergic pathways correlate better with clinical features than do atrophy of cholinergic cell bodies [63]. Atrophy of the basal forebrain is closely linked to reduced integrity of its cortical cholinergic projections and cortical cholinergic signal, but it is loss of the projecting axons that is important in the pathological process [64].
Preclinical Changes in Cholinergic Axons
The number of cholinergic neurons in the basal forebrain decreases with age, particularly when transitioning from preclinical to MCI stages of DLB and AD. Morphological abnormalities in the cholinergic axons occur at very early stages of AD. In healthy young brains without AD pathology, the cholinergic axons are thin and homogenous with small uniform varicosities. In middle-aged non-demented persons, axonal abnormalities start to be present. These can include swollen axons, ballooned terminals, less branching, and fewer terminals. The abnormalities increase in non-demented elderly and then decrease in severe ADD, suggesting that the abnormal cholinergic axons atrophy in ADD [65].
The abnormal swellings of the cholinergic axons contain abundant AChE and ChAT. This could explain why some studies find preserved or even locally increased levels of cholinergic markers in prodromal and early disease stages [66, 67]. Also, it suggests that the cholinergic system may be functionally impaired despite preserved or increased levels of cholinergic markers [65]. A post-mortem study found that the increase in cholinergic markers could not be explained by increased number of cholinergic fibers or varicosities [68]. A more likely explanation appears to be an upregulation of proteins and enzymes involved in production and delivery of acetylcholine. This phenomenon has been documented in post-mortem studies [69, 70].
ChAT and VAChT are co-regulated and co-located on the same gene [71]. A recent PET-study found that VAChT may also be upregulated locally in iRBD [72]. The most marked areas of upregulation were found to be in the brainstem which receives most of its cholinergic projections from the PPN/LDT complex on the pontomesencephalic junction. This implies that, in iRBD, the PPN/LDT complex is affected by pathology. This seems reasonable, as iRBD is caused in part by pathology in the nearby subcerulean nucleus. It should be mentioned, however, that the results were based on only five patients with iRBD that were also younger and performed better on cognitive measures compared to their comparison group.
Another study found increased binding of VAChT in the hippocampi of PD patients with normal cognition while PD patients with MCI had normal levels of binding [66]. A possible explanation could be a local upregulation of VAChT due to dysfunctional cholinergic projections from the medial septum and horizontal band to the hippocampus. Local increases in tracer activity have been reported in studies using substrate-tracer for AChE in patients predisposed to PD [73], ligand-tracers for nicotinic receptors in patients with MCI and patients with AD [74, 75], and tracers for muscarinic receptors in patients with PD [76].
Interestingly, axonal swellings may appear prior to accumulation of pathological protein. Reducing axonal transport by genetic modification in mice led to increased levels of axonal swellings and amyloid deposition [77]. Also, reduced expression of VAChT can facilitate Alzheimer pathology in mice [78]. Evidence suggest that similar mechanisms may be at play in humans [65]. Intriguingly, this evidence suggests that cholinergic dysfunction can lead to the accumulation of pathological protein. However, the prevailing understanding is still that it is the accumulation of pathology that causes neuronal dysfunction and degeneration, not the reverse.
To conclude, cholinergic molecular in vivo imaging has made it possible to study early disease stages of dementia and follow progression over time. The studies of neurodegenerative dementias are no longer confined to investigating the cortical cholinergic changes associated with cognitive deficits. Today, we understand that these disorders affect multiple systems that cause a wide range of symptoms and signs, and that changes in the cholinergic system are involved in several of these functions. There is evidence of cholinergic dysfunction in the prodromal stages of ADD and LBD. These changes likely begin during the preclinical stages, but such early patient cases are currently hard to identify. In AD and RBD-negative Lewy body disorders, the earliest changes may occur in cholinergic axons or cell bodies of the basal forebrain. In RBD-positive LBD, the earliest changes most likely occur in cholinergic parasympathetic and intrinsic neurons innervating the internal organs. Overall, this makes cholinergic molecular imaging one of the most interesting, versatile, and promising fields within dementia research.
References
Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 2012;76(1):116–29. https://doi.org/10.1016/j.neuron.2012.08.036.
Sarter M, Bruno JP. Cognitive functions of cortical acetylcholine: toward a unifying hypothesis. Brain Res Rev. 1997;23(1–2):28–46.
Roy R, Niccolini F, Pagano G, Politis M. Cholinergic imaging in dementia spectrum disorders. Eur J Nucl Med Mol Imaging. 2016;43(7):1376–86. https://doi.org/10.1007/s00259-016-3349-x.
Bohnen NI, Grothe MJ, Ray NJ, Müller ML, Teipel SJ. Recent advances in cholinergic imaging and cognitive decline—revisiting the cholinergic hypothesis of dementia. Curr Geriatr Rep. 2018;7(1):1–11.
Hampel H, Mesulam MM, Cuello AC, Farlow MR, Giacobini E, Grossberg GT, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain. 2018;141(7):1917–33. https://doi.org/10.1093/brain/awy132.
Hampel H, Mesulam MM, Cuello AC, Khachaturian AS, Vergallo A, Farlow MR, et al. Revisiting the cholinergic hypothesis in Alzheimer's disease: emerging evidence from translational and clinical research. J Prev Alzheimers Dis. 2019;6(1):2–15. https://doi.org/10.14283/jpad.2018.43.
Pasquini J, Brooks DJ, Pavese N. The cholinergic brain in Parkinson's disease. Mov Disord Clin Pract. 2021;8(7):1012–26. https://doi.org/10.1002/mdc3.13319.
Bohnen NI, Yarnall AJ, Weil RS, Moro E, Moehle MS, Borghammer P, et al. Cholinergic system changes in Parkinson's disease: emerging therapeutic approaches. Lancet Neurol. 2022;21:381. https://doi.org/10.1016/s1474-4422(21)00377-x.
Richter N, Beckers N, Onur OA, Dietlein M, Tittgemeyer M, Kracht L, et al. Effect of cholinergic treatment depends on cholinergic integrity in early Alzheimer's disease. Brain. 2018;141(3):903–15. https://doi.org/10.1093/brain/awx356.
Kuhl DE, Minoshima S, Fessler JA, Ficaro EP, Wieland DM, Koeppe RA, et al. In vivo mapping of cholinergic terminals in normal aging, Alzheimer's disease, and Parkinson's disease. Ann Neurol. 1996;40(3):399–410.
Staer K, Iranzo A, Stokholm MG, Ostergaard K, Serradell M, Otto M, et al. Cortical cholinergic dysfunction correlates with microglial activation in the substantia innominata in REM sleep behavior disorder. Parkinsonism Relat Disord. 2020;81:89–93. https://doi.org/10.1016/j.parkreldis.2020.10.014.
Aghourian M, Legault-Denis C, Soucy JP, Rosa-Neto P, Gauthier S, Kostikov A, et al. Quantification of brain cholinergic denervation in Alzheimer's disease using PET imaging with [(18)F]-FEOBV. Mol Psychiatry. 2017;22(11):1531–8. https://doi.org/10.1038/mp.2017.183.
Bohnen N, Mueller ML, Kuwabara H, Constantine G, Studenski S. Age-associated leukoaraiosis and cortical cholinergic deafferentation. Neurology. 2009;72(16):1411–6.
Kanel P, Bedard MA, Aghourian M, Rosa-Neto P, Soucy JP, Albin RL, et al. Molecular imaging of the cholinergic system in Alzheimer and Lewy body dementias: expanding views. Curr Neurol Neurosci Rep. 2021;21(10):52. https://doi.org/10.1007/s11910-021-01140-z.
Berg D, Borghammer P, Fereshtehnejad S-M, Heinzel S, Horsager J, Schaeffer E, et al. Prodromal Parkinson disease subtypes—key to understanding heterogeneity. Nat Rev Neurol. 2021;17(6):349–61.
Prado VF, Roy A, Kolisnyk B, Gros R, Prado MA. Regulation of cholinergic activity by the vesicular acetylcholine transporter. Biochem J. 2013;450(2):265–74.
Nejad-Davarani S, Koeppe RA, Albin RL, Frey KA, Müller ML, Bohnen NI. Quantification of brain cholinergic denervation in dementia with Lewy bodies using PET imaging with [18 F]-FEOBV. Mol Psychiatry. 2018;1:322.
van der Zee S, García DV, Elsinga PH, Willemsen AT, Boersma HH, Gerritsen MJ, et al. [18 F] Fluoroethoxybenzovesamicol in Parkinson’s disease patients: quantification of a novel cholinergic positron emission tomography tracer. Mov Disord. 2019;34(6):924–6.
Kikuchi T, Okamura T, Zhang MR, Irie T. PET probes for imaging brain acetylcholinesterase. J Labelled Comp Radiopharm. 2013;56(3–4):172–9. https://doi.org/10.1002/jlcr.3002.
Nathanson NM. Synthesis, trafficking, and localization of muscarinic acetylcholine receptors. Pharmacol Ther. 2008;119(1):33–43.
Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729.
Tiepolt S, Becker G-A, Wilke S, Cecchin D, Rullmann M, Meyer PM, et al. (+)-[18F] Flubatine as a novel α4β2 nicotinic acetylcholine receptor PET ligand—results of the first-in-human brain imaging application in patients with β-amyloid PET-confirmed Alzheimer’s disease and healthy controls. Eur J Nucl Med Mol Imaging. 2021;48(3):731–46.
Sultzer DL, Lim AC, Gordon HL, Yarns BC, Melrose RJ. Cholinergic receptor binding in unimpaired older adults, mild cognitive impairment, and Alzheimer’s disease dementia. Alzheimers Res Ther. 2022;14(1):25. https://doi.org/10.1186/s13195-021-00954-w.
Kadir A, Darreh-Shori T, Almkvist O, Wall A, Grut M, Strandberg B, et al. PET imaging of the in vivo brain acetylcholinesterase activity and nicotine binding in galantamine-treated patients with AD. Neurobiol Aging. 2008;29(8):1204–17.
Zubieta JK, Koeppe RA, Frey KA, Kilbourn MR, Mangner TJ, Foster NL, et al. Assessment of muscarinic receptor concentrations in aging and Alzheimer disease with [11C] NMPB and PET. Synapse. 2001;39(4):275–87.
Liu AKL, Chang RC-C, Pearce RK, Gentleman SM. Nucleus basalis of meynert revisited: anatomy, history and differential involvement in Alzheimer’s and Parkinson’s disease. Acta Neuropathol. 2015;129(4):527–40.
Mesulam MM. Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. J Comp Neurol. 2013;521(18):4124–44. https://doi.org/10.1002/cne.23415.
Selden NR, Gitelman DR, Salamon-Murayama N, Parrish TB, Mesulam M-M. Trajectories of cholinergic pathways within the cerebral hemispheres of the human brain. Brain J Neurol. 1998;121(12):2249–57.
Richter N, Michel A, Onur OA, Kracht L, Dietlein M, Tittgemeyer M, et al. White matter lesions and the cholinergic deficit in aging and mild cognitive impairment. Neurobiol Aging. 2017;53:27–35. https://doi.org/10.1016/j.neurobiolaging.2017.01.012.
Iranzo A, Fernández-Arcos A, Tolosa E, Serradell M, Molinuevo JL, Valldeoriola F, et al. Neurodegenerative disorder risk in idiopathic REM sleep behavior disorder: study in 174 patients. PLoS One. 2014;9(2):e89741.
Aarsland D, Andersen K, Larsen JP, Lolk A. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol. 2003;60(3):387–92.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–9. https://doi.org/10.1016/j.jalz.2011.03.008.
Mazère J, Lamare F, Allard M, Fernandez P, Mayo W. 123I-Iodobenzovesamicol SPECT imaging of cholinergic systems in dementia with Lewy bodies. J Nucl Med. 2017;58(1):123–8.
Shimada H, Hirano S, Shinotoh H, Aotsuka A, Sato K, Tanaka N, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. 2009;73(4):273–8.
Bohnen NI, Kaufer DI, Ivanco LS, Lopresti B, Koeppe RA, Davis JG, et al. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol. 2003;60(12):1745–8.
Shimada H, Hirano S, Sinotoh H, Ota T, Tanaka N, Sato K, et al. Dementia with Lewy bodies can be well-differentiated from Alzheimer’s disease by measurement of brain acetylcholinesterase activity-a [11C]MP4A PET study. Int J Geriatr Psychiatry. 2015;30(11):1105–13. https://doi.org/10.1002/gps.4338.
Haense C, Kalbe E, Herholz K, Hohmann C, Neumaier B, Krais R, et al. Cholinergic system function and cognition in mild cognitive impairment. Neurobiol Aging. 2012;33(5):867–77. https://doi.org/10.1016/j.neurobiolaging.2010.08.015.
Rinne JO, Kaasinen V, Järvenpää T, Någren K, Roivainen A, Yu M, et al. Brain acetylcholinesterase activity in mild cognitive impairment and early Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74(1):113–5.
Marcone A, Garibotto V, Moresco RM, Florea I, Panzacchi A, Carpinelli A, et al. [11C]-MP4A PET cholinergic measurements in amnestic mild cognitive impairment, probable Alzheimer’s disease, and dementia with Lewy bodies: a Bayesian method and voxel-based analysis. J Alzheimers Dis. 2012;31(2):387–99. https://doi.org/10.3233/JAD-2012-111748.
Kendziorra K, Wolf H, Meyer PM, Barthel H, Hesse S, Becker GA, et al. Decreased cerebral α4β2* nicotinic acetylcholine receptor availability in patients with mild cognitive impairment and Alzheimer’s disease assessed with positron emission tomography. Eur J Nucl Med Mol Imaging. 2011;38(3):515–25.
Sabri O, Kendziorra K, Wolf H, Gertz H-J, Brust P. Acetylcholine receptors in dementia and mild cognitive impairment. Eur J Nucl Med Mol Imaging. 2008;35(1):30–45.
Vogels O, Broere C, Ter Laak H, Ten Donkelaar H, Nieuwenhuys R, Schulte B. Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer’s disease. Neurobiol Aging. 1990;11(1):3–13.
Herholz K, Weisenbach S, Kalbe E, Diederich NJ, Heiss W-D. Cerebral acetylcholine esterase activity in mild cognitive impairment. Neuroreport. 2005;16(13):1431–4.
Wilson RS, Leurgans SE, Boyle PA, Bennett DA. Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment. Arch Neurol. 2011;68(3):351–6.
Grothe M, Heinsen H, Teipel SJ. Atrophy of the cholinergic basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biol Psychiatry. 2012;71(9):805–13. https://doi.org/10.1016/j.biopsych.2011.06.019.
Grothe MJ, Ewers M, Krause B, Heinsen H, Teipel SJ, Alzheimer’s disease neuroimaging Initiative. Basal forebrain atrophy and cortical amyloid deposition in nondemented elderly subjects. Alzheimers Dement. 2014;10(5 Suppl):S344–53. https://doi.org/10.1016/j.jalz.2013.09.011.
Grothe MJ, Heinsen H, Amaro E Jr, Grinberg LT, Teipel SJ. Cognitive correlates of basal forebrain atrophy and associated cortical hypometabolism in mild cognitive impairment. Cereb Cortex. 2016;26(6):2411–26. https://doi.org/10.1093/cercor/bhv062.
Schmitz TW, Nathan Spreng R, Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat Commun. 2016;7:13249. https://doi.org/10.1038/ncomms13249.
Gersel Stokholm M, Iranzo A, Ostergaard K, Serradell M, Otto M, Bacher Svendsen K, et al. Cholinergic denervation in patients with idiopathic rapid eye movement sleep behaviour disorder. Eur J Neurol. 2020;27(4):644–52. https://doi.org/10.1111/ene.14127.
Stokholm MG, Iranzo A, Østergaard K, Serradell M, Otto M, Svendsen KB, et al. Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a case-control study. Lancet Neurol. 2017;16(10):789–96. https://doi.org/10.1016/s1474-4422(17)30173-4.
Braak H, Del Tredici K, Rüb U, De Vos RA, Steur ENJ, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211.
Knudsen K, Fedorova TD, Hansen AK, Sommerauer M, Otto M, Svendsen KB, et al. In-vivo staging of pathology in REM sleep behaviour disorder: a multimodality imaging case-control study. Lancet Neurol. 2018;17:618.
Horsager J, Andersen KB, Knudsen K, Skjærbæk C, Fedorova TD, Okkels N, et al. Brain-first versus body-first Parkinson’s disease: a multimodal imaging case-control study. Brain. 2020;143(10):3077–88.
Giannini G, Calandra-Buonaura G, Asioli GM, Cecere A, Barletta G, Mignani F, et al. The natural history of idiopathic autonomic failure: the IAF-BO cohort study. Neurology. 2018;91(13):e1245–e54. https://doi.org/10.1212/WNL.0000000000006243.
van de Beek M, van Steenoven I, van der Zande J, Porcelijn I, Barkhof F, Stam C, et al. Characterization of symptoms and determinants of disease burden in dementia with Lewy bodies: DEvELOP design and baseline results. Alzheimers Res Ther. 2021;13(1):1–13.
McKeith IG, Ferman TJ, Thomas AJ, Blanc F, Boeve BF, Fujishiro H, et al. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology. 2020;94(17):743–55.
Kantarci K, Nedelska Z, Chen Q, Senjem ML, Schwarz CG, Gunter JL, et al. Longitudinal atrophy in prodromal dementia with Lewy bodies points to cholinergic degeneration. Brain Commun. 2022;4:fcac013.
Grothe MJ, Labrador-Espinosa MA, Jesus S, Macias-Garcia D, Adarmes-Gomez A, Carrillo F, et al. In vivo cholinergic basal forebrain degeneration and cognition in Parkinson’s disease: imaging results from the COPPADIS study. Parkinsonism Relat Disord. 2021;88:68–75. https://doi.org/10.1016/j.parkreldis.2021.05.027.
Pereira JB, Hall S, Jalakas M, Grothe MJ, Strandberg O, Stomrud E, et al. Longitudinal degeneration of the basal forebrain predicts subsequent dementia in Parkinson’s disease. Neurobiol Dis. 2020;139:104831. https://doi.org/10.1016/j.nbd.2020.104831.
Ray NJ, Bradburn S, Murgatroyd C, Toseeb U, Mir P, Kountouriotis GK, et al. In vivo cholinergic basal forebrain atrophy predicts cognitive decline in de novo Parkinson’s disease. Brain. 2018;141(1):165–76.
Schumacher J, Taylor JP, Hamilton CA, Firbank M, Cromarty RA, Donaghy PC, et al. In vivo nucleus basalis of Meynert degeneration in mild cognitive impairment with Lewy bodies. Neuroimage Clin. 2021;30:102604. https://doi.org/10.1016/j.nicl.2021.102604.
Craig CE, Ray NJ, Muller M, Bohnen NI. New developments in cholinergic imaging in Alzheimer and Lewy body disorders. Curr Behav Neurosci Rep. 2020;7(4):278–86. https://doi.org/10.1007/s40473-020-00221-6.
Schumacher J, Ray NJ, Hamilton CA, Donaghy PC, Firbank M, Roberts G, et al. Cholinergic white matter pathways in dementia with Lewy bodies and Alzheimer’s disease. Brain. 2021;145:1773. https://doi.org/10.1093/brain/awab372.
Schmitz TW, Mur M, Aghourian M, Bedard MA, Spreng RN, Alzheimer’s disease Neuroimaging Initiative. Longitudinal Alzheimer’s degeneration reflects the spatial topography of cholinergic basal forebrain projections. Cell Rep. 2018;24(1):38–46. https://doi.org/10.1016/j.celrep.2018.06.001.
Geula C, Nagykery N, Nicholas A, Wu C-K. Cholinergic neuronal and axonal abnormalities are present early in aging and in Alzheimer disease. J Neuropathol Exp Neurol. 2008;67(4):309–18.
Legault-Denis C, Aghourian M, Soucy J-P, Rosa-Neto P, Dagher A, Wickens R, et al. Normal cognition in Parkinson’s disease may involve hippocampal cholinergic compensation: a PET imaging study with [18F]-FEOBV. Parkinsonism Relat Disord. 2021;91:162. https://doi.org/10.1016/j.parkreldis.2021.09.018.
Sanchez-Catasus C, Bohnen NI, D’Cruz N, Muller M. Striatal acetylcholine-dopamine imbalance in Parkinson’s disease: in vivo neuroimaging study with dual-tracer PET and dopaminergic PET-informed correlational tractography. J Nucl Med. 2021;62:545. https://doi.org/10.2967/jnumed.121.261939.
Ikonomovic MD, Abrahamson EE, Isanski BA, Wuu J, Mufson EJ, DeKosky ST. Superior frontal cortex cholinergic axon density in mild cognitive impairment and early Alzheimer disease. Arch Neurol. 2007;64(9):1312–7.
Davis KL, Mohs RC, Marin D, Purohit DP, Perl DP, Lantz M, et al. Cholinergic markers in elderly patients with early signs of Alzheimer disease. JAMA. 1999;281(15):1401–6.
DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002;51(2):145–55.
Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37(6):475–524.
Bedard M-A, Aghourian M, Legault-Denis C, Postuma RB, Soucy J-P, Gagnon J-F, et al. Brain cholinergic alterations in idiopathic REM sleep behaviour disorder: a PET imaging study with 18F-FEOBV. Sleep Med. 2019;58:35–41.
Liu S-Y, Wile DJ, Fu JF, Valerio J, Shahinfard E, McCormick S, et al. The effect of LRRK2 mutations on the cholinergic system in manifest and premanifest stages of Parkinson’s disease: a cross-sectional PET study. Lancet Neurol. 2018;17(4):309–16. https://doi.org/10.1016/s1474-4422(18)30032-2.
Coughlin JM, Rubin LH, Du Y, Rowe SP, Crawford JL, Rosenthal HB, et al. High availability of the alpha7-nicotinic acetylcholine receptor in brains of individuals with mild cognitive impairment: a pilot study using (18)F-ASEM PET. J Nucl Med. 2020;61(3):423–6. https://doi.org/10.2967/jnumed.119.230979.
Ellis J, Villemagne VL, Nathan PJ, Mulligan RS, Gong SJ, Chan JG, et al. Relationship between nicotinic receptors and cognitive function in early Alzheimer’s disease: a 2-[18F] fluoro-A-85380 PET study. Neurobiol Learn Mem. 2008;90(2):404–12.
Colloby SJ, Nathan PJ, Bakker G, Lawson RA, Yarnall AJ, Burn DJ, et al. Spatial covariance of cholinergic muscarinic M1/M4 receptors in Parkinson’s disease. Mov Disord. 2021;36(8):1879–88. https://doi.org/10.1002/mds.28564.
Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282.
Kolisnyk B, Al-Onaizi M, Soreq L, Barbash S, Bekenstein U, Haberman N, et al. Cholinergic surveillance over hippocampal RNA metabolism and Alzheimer’s-like pathology. Cereb Cortex. 2017;27(7):3553–67. https://doi.org/10.1093/cercor/bhw177.
Terrière E, Dempsey MF, Herrmann LL, Tierney KM, Lonie JA, O’Carroll RE, et al. 5-123I-A-85380 binding to the α4β2-nicotinic receptor in mild cognitive impairment. Neurobiol Aging. 2010;31(11):1885–93.
Richter N, Nellessen N, Dronse J, Dillen K, Jacobs HIL, Langen KJ, et al. Spatial distributions of cholinergic impairment and neuronal hypometabolism differ in MCI due to AD. Neuroimage Clin. 2019;24:101978. https://doi.org/10.1016/j.nicl.2019.101978.
Xia Y, Eeles E, Fripp J, Pinsker D, Thomas P, Latter M, et al. Reduced cortical cholinergic innervation measured using [(18)F]-FEOBV PET imaging correlates with cognitive decline in mild cognitive impairment. Neuroimage Clin. 2022;34:102992. https://doi.org/10.1016/j.nicl.2022.102992.
Acknowledgement
To Michel J. Grothe, PhD., Movement Disorders Group, Instituto de Biomedicina de Sevilla (iBiS), for commenting on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Okkels, N., Horsager, J., Pavese, N., Brooks, D.J., Borghammer, P. (2023). Cholinergic Imaging and Dementia. In: Cross, D.J., Mosci, K., Minoshima, S. (eds) Molecular Imaging of Neurodegenerative Disorders. Springer, Cham. https://doi.org/10.1007/978-3-031-35098-6_8
Download citation
DOI: https://doi.org/10.1007/978-3-031-35098-6_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-35097-9
Online ISBN: 978-3-031-35098-6
eBook Packages: MedicineMedicine (R0)