
Abstract
The past few years observed a breakthrough of genome sequences of bacteria of Rhodococcus genus with significant biodegradation abilities. Invaluable knowledge from genome data and their functional analysis can be applied to develop and design strategies for attenuating damages caused by hydrocarbon contamination. With the advent of high-throughput -omic technologies, it is currently possible to utilize the functional properties of diverse catabolic genes, analyze an entire system at the level of molecule (DNA, RNA, protein, and metabolite), simultaneously predict and construct catabolic degradation pathways. In this review, the genes involved in the biodegradation of hydrocarbons and several emerging plasticizer compounds in Rhodococcus strains are described in detail (aliphatic, aromatics, PAH, phthalate, polyethylene, and polyisoprene). The metabolic biodegradation networks predicted from omics-derived data along with the catabolic enzymes exploited in diverse biotechnological and bioremediation applications are characterized.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rhodococcus genus is a heterogeneous group of microorganisms taxonomically associated with the Actinobacteria phylum, one of the largest within the bacteria domain (Ludwig et al. 2012). This phylum represents Gram-positive bacteria with high G-C content, often widespread in aquatic and terrestrial ecosystems. Bacteria of this group exhibit remarkable catabolic versatility and an array of unique enzymatic capabilities that reveal their environmental and biotechnological importance (Barka et al. 2016). For instance, they can degrade a large number of organic and xenobiotic compounds often toxic and recalcitrant categorized in different groups such as aliphatic, aromatic, and polyaromatic hydrocarbons and heterocyclic, halogenated, and nitro-substituted compounds (Martínková et al. 2009). Additional attributes of rhodococci are the modification of steroid, the enantio-selective synthesis, the production of amides from nitriles, the conversion of many plant secondary metabolites found in soil and rhizosphere, such as alkaloids, terpenes, and sterols, and the elimination of sulfur from coal and petroleum products (Larkin et al. 2006; Van Der Geize and Dijkhuizen 2004).
The genus of Rhodococcus represents an important reservoir of physiological and functional diversity. Their metabolites have a biotechnological and industrial significance. Indeed, they can produce carotenoids, wax esters, oils, biosurfactants, bioflocculation agents, and acrylamide (Jones and Goodfellow 2010). For instance, Rhodococcus opacus PD630 was shown to accumulate lipids in its cytosol due to its natural ability to tolerate and to utilize phenolic compounds, which emerges this strain as a promising microbial host for lignocellulose conversion into value-added products (Yoneda et al. 2016).
Moreover, rhodococci were characterized for their ability to thrive harsh environmental conditions and for their biodegradative potential with respect to very toxic and recalcitrant compounds. In this regard, Rhodococcus jostii RHA1 was assessed for its resistance to desiccation and nutrient starvation (LeBlanc et al. 2008; Patrauchan et al. 2012) by evaluating genes’ expression through a proteomic approach while Rhodococcus erythropolis DCL14 was studied for its ability to tolerate various concentrations of water miscible (ethanol, butanol, and dimethylformamide, up to 50% v/v) and water immiscible solvents (dodecane, bis(2-ethylhexyl) phthalate, and toluene, up to 5% v/v) (De Carvalho et al. 2004).
The recent awareness that Rhodococcus genus represents a promising potential in bioremediation, biotransformations, and biocatalysis applications is prompting research and interest from the scientific community in many countries of the world. Indeed, the number of publications and patents on rhodococci has increased during the last several years; in particular, research on the biotechnological exploitation of rhodococci has intensified significantly within the last 37 years, as illustrated by citation analysis (Fig. 1). Genome sequencing is the milestone in unveiling the bioremediation and biotechnological potential in bacteria. In fact, since the advent of genome sequencing two decades ago (Fleischmann et al. 1995), the technical and biological knowledge have increased. Nowadays, sequencing of bacterial genomes is a standard procedure, and the information from tens of thousands of bacterial genomes has had a major impact on our views of the bacterial world (Land et al. 2015) (Table 1). In regard to Rhodococcus genus, several public and private genome projects involving Rhodococcus members are now in progress due to their genome complexity, demonstrated by a multiplicity of catabolic genes and a high genetic redundancy of biosynthetic pathways often connected to a sophisticated regulatory network. Thus, combining functional genome studies with biochemical and physiological knowledge could enhance the exploitation of rhodococcal biotechnological use (Alvarez 2010).

Number of publications on Rhodococcus strains
This review intends to report new insights into the biodegradation potential of Rhodococcus genus based on genome analyses and “-omics” approaches. It summarizes the main characteristics of members of Rhodococcus genus and presents a collection of catabolic functions of different Rhodococcus species involved in the degradation and/or use of several toxic compounds, from hydrocarbons to several emerging contaminants as plasticizer compounds.
Genomics of Rhodococcus and “-omics studies”
This review presents an update collection of Rhodococcus genes and metabolic pathways involved in the biodegradation of main group of toxic compounds basing on “-omic” approaches. The suffix “-omics” mainly indicates studies undertaken on a genome-wide scale, including the genome itself (genomic), RNA transcription (transcriptomic), protein (proteomic), and metabolic products (metabolomic), relying on bioinformatic data.
Particularly, bacterial genomic information could be exploited on at least two levels, to (i) elucidate gene function of unknown enzymes and to (ii) understand the metabolic network of strains endowed with a broad catabolic diversity (Vilchez-Vargas et al. 2010). Hence, genomic analyses constitute an immense source for discovering and exploiting novel biocatalysts.
To date, among the bacterial genome completely sequenced and listed at https://www.ncbi.nlm.nih.gov/genome/browse/?report=2, 236 genome sequences belong to Rhodococcus sp. strains, of which 82 are fully sequenced. Accordingly, almost 80 publications on genome sequencing projects of Rhodococcus strains were released in the last four years in eminent journals, Genome Announcement. Furthermore, transcriptomic, proteomic, and metabolomic projects are in continuous increase.
Omics era elucidated the biotechnological potential of Rhodococcus genus in bioremediation and ecological applications. Since the first completely sequenced genome of R. jostii RHA1, Rhodococcus spp. showed to possess complex genomes that are among the largest available sequenced genomes in prokaryotes. In particular, RHA1 9.7 Mb genome is arranged in four linear replicons: one chromosome and three linear megaplasmids pRHL1, pRHL2, and pRHL3 (McLeod et al. 2006).
Other Rhodococcus strains including Rhodococcus opacus R7 (Di Gennaro et al. 2014), Rhodococcus opacus M213 (Pathak et al. 2016), and R. opacus PD630 (Holder et al. 2011) have been also described for their large genomes (10.1, 9.19, and 9.17 Mbp, respectively) and their numerous plasmids (5 in R7, 2 in M213, and even 9 in PD630).
Moreover, it has been reported that a significant component (6%) of the biotransformations with potential biotechnological application (primarily xenobiotic reactions) recorded in the Minnesota biocatalysis/biodegradation database (http://http://umbbd.msi.umn.edu/) are assigned to Rhodococcus spp. This biotransformation activity in Rhodococcus genus is second only to Pseudomonas and it is achieved through numerous catabolic pathways.
In fact, a large number of enzymatic classes have been predicted to be involved in the degradation of aromatic compounds in RHA1; indeed genomic analyses revealed 203 oxygenases, of which 86 dioxygenases and 88 putative flavoprotein monooxygenases. Additionally, 50 hydroxylases, of which 28 putative cytochrome P450 hydroxylases putatively involved in aromatic and steroids compounds, have been also reported (McLeod et al. 2006).
The immense catabolic diversity shown by the members of this genus lies hidden in the multiplicity of catabolic genes, the high genetic redundancy of biosynthetic pathways, and the sophisticated regulatory networks of their genomes (Alvarez 2010). In this context, Rhodococcus erythropolis PR4 and Rhodococcus sp. DK17 showed a broad degradative abilities towards aliphatic and monoaromatic hydrocarbons, respectively (Laczi et al. 2015; Kim et al. 2002), while R. opacus R7 is capable of degrading the latter two as well as polycyclic aromatic hydrocarbons (Di Gennaro et al. 2010; Zampolli et al. 2014; Orro et al. 2015; Di Canito et al. 2018). Recently, -omic studies ascribed other metabolic traits to Rhodococcus members, such as the degradation of PCBs and steroids (chlorate and cholesterol) in R. jostii RHA1, R. erythropolis PR4, Rhodococcus opacus B4, and Rhodococcus aetherivorans I24 (Goncalves et al. 2006; Swain et al. 2012; Alvarez 2010; Puglisi et al. 2010), the metabolism of isoprene (Crombie et al. 2015), short-chain alkanes, and chloroform (Ciavarelli et al. 2012; Cappelletti et al. 2015), and herbicides (Fang et al. 2016) in Rhodococcus sp. AD45, Rhodococcus aetherivorans BCP1, and Rhodococcus sp. MET, respectively.
Besides, intriguing degradative capabilities towards natural and synthetic polymers, organic sulfur compound as 4,4-dithiodibutyric acid (DTDB), and biodesulfurization of petroleum oil have been reported in Rhodococcus rhodochrous RPK1, Rhodococcus erythropolis MI2, and Rhodococcus erythropolis XP (Watcharakul et al. 2016; Gravouil et al. 2017; Khairy et al. 2016; Tao et al. 2011).
The huge repertoire of catabolic abilities of this genus could be also explained by horizontal gene transfer and gene duplication phenomena. In particular, the high frequency of recombination (homologous and illegitimate recombination) may trigger the flexibility of Rhodococcus genomes to easily acquire new functions (Larkin et al. 2006; Larkin et al. 2010). Indeed, catabolic genes, often identified on linear plasmids, have been found to contribute to degradation pathways together with genes located on the chromosome (Goncalves et al. 2006).
It is worth to mention that many Rhodococcus functional traits were unveiled combining omics data with genome comparative approaches providing insights in terms of phenotypes, metabolic capacities, and cellular response to different stress conditions. This has also contributed to elucidate phylogeny and evolutionary concepts and their relation.
Biodegradation of hydrocarbon compounds
Hydrocarbon compounds are widespread in the environment and originate from natural sources and anthropogenic activities. Large amounts of aromatic compounds were derived from decaying plant material (e.g., from lignin), soil weathering processes, and volcano emissions. Moreover, anthropogenic sources, in particular spillage of petroleum products, discharge of industrial effluents, and transport accidents, release significant amount of hydrocarbons into the environment (Martínková et al. 2009).
Mechanisms underpinning biodegradation of hydrocarbons have been described in several bacterial strains (Pérez-Pantoja et al. 2009; Juarkar et al. 2010). In particular, the aerobic biodegradation of hydrocarbon compounds has been well elucidated in several Rhodococcus strains (Field and Sierra-Alvarez 2004; Alvarez 2010). It was well established that aromatic compounds are catabolized through many upper and/or lower pathways leading to few central intermediates (catechol, protocatechuate, gentisate) (Fig. 2). The generated metabolites are then degraded to compounds involved in the TCA cycle through central pathways. The biodegradation of each class of hydrocarbons requires a specific enzyme due to the high diversity of the molecular structures of these compounds (Abbasian et al. 2016).

Scheme of biodegradation pathways for aromatic hydrocarbon compounds in Rhodococcus
Aliphatic hydrocarbons
Rhodococcus strains are able to oxidize n-alkanes. Basically, the first step of oxidation is mainly performed by an alkane1-monooxygenase encoded by the alkB gene. This gene has been identified in well-known members of the Rhodococcus genus (e.g., R. erythropolis, R. ruber, R. opacus, R. equi, and R. jostii) and can be employed as a marker of this metabolism (Táncsics et al. 2014).
A multiplicity of alkB is reported in several Rhodococcus strains; indeed, it was identified at least four homologous alkane monooxygenase genes (alkB1, alkB2, alkB3, and alkB4) in two different rhodococcal strains (Rhodococcus strains Q15 and NRRLB-16531) (Whyte et al. 2002) and two different alkane hydroxylase systems (alkBa and alkBb) in Rhodococcus ruber SP2B (Amouric et al. 2010). Although the multiplicity of alkB in the genome of Rhodococcus spp. is frequently observed, this phenomenon is not universal for all members of the genus (Táncsics et al. 2014). Indeed, only one copy of alkB gene was identified in the genomes of R. jostii RHA1, (McLeod et al. 2006), R. opacus B4 (Sameshima et al. 2008), R. aetherinovorans BCP1 (Cappelletti et al. 2011; Cappelletti et al. 2013), and R. opacus R7 (Zampolli et al. 2014).
In addition, other alkane hydroxylase systems have been described in Rhodococcus, including (i) P450 cytochrome alkane monooxygenase systems (CYP153) that hydroxylate C5–C16n-alkanes (van Beilen et al. 2006) and (ii) short-alkane monooxygenases that are involved in the oxidation of C1–C6n-alkanes (Cappelletti et al. 2011). Cytochromes P450 (CYPs) are hemoproteins that oxidize a large number of chemical compounds, such as xenobiotics, antibiotics, steroids, terpenes, alkanes, fatty acids, and alkaloids in aerobiosis (Bernhardt and Urlacher 2014; Larkin et al. 2006). In fact, Actinobacteria display several P450 cytochromes distributed over 220 families. For instance, 25, 26, and 45 P450 cytochromes were reported in R. jostii RHA1 (McLeod et al. 2006), R. opacus R7 (Di Gennaro et al. 2014), and R. aetherinovorans BCP1, respectively (Cappelletti et al. 2013). Moreover, a cyp125 gene was identified in R. jostii RHA1 genome. This gene is highly upregulated during growth on cholesterol and encodes a CYP125 that catalyzes the hydroxylation of the C26 atom of sterols through side-chain oxidation leading to the C26-carboxylic acid formation (Rosłoniec et al. 2013). Transcriptomic analysis of this strain suggested that the CYP257A1 (novel cytochrome P450 enzyme family) (Rosłoniec et al. 2009) is involved in sterol metabolism, while protein analysis showed that it N-demethylates the alkaloid substrate, i.e., dextromethorphan (Kulig et al. 2015). Another cytochrome P450 able to hydroxylate octane was described in Corynebacterium sp. 7E1C (in recent literature known as Rhodococcus rhodochrous) (Cardini and Jurtshuk 1970).
Besides, aerobic bacterial metabolism of gaseous and short-chain n-alkanes (ranged from ethane to n-butane) was reported in Rhodococcus genus. For instance, R. aetherivorans BCP1 was described for its ability to grow on gaseous n-alkanes as inducing condition for the co-metabolism with low chlorinated compounds. This strain presents two prm gene clusters: the first, prm gene cluster (prmA,C,B,D genes), is located on the chromosome and implicated in the degradation of propane and butane, and the second, smo gene cluster (soluble di-iron monooxygenase) (smoA,B,D,C genes), is positioned on the pBMC2 plasmid and involved in the degradation of short-chain n-alkanes (Cappelletti et al. 2015).
Aromatic hydrocarbons
The degradation of aromatic hydrocarbons employs the conversion of such substrates into a number of central intermediates, which are consequently subjected to ring cleavage. The reactions of the pathways consist in activation of the aromatic ring through oxygenases generating di- or hydroxylated intermediates. The subsequent ring cleavage of these intermediates can be catalyzed by either intradiol or extradiol dioxygenases (Martínková et al. 2009).
The BTEX system, comprising benzene, toluene, ethylbenzene, and xylene isomers, is listed as priority pollutants. Among BTEX-degrading bacteria, Rhodococcus sp. strain DK17, capable of degrading o-xylene and toluene, has been studied (Kim et al. 2002, 2010). The sequenced genome of this strain revealed that akb gene cluster is involved in the metabolism of these hydrocarbons. This cluster is composed of two genes encoding for the large subunit of an oxygenase (designated akbA1a and akbA1b) followed respectively by other two genes encoding for the small subunit (designated akbA2a and akbA2b). Within this cluster, three other genes akbA3, akbA4, and akbB encoding respectively for a reductase (AkbA4), a ferredoxin component (AkbA3), and a dehydrogenase (AkbB) were identified. Besides, in the same genomic region, akbCDEF genes encoding proteins involved in the ring cleavage (lower pathway) were reported. The latter genes are capable to hydroxylate aromatic compounds (i.e., ethylbenzene) to 2,3- and 3,4-cis-dihydrodiols and able to perform benzylic and aryl hydroxylations on m-xylene (Kim et al. 2002).
Another studied BTEX-degrading bacteria is R. jostii RHA1 (Seto et al. 1995). This strain efficiently assimilates ethylbenzene (ETB), isopropylbenzene (IPB), and biphenyl (BPH) via a common pathway. Transcriptomic analysis revealed that three gene clusters bph, etb1, and etb2, identified on large linear plasmids, are induced by BPH, ETB, and IPB, which indicates that all these hydrocarbons are catabolized by the same enzymes and the genes are controlled by a common regulatory system.
Basically, these compounds are converted to benzoate and 2-hydroxypenta-2,4-dienoate (HPD) in four successive steps catalyzed by (i) four-component biphenyl dioxygenase (ferredoxin reductase, ferredoxin, and two-subunit terminal oxygenase coded by bphA1A2A3A4, etbAa1Ab1, and etbAa2Ab2Ac; (ii) dihydrodiol dehydrogenase (bphB1 and bphB2); (iii) 2,3-dihydroxybiphenyl dioxygenase (bphC1 and etbC); and finally 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoatehydrolase (bphD1). ETB degradation results in propionate and HPD. HPD is further metabolized to pyruvate and acetyl coenzyme A (CoA) through the lower biphenyl pathway divided into three steps (encoded by bphGFE in R. jostii RHA1) (Martínková et al. 2009; Goncalves et al. 2006) while benzoate is metabolized to succinate and acetyl-CoA through the benzoate pathway.
It was found that bphA1A2A3A4 and etbAa1Ab1 genes are under the control of a two-component system encoded by bphS and bphT genes, which encode the sensor histidine kinase and response regulator, respectively. Indeed, the transcriptional induction of the bphA1 promoter by biphenyl, benzene, alkylbenzenes, and chlorinated benzenes requires bphS1T1 genes. In a further study, Takeda et al. showed that the inducing-substrate spectrum of BphS1 includes substrates of BphS2. However, the only difference between bphS1T1 and bphS2T2 systems is that BPH is an inducing substrate only for the bphS1T1 (Takeda et al. 2004). Therefore, they suggested that in the presence of a single-ring aromatic compound, such as ethylbenzene, both bphS1 and bphS2 are responsible for the transcriptional activation of degradation genes in RHA1.
Recently, a particular BTEX degradation pathway was found in R. opacus R7. This strain was found to metabolize o-xylene with the same dioxygenase system previously identified in DK17 (akb) and in RHA1 (etb) strains (allocated on two plasmids, pPDG5 and pPDG2). Furthermore, R7 was shown to possess numerous monooxygenases/phenol hydroxylases included in the o-xylene degradation pathway, highlighting the redundancy of oxygenases genes in this strain (Di Canito et al. 2018).
Phenols
Phenolic compounds (e.g., cresols), released and accumulated in the environment, pose serious health hazards to living organisms (Michałowicz and Duda 2007).
A wide range of phenols are degraded by Rhodococcus strains. The degradation begins by the conversion of phenol compound to catechol by phenol hydroxylase, which is further metabolized through ortho- or meta-cleavage. Basically, catechol 1,2-dioxygenase, an ortho-cleaving enzyme, and catechol 2,3-dioxygenase, a meta-cleaving enzyme, are proteins involved in the β-ketoadipate pathway in which the bacterial degradation of catechol to central metabolic intermediates occurs (Guzik et al. 2011; Szőköl et al. 2014; Zídková et al. 2013). In Rhodococcus erythropolis CCM2595, the phenol hydroxylase enzyme was described; it consists of two subunits encoded by the pheA1 and pheA2 genes clustered with the pheR gene, coding for an AraC-type transcriptional regulator (activator) (Zídková et al. 2013). These genes were found to be induced by phenol and other aromatic compounds such as p-chlorophenol, p-nitrophenol, resorcinol, and p-cresol (Fialova et al. 2003). Moreover, this gene cluster was found in R. jostii RHA1 in two copies, on the chromosome and on the plasmid pRHL1 (McLeod et al. 2006).
Other Rhodococcus strains with phenol degradation ability were reported. For instance, Rhodococcus erythropolis UPV-1 is efficiently capable of degrading PAHs, phenol, and a mixture of o-, m-, and p-cresols (Irvine et al. 2000). In addition, Rhodococcus opacus 1CP is able of degrading p-cresols through 4-methylcatechol and 3-methyl-cis,cis-muconate via ortho-pathway. This strain possesses also another catechol-1,2dioxygenase activity, in addition to dioxygenases with specificities for 4-chlorophenol and p-toluate (Kolomytseva et al. 2007).
Interestingly, an important biotechnological trait attributed to Rhodococcus genus is the capability of accumulating lipids to high extent in their cytosol, up to 80% in R. opacus PD630 (Holder et al. 2011). Recent study employing omics techniques has shown a positive correlation between phenolic compound degradation and lipid production in Rhodococcus rhodochrous. This could explain the capacity of Rhodococcus strains to utilize recalcitrant compounds including complex polyaromatic structure (Shields-Menard et al. 2017).
Polyaromatic hydrocarbons
A wide range of PAH compounds are catabolized by bacteria of Rhodococcus genus, i.e., fluoranthene, pyrene, and chrysene (Xu-Xiang et al. 2006). Basically, PAH degradation initiates in the cytosol by the action of intracellular dioxygenases. Subsequently, PAHs are oxidized to cis-dihydrodiols, which in turn are re-oxidized to aromatic dihydroxy compounds (catechols) and eventually channeled through the ortho or meta-cleavage pathways (Cerniglia 1984; Smith 1990). PAH metabolic pathway has been investigated in many Rhodococcus strains by growing the bacterium on naphthalene (the simplest and most soluble PAH). The latter is considered a model compound to investigate the ability of bacteria to degrade PAHs. For instance, R. opacus R7 degrades naphthalene through the dioxygenation of the aromatic ring, via 1,2-dihydro-1,2-dihydroxynaphthalene, that is further oxidized to salicylate and gentisate (Di Gennaro et al. 2010). In this pathway, two genes, narAa and narAb, encoding respectively for the large (NarAa) and the small (NarAb) components of the naphthalene dioxygenase are involved. These genes are clustered with rub1, rub2, rub1bis, narB, and orf (orf7) genes, encoding for three rubredoxins, a naphthalene dihydrodiol dehydrogenase, and a protein of unknown function, respectively. In addition, two regulatory genes (narR1, narR2) were described in R7 encoding for putative regulatory proteins NarR1 and NarR2, belonging to GntR and NtrC families, accordingly. Nonetheless, no LysR-type regulatory gene was reported. The final aromatic ring cleavage in this strain proceeds through the gentisate pathway, encoded by gen and sal genes (Di Gennaro et al. 2010). Moreover, the lower pathway of naphthalene degradation was found in two copies, the first is on pPDG4 plasmid and the second on pPDG1 plasmid, which lacks genL gene (Orro et al. 2015). Besides, salicylate metabolism in R7 involves genC, genB, and genA genes located upstream the genes associated with gentisate degradation. In particular, genA, genB, genC, genH, genI, and genL encode for salicylate CoA ligase, salicylate CoA synthetase, salicylate hydroxylase, gentisate 1,2-dioxygenase, 3-maleylpyruvate isomerase, and a protein of unknown function, respectively.
Naphthalene degradation pathway has been also described in Rhodococcus sp. NCIMB12038. In particular, nar region is involved in this degradation; it is composed of narA and narB genes which are transcribed in several units. Contrariwise, these genes are transcribed as single units in P200 and P400 strains (Kulakov et al. 2005).
Other genetic determinants that likely participate in naphthalene (and o-xylene) degradation were reported by Martínková et al. (2009). For instance, in Rhodococcus opacus TKN14 (Kulakov et al. 2000), nid gene cluster was identified; it is composed of nidAB, nidE, and nidF genes encoding for the subunits of oxygenase, rubredoxin, and auxiliary protein, respectively (Maruyama et al. 2005). nid genes were found to be induced by o-xylene and are supposed to be involved in the degradation pathways of a wide range of aromatic hydrocarbons in Rhodococcus species.
Thanks to -omics approach a peculiar naphthalene metabolism has been described in R. opacus M213, which occurs via o-phthalate and salicylate pathways. Conversely to naphthalene pathways previously described in other rhodococci, which occur via gentisate pathway in NCIMB 12038, B4 and R7 and through catechol pathway in P200 and P400 strains. The metabolism of naphthalene in M213 occurs via the cyclization of 2-hydroxycinnamic acid or 2-carboxycinnamic acid and eventually oxidizing into phthalic anhydride or phthalic acid. It is particularly interesting that several genes associated with this peculiar biodegradation pathway were found on genome islands (GEIs), such as small and large subunits of naphthalene dioxygenase (NDO), cis-naphthalene dihydrodiol dehydrogenase gene, and a putative naphthalene degradation regulatory protein (Pathak et al. 2016).
Degradation of plasticizer compounds
Plasticizers and their derivatives are among the prime examples of emerging compounds. A large amount of these compounds derives from anthropogenic sources, in particular discharge of industrial activities (Gravouil et al. 2017; Koutny et al. 2006). In this paragraph, we focused on -omic studies related to the degradation of phthalate, phthalate esters (PEs), polyisoprene, and 4,4-dithiodibutyric acid (DTDB) in Rhodococcus strains (Table 2).
Phthalate
After decades of global industrial use as plasticizers, phthalate esters are now recognized as ubiquitous toxicologically significant environmental pollutants (Hara et al. 2007).
Among strains of Rhodococcus genus, R. jostii RHA1 is able to grow on a variety of monoalkyl PEs, including methyl, butyl, hexyl, 2-ethylhexyl phthalates, and terephthalate (Hara et al. 2010). This strain is able to degrade phthalate (PTH), terephthalate, and 4-hydroxybenzoate via protocatechuate ortho-cleavage pathway (Patrauchan et al. 2005). Nevertheless, it can also degrade terephthalate via the catechol branch of the 3-oxoadipate pathway (Hara et al. 2007).
Transcriptomic analysis revealed the metabolic pathway of PTH in RHA1. Two identical functional copies of pad genes carried on two large linear plasmids, pRHL1 and pRHL2, are employed. These genes encode a ring-hydroxylating 3,4-dioxygenase system (PadAaAbAcAd), a dihydrodiol dehydrogenase (PadB), a decarboxylase (PadC), and a regulatory protein (PadR). Moreover, close to pad genes, pat genes were identified, which encode an ABC transport system (PatABCD) and an ester hydrolase (PatE) (Patrauchan et al. 2012) and are highly upregulated in presence of phthalate rather than terephthalate. Interestingly, in a further study involving patB gene knockout, ATP-binding cassette (ABC) transporter was identified necessary for the uptake of phthalate and monoalkyl phthalate esters in RHA1. Moreover, in the same study, it was demonstrated that PatE which is a member of monoalkyl PE hydrolase family is able to specifically hydrolyze monoalkyl PEs to phthalate, rather than terephthalate or other PEs (Hara et al. 2010). In addition, other genes named tpa gene were identified to be putatively involved in terephthalate (TPA) uptake and degradation. The tpa genes encode a transport protein (TpaK), the large and small subunits plus the reductase of a ring-hydroxylating 1,2-dioxygenase system (TpaAaAbB), a dihydrodiol dehydrogenase (TpaC), and a regulatory protein (TpaR). Interestingly, the nearly identical regions of pRHL1 and pRHL2 were also identified to nearly identical regions that are duplicated in pDK2 and pDK3 in Rhodococcus sp. strain DK17 (Choi et al. 2005), which have also been shown to encode PTH and TPA degradation proteins.
Polyethylene
Polyethylene (PE) is a notable synthetic polymer used worldwide, particularly in the packaging industry. PE is stable and tends to accumulate in natural environment, representing by itself the majority of all plastic wastes (Gravouil et al. 2017). The degradation of PE in nature is a complex procedure in which hydrocarbon chains are oxidized into short aliphatic fragments (e.g., complex mixture of alkanes, alkenes, ketones, aldehydes, alcohols, carboxylic acids); subsequently, these fragments are mineralized by specific microorganisms from the environment (Koutny et al. 2006).
Species of Rhodococcus genus have been associated with PE degradation in several studies (Gravouil et al. 2017; Santo et al. 2013). For instance, Sivan and colleagues reported their ability to grow as biofilm on PE-oxidized films. Nevertheless, the molecular mechanism of PE consumption has not been revealed (Sivan et al. 2006).
Recent study based on transcriptomics and lipidomics approaches pointed out the consumption pathways of polyethylene degradation in Rhodococcus ruber C208. In this context, non-oxidized short-chain PE and oxidized PE samples resulting from abiotic treatment induce the pathways related to alkane degradation and β-oxidation of fatty acids in R. ruber C208. This observation revealed these pathways as being the bulk for the consumption of PE. The induction of these pathways could be explained by the presence of short aliphatic fragments in the samples, which could be recognized as natural inducers (Gravouil et al. 2017).
Abiotic treatment of PE (thermo and photo-oxidization) releases mixture of fragments displaying a wide range of lengths and oxidation levels. Fragments of low molecular weight are more likely to be consumed than PE films by Rhodococcus species.
Santo and colleagues have reported the role of laccase enzyme in catalyzing the biotic oxidation of PE and consequently in increasing the amount of metabolized short fragments. Indeed, the authors identified a copper-binding laccase in R. ruber C208, which is able, when overproduced, to induce a reduction of the PE Mw and Mn of 20% and 15%, respectively (Santo et al. 2013). In addition, Gravouil and colleagues identified three close homologs to laccase in the RNA library of R. ruber C208, but interestingly, none of them were shown to be significantly upregulated in the presence of the PE samples, regardless their oxidation levels (Gravouil et al. 2017).
Polyisoprene
Natural rubber is synthetized in large amount by many plants and is used in the production of tires, sealings, and latex gloves. The main component of rubber is the poly(cis1,4-isoprene) (Sharkey 1996). Nonetheless, the economic importance of rubber and the enormous amount of rubber waste materials released into the environment, only a limited number of studies have investigated its biodegradation in bacteria.
Van Hylckama Vlieg and colleagues revealed the genes involved in isoprene utilization in R. sp. AD45. In this strain, the consumption of isoprene includes its oxidation to epoxide, conjugation with glutathione, and dehydrogenation steps. The authors identified the presence of 10 genes, where two of them encode enzymes involved in isoprene degradation: a glutathione S-transferase with activity towards 1,2-epoxy-2-methyl-3-butene (isoI) and a 1-hydroxy-2-glutathionyl-2-methyl-3-butene dehydrogenase (isoH) (Van Hylckama Vlieg et al. 2000). Another glutathione S-transferase gene (isoJ) was reported. The latter acts with 1-chloro-2,4-dinitrobenzene and 3,4-dichloro-1-nitrobenzene, but not with 1,2-epoxy-2-methyl-3-butene. In addition, the authors identified downstream of isoJ, six genes (isoABCDEF) that encode for a putative alkene monooxygenase, and they figured out an amino acid sequence encoded by an additional gene (isoG) which shows high homology with respect to α-methylacyl-coenzyme A racemase (Van Hylckama Vlieg et al. 2000).
Subsequently, Crombie and colleagues have identified the complete set of inducible genes responsible for isoprene degradation. The authors reported the genome of R. sp. AD45. They found that genes involved in isoprene metabolism are concentrated in a small region on a megaplasmid, containing a relatively large number of transposases (Crombie et al. 2015). Their analyses showed high level of transcription of 22 contiguous genes when induced by isoprene or epoxyisoprene, suggesting their involvement in isoprene metabolism, including the monooxygenase, glutathione transferase, dehydrogenase (IsoH), and glutathione biosynthesis genes. In addition, the authors showed that only two aldehyde dehydrogenases, a disulfide reductase, and hypothetical protein are induced by isoprene, suggesting that this cluster may contain all the genes specific to this metabolism. Finally, they demonstrated that the previous identified genes were only induced by isoprene in a strain able to oxidize isoprene to epoxyisoprene, demonstrating that isoprene itself was not the inducing molecule. Hence, they suggested that isoGHIJABCDEF are co-transcribed as an operon with a promoter upstream of isoG (Crombie et al. 2015).
Watcharakul and colleagues identified a gene coding for the latex-clearing protein (lcp) in Rhodococcus rhodochrous strain RPK1, which is putatively involved in the initial oxidative attack on the polyisoprene polymer. This gene was also found in many Actinobacteria and all known rubber-degrading Actinobacteria for which the genome sequences are available (Nanthini et al. 2015; Rose et al. 2005). In addition, a hypothetical lcp gene in the genomes of Rhodococcus rhodochrous strain MTCC11081, Rhodococcus sp. MK3027, and Rhodococcus sp. ARG-BN062 was identified. Particularly, the predicted Lcp amino acid sequence of this gene includes the DUF2236 domain which constitutes the central part of most Lcp proteins. Moreover, by isolating and characterizing the Lcp protein of R. rhodochrous RPK1, the authors pointed out the presence of different Lcp subgroups which vary among them by metal ion dispositions and spectroscopic properties (Watcharakul et al. 2016).
4,4-Dithiodibutyric acid
4,4-Dithiodibutyric acid (DTDB) is a synthetic organic sulfur compound employed in diverse biochemical fields (Imbernon et al. 2015; Jang and Keng 2006). However, the biotechnological significance of this compound is its application in studies for the improvement of polythioester (PTE) synthesis. Although DTDB is non-toxic compound, its biodegradation results in the formation of very harmful metabolite: the poly(4-mercaptobutyric acid) (4MB), which is a PTE precursor substrate (Khairy et al. 2016). Hence, investigating DTDB catabolism in bacteria is necessary for the generation of metabolically engineered PTE production strains. R. erythropolis strain MI2 has been investigated for its rare ability to use DTDB as sole carbon source and electron donor for aerobic growth (Khairy et al. 2016). Genome and proteome analysis of this strain has clarified the DTDB degradation by elucidating the 4MB catabolism pathway. Basically, DTDB is cleaved by the action of Nox (RERY_03780) to 4MB, which is then oxidized forming 4-oxo-4-sulfanylbutyric acid via the LLMMI2 F420-dependent enzyme (RERY_05640) encoded by one of the 126 monooxygenase-encoding genes found in MI2 genome. The ultimate step is supposed to be a desulfurization reaction. Thus, 4-oxo-4-sulfanylbutyric could either be degraded by SQRMI2 (RERY_02710) or the putative desulfhydrase (RERY_06500) which forms succinic acid and volatile hydrogen sulfide. H2S could be oxidized by SQR and then reacts with a free thiol group forming a sulfurhydrate-containing compound. The latter may be oxidized forming thiosulfite by protein encoded by RERY_02720, which is then oxidized by the putative rhodanese-related sulfurtransferase (RERY_02740), giving sulfite and succinic acid as final products. Sulfite could be oxidized to sulfate prior to be transported outside the cell through the transmembrane exporter protein (RERY_02750), while succinic acid is most probably utilized for growth (Khairy et al. 2016). More studies are needed to verify the postulated enzymatic reactions.
Conclusions
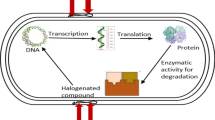
In the last decades, -omics data have increased our understanding of the biological aspects, genetics, and metabolic pathways that could be exploited for biotechnological and bioremediation applications (Khairy et al. 2016; Juwarkar et al. 2010). Traditional approaches of genetics and biochemistry have been applied to Rhodococcus bacteria. Nonetheless, understanding the mechanisms and pathways of hydrocarbons biodegradation have been limited. Nowadays, omics techniques with advanced high-throughput analytical technologies have a special impact in the wake of predicting and constructing metabolic pathways and since the possibility to exploit constructed pathways and gene functions in biodegradation and bacterial biotechnological potential (Fig. 3) (Pathak et al. 2016; Yoneda et al. 2016). With this aim, combination of molecular tools and markers could be useful in bioremediation processes.

Schematic representation of relationship between omics technologies and Rhodococcus biodegradative pathways
Recent achievements of combining genomic, proteomic, and bioinformatics approaches provided insights not only on gene functions, degradation pathways, and molecular mechanisms, but also in the genome-wide gene expression of Rhodococcus bacteria in various environmental conditions (Kim et al. 2018). However, further researches (i.e., cell physiology and regulatory studies, environmental stress response) are required in the field of hydrocarbon degradation in Rhodococcus. The analysis of generated data from currently available sequenced genomes along with genetics and molecular biology approaches will allow to investigate in depth both biodegradation properties and biotechnological potential of these bacteria, which have been recognized as a untapped source of genetic and functional diversity.
References
Abbasian F, Palanisami T, Megharaj M, Naidu R, Lockington R, Ramadass K (2016) Microbial diversity and hydrocarbon degrading gene capacity of a crude oil field soil as determined by metagenomics analysis. Biotechnol Prog 32:638–648
Alvarez HM (2010) Central metabolism of the species of the genus Rhodococcus. In: Alvarez HM (ed) Biology of Rhodococcus. Springer-Verlag, Berlin Heidelber, pp 91–108
Amouric A, Quéméneur M, Grossi V, Liebgott PP, Auria R, Casalot L (2010) Identification of different alkane hydroxylase systems in Rhodococcus ruber strain SP2B, an hexane-degrading actinomycete. J Appl Microbiol 108:1903–1916
Barka EA, Vatsa P, Sanchez L, Gaveau-Vaillant N, Jacquard C, Klenk H-P, Clément C, Ouhdouch Y, van Wezel GP (2016) Taxonomy, physiology, and natural products of Actinobacteria. Microbiol Mol Biol Rev 80:1–43
Bernhardt R, Urlacher VB (2014) Cytochromes P450 as promising catalysts for biotechnological application: chances and limitations. Appl Microbiol Biotechnol 98:6185–6203
Cappelletti M, Fedi S, Frascari D, Ohtake H, Turner RJ, Zannoni D (2011) Analyses of both the alkB gene transcriptional start site and alkB promoter-inducing properties of Rhodococcus sp. strain BCP1 grown on n-alkanes. Appl Environ Microbiol 77:1619–1627
Cappelletti M, Di Gennaro P, D’Ursi P, Orro A, Mezzelani A, Landini M, Fedi S, Frascari D, Presentato A, Zannoni D, Milanesi L (2013) Genome sequence of Rhodococcus sp. strain BCP1, a biodegrader of alkanes and chlorinated compounds. Genome Announc 1:e00657–e00613
Cappelletti M, Presentato A, Milazzo G, Turner RJ, Fedi S, Frascari D, Zannoni D (2015) Growth of Rhodococcus sp. strain BCP1 on gaseous n-alkanes: new metabolic insights and transcriptional analysis of two soluble di-iron monooxygenase genes. Front Microbiol https://doi.org/10.3389/fmicb.2015.00393
Cardini G, Jurtshuk P (1970) The enzymatic hydroxylation of n-octane by Corynebacterium sp. strain 7E1C. J Biol Chem 245:2789–2796
Cerniglia CE (1984) Microbial metabolism of polycyclic aromatic hydrocarbons. Adv Appl Microbiol 30:31–71
Choi KY, Kim D, Sul WJ, Chae J-C, Zylstra GJ, Kim YM, Kim E (2005) Molecular and biochemical analysis of phthalate and terephthalate degradation by Rhodococcus sp. strain DK17. FEMS Microbiol Lett 252:207–213
Ciavarelli R, Cappelletti M, Fedi S, Pinelli D, Frascari D (2012) Chloroform aerobic cometabolism by butane-growing Rhodococcus aetherivorans BCP1 in continuous-flow biofilm reactors. Bioprocess Biosyst Eng 35:667–681
Crombie AT, El Khawand M, Rhodius VA, Fengler KA, Miller MC, Whited GM, Mcgenity TJ, Murrell JC (2015) Regulation of plasmid-encoded isoprene metabolism in Rhodococcus, a representative of an important link in the global isoprene cycle. Environ Microbiol 17:3314–3329
De Carvalho CCCR, Da Cruz AARL, Pons MN, Pinheiro HMRV, Cabral JMS, Da Fonseca MMR, Ferreira BS, Fernandes P (2004) Mycobacterium sp., Rhodococcus erythropolis, and Pseudomonas putida behavior in the presence of organic solvents. Microsc Res Tech 64:215–222
Di Canito A, Zampolli J, Orro A, D’Ursi P, Milanesi L, Sello G, Steinbüchel A, Di Gennaro P (2018) Genome-based analysis for the identification of genes involved in o-xylene degradation in Rhodococcus opacus R7. BMC Genomics 19:587
Di Gennaro P, Terreni P, Masi G, Botti S, De Ferra F, Bestetti G (2010) Identification and characterization of genes involved in naphthalene degradation in Rhodococcus opacus R7. Appl Microbiol Biotechnol 87:297–308
Di Gennaro P, Zampolli J, Presti I, Cappelletti M, D’Ursi P, Orro A, Mezzelani A, Milanesi L (2014) Genome sequence of Rhodococcus opacus strain R7, a biodegrader of mono- and polycyclic aromatic hydrocarbons. Genome Announc 2:e00827
Fang Y, Du Y, Hu L, Xu J, Long Y, Shen D (2016) Effects of sulfur-metabolizing bacterial community diversity on H2S emission behavior in landfills with different operation modes. Biodegradation 27:237–246
Fialova A, Cejkova A, Masak J, Jirku V (2003) Comparison of yeast (Candida maltosa) and bacterial (Rhodococcus erythropolis) phenol hydroxylase activity and its properties in the phenolic compounds biodegradation. Commun Agric Appl Biol Sci 68:155–158
Field JA, Sierra-Alvarez R (2004) Biodegradability of chlorinated solvents and related chlorinated aliphatic compounds. Rev Environ Sci Biotechnol 3:185–254
Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, McKenney K, Sutton G, FitzHugh W, Fields C, Gocayne JD, Scott J, Shirley R, Liu LI, Glodek A, Kelley JM, Weidman JF, Phillips CA, Spriggs T, Hedblom E, Cotton MD, Utterback TR, Hanna MC, Nguyen DT, Saudek DM, Brandon RC, Fine LD, Fritchman JL, Fuhrmann JL, Geoghagen NSM, Gnehm CL, McDonald LA, Small KV, Fraser CM, Smith HO, Venter JC (1995) Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496–512
Goncalves ER, Hara H, Miyazawa D, Davies JE, Eltis LD, Mohn WW (2006) Transcriptomic assessment of isozymes in the biphenyl pathway of Rhodococcus sp. strain RHA1. Appl Environ Microbiol 72:6183–6193
Gravouil K, Ferru-Clément R, Colas S, Helye R, Kadri L, Bourdeau L, Moumen B, Mercier A, Ferreira T (2017) Transcriptomics and lipidomics of the environmental strain Rhodococcus ruber point out consumption pathways and potential metabolic bottlenecks for polyethylene degradation. Environ Sci Technol 51:5172–5181
Guzik U, Greń I, Hupert-Kocurek K, Wojcieszyńska D (2011) Catechol 1,2-dioxygenase from the new aromatic compounds-degrading Pseudomonas putida strain N6. Int Biodeterior Biodegrad 65:504–512
Hara H, Eltis LD, Davies JE, Mohn WW (2007) Transcriptomic analysis reveals a bifurcated terephthalate degradation pathway in Rhodococcus sp. strain RHA1. J Bacteriol 189:1641–1647
Hara H, Stewart GR, Mohn WW (2010) Involvement of a novel ABC transporter and monoalkyl phthalate ester hydrolase in phthalate ester catabolism by Rhodococcus jostii RHA1. Appl Environ Microbiol 76:1516–1523
Holder JW, Ulrich JC, DeBono AC, Godfrey PA, Desjardins CA, Zucker J, Zeng Q, Leach ALB, Ghiviriga I, Dancel C, Abeel T, Gevers D, Kodira CD, Desany B, Affourtit JP, Birren BW, Sinskey AJ (2011) Comparative and functional genomics of Rhodococcus opacus PD630 for biofuels development. PLoS Genet 7:e1002219
Imbernon L, Oikonomou EK, Norvez S, Leibler L (2015) Chemically crosslinked yet reprocess able epoxidized natural rubber via thermo-activated disulfide rearrangements. Polym Chem 6:4271–4278
Irvine VA, Kulakov LA, Larkin MJ (2000) The diversity of extradiol dioxygenase “edo” genes in cresol degrading rhodococci from a creosote-contaminated site that express a wide range of degradative abilities. Antonie van Leeuwenhoek, Int J Gen Mol Microbiol 78:341–352
Jang L, Keng H (2006) Development and characterization of as a monolayer for protein chips. Sens Mater 18:367–380
Jones A, Goodfellow M (2010) Genus II. Rhodococcus (Zopf 1891) emend Goodfellow et al. 1998. In: Bergey’s Manual of Systematic Bacteriology, vol 4, 2nd edn. Springer, Berlin, pp 1–65
Juwarkar AA, Singh SK, Mudhoo A (2010) A comprehensive overview of elements in bioremediation. Rev Environ Sci Biotechnol 9:215–288
Khairy H, Meinert C, Wübbeler JH, Poehlein A, Daniel R, Voigt B, Riedel K, Steinbüchel A (2016) Genome and proteome analysis of Rhodococcus erythropolis MI2: elucidation of the 4,4′-dithiodibutyric acid catabolism. PLoS One 11:e0167539
Kim D, Kim Y-S, Kim S-K, Kim SW, Zylstra GJ, Kim YM, Kim E (2002) Monocyclic aromatic hydrocarbon degradation by Rhodococcus sp. strain DK17. Appl Environ Microbiol 68:3270–3278
Kim SH, Han HY, Lee YJ, Kim CW, Yang JW (2010) Effect of electrokinetic remediation on indigenous microbial activity and community within diesel contaminated soil. Sci Total Environ 408:3162–3168
Kim D, Choi KY, Yoo M, Zylstra GJ, Kim E (2018) Biotechnological potential of Rhodococcus biodegradative pathways. J Microbiol Biotechnol 28:1037–1051
Kolomytseva MP, Baskunov BP, Golovleva LA (2007) Intradiol pathway of para-cresol conversion by Rhodococcus opacus 1CP. Biotechnol J 2:886–893
Koutny M, Sancelme M, Dabin C, Pichon N, Delort AM, Lemaire J (2006) Acquired biodegradability of polyethylenes containing pro-oxidant additives. Polym Degrad Stab 91:1495–1503
Kulakov LA, Allen CCR, Lipscomb DA, Larkin MJ (2000) Cloning and characterization of a novel cis-naphthalene dihydrodiol dehydrogenase gene (narB) from Rhodococcus sp. NCIMB12038. FEMS Microbiol Lett 182:327–331
Kulakov LA, Chen S, Allen CCR, Larkin MJ (2005) Web-type evolution of Rhodococcus gene clusters associated with utilization of naphthalene. Appl Environ Microbiol 71:1754–1764
Kulig JK, Spandolf C, Hyde R, Ruzzini AC, Eltis LD, Grönberg G, Hayes MA, Grogan G (2015) A P450 fusion library of heme domains from Rhodococcus jostii RHA1 and its evaluation for the biotransformation of drug molecules. Bioorganic Med Chem 23:5603–5609
Laczi K, Kis Á, Horváth B, Maróti G, Hegedüs B, Perei K, Rákhely G (2015) Metabolic responses of Rhodococcus erythropolis PR4 grown on diesel oil and various hydrocarbons. Appl Microbiol Biotechnol 99:9745–9759
Land M, Hauser L, Jun SR, Nookaew I, Leuze MR, Ahn TH, Karpinets T, Lund O, Kora G, Wassenaar T, Poudel S, Ussery DW (2015) Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics 15:141–161
Larkin MJ, Kulakov LA, Allen CC (2006) Biodegradation by members of the genus Rhodococcus: biochemistry, physiology, and genetic adaptation. Adv Appl Microbiol 59:1–29
Larkin MJ, Kulakov LA, Allen CC (2010) Genomes and plasmids in Rhodococcus. In: Alvarez HM (ed) Biology of Rhodococcus. Springer, Berlin, pp 73–90
LeBlanc JC, Gonçalves ER, Mohn WW (2008) Global response to desiccation stress in the soil actinomycete Rhodococcus jostii RHA1. Appl Environ Microbiol 74:2627–2636
Ludwig W, Euzéby J, Schumann P, Buss HJ, Trujillo ME, Kämpfer P, Whiteman WB (2012) Road map of the phylum Actinobacteria. In: Goodfellow M, Kämpfer P, Busse HJ, Trujillo ME, Suzuki KI, Ludwig W, Whitman WB (eds) Bergey’s manual of systematic bacteriology. Springer-Verlag, New York, pp 1–28
Martínková L, Uhnáková B, Pátek M, Nešvera J, Křen V (2009) Biodegradation potential of the genus Rhodococcus. Environ Int 35:162–177
Maruyama T, Ishikura M, Taki H, Shindo K, Kasai H, Haga M, Inomata Y, Misawa N (2005) Isolation and characterization of o-xylene oxygenase genes from Rhodococcus opacus TKN14. Appl Environ Microbiol 71:7705–7715
McLeod MP, Warren RL, Hsiao WWL, Araki N, Myhre M, Fernandes C, Miyazawa D, Wong W, Lillquist AL, Wang D, Dosanjh M, Hara H, Petrescu A, Morin RD, Yang G, Stott JM, Schein JE, Shin H, Smailus D, Siddiqui AS, Marra MA, Jones SJM, Holt R, Brinkman FSL, Miyauchi K, Fukuda M, Davies JE, Mohn WW, Eltis LD (2006) The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc Natl Acad Sci USA 103:15582–15587
Michałowicz J, Duda W (2007) Phenols-sources and toxicity. Polish J Environ Stud 6:347–362
Nanthini J, Chia KH, Thottathil GP, Taylor TD, Kondo S, Najimudin N, Baybayane P, Singh S, Sudesh K (2015) Complete genome sequence of Streptomyces sp. strain CFMR 7, a natural rubber degrading actinomycete isolated from Penang, Malaysia. J Biotechnol 214:47–48
Orro A, Cappelletti M, D’Ursi P, Milanesi L, Di Canito A, Zampolli J, Collina E, Decorosi F, Viti C, Fedi S, Presentato A, Zannoni D, Di Gennaro P (2015) Genome and phenotype microarray analyses of Rhodococcus sp. BCP1 and Rhodococcus opacus R7: genetic determinants and metabolic abilities with environmental relevance. PLoS One 10:e0139467
Pathak A, Chauhan A, Blom J, Indest KJ, Jung CM, Stothard P, Bera G, Green SJ, Ogram A (2016) Comparative genomics and metabolic analysis reveals peculiar characteristics of Rhodococcus opacus strain M213 particularly for naphthalene degradation. PLoS One 11:e0161032
Patrauchan MA, Florizone C, Dosanjh M, Mohn WW, Davies J, Eltis LD (2005) Catabolism of benzoate and phthalate in Rhodococcus sp. strain RHA1: redundancies and convergence. J Bacteriol 187:4050–4063
Patrauchan MA, Miyazawa D, LeBlanc JC, Aiga C, Florizone C, Dosanjh M, Davies J, Eltis LD, Mohn WW (2012) Proteomic analysis of survival of Rhodococcus jostii RHA1 during carbon starvation. Appl Environ Microbiol 78:6714–6725
Pérez-Pantoja D, Donoso R, Junca H, González D, Pieper H (2009) Phylogenomics of aerobic bacterial degradation of aromatics. Handbook of hydrocarbon and lipid microbiology, In, pp 1355–1397
Puglisi E, Cahill MJ, Lessard PA, Capri E, Sinskey AJ, Archer JAC, Boccazzi P (2010) Transcriptional response of Rhodococcus aetherivorans I24 to polychlorinated biphenyl-contaminated sediments. Microb Ecology 60:505–515
Rose K, Tenberge KB, Steinbüchel A (2005) Identification and characterization of genes from Streptomyces sp. strain K30 responsible for clear zone formation on natural rubber latex and poly(cis-1,4-isoprene) rubber degradation. Biomacromolecules 6:180–188
Rosłoniec KZ, Wilbrink MH, Capyk JK, Mohn WW, Ostendorf M, Van Der Geize R, Dijkhuizen L, Eltis LD (2009) Cytochrome P450 125 (CYP125) catalyses C26-hydroxylation to initiate sterol side-chain degradation in Rhodococcus jostii RHA1. Mol Microbiol 74:1031–1043
Rosłoniec KZ, van der Geize R, Dijkhuizen L (2013) CYP257A1 of Rhodococcus jostii strain RHA1 represents a novel cytochrome P450 enzyme family with demethylase activity and a putative physiological role in sterol metabolism. Dissertation, University of Groningen
Sameshima Y, Honda K, Kato J, Omasa T, Ohtake H (2008) Expression of Rhodococcus opacus alkB genes in anhydrous organic solvents. J Biosci Bioeng 106:199–203
Santo M, Weitsman R, Sivan A (2013) The role of the copper-binding enzyme-laccase-in the biodegradation of polyethylene by the actinomycete Rhodococcus ruber. Int Biodeterior Biodegrad 84:204–210
Sekine M, Tanikawa S, Omata S, Saito M, Fujisawa T, Tsukatani N, Tajima T, Sekigawa T, Kosugi H, Matsuo Y, Nishiko R, Imamura K, Ito M, Narita H, Tago S, Fujita N, Harayama S (2006) Sequence analysis of three plasmids harboured in Rhodococcus erythropolis strain PR4. Environ Microbiol 8 (2):334–346
Seto M, Kimbara K, Shimura M, Hatta T, Fukuda M, Yano K (1995) A novel transformation of polychlorinated biphenyls by Rhodococcus sp. strain RHA1. Appl Environ Microbiol 61:3353–3358
Sharkey TD (1996) Isoprene synthesis by plants and animals. Endeavour 20:74–78
Shields-Menard SA, AmirSadeghi M, Green M, Womack E, Sparks DL, Blake J, Edelmann M, Ding X, Sukhbaatar B, Hernandez R, Donaldson JR, French T (2017) The effects of model aromatic lignin compounds on growth and lipid accumulation of Rhodococcus rhodochrous. Int Biodeterior Biodegrad 121:79–90
Sivan A, Szanto M, Pavlov V (2006) Biofilm development of the polyethylene-degrading bacterium Rhodococcus ruber. Appl Microbiol Biotechnol 72:346–352
Smith MR (1990) The biodegradation of aromatic hydrocarbons by bacteria. Biodegradation 1:191–206
Swain K, Casabon I, Eltis LD, Mohn WW (2012) Two transporters essential for reassimilation of novel cholate metabolites by Rhodococcus jostii RHA1. J Bacteriol 194:6720–6727
Szőköl J, Rucká L, Šimčíková M, Halada P, Nešvera J, Pátek M (2014) Induction and carbon catabolite repression of phenol degradation genes in Rhodococcus erythropolis and Rhodococcus jostii. Appl Microbiol Biotechnol 98:8267–8279
Takeda H, Yamada A, Miyauchi K, Masai E, Fukuda M (2004) Characterization of transcriptional regulatory genes for biphenyl degradation in Rhodococcus sp. strain RHA1. J Bacteriol 186:2134–2146
Táncsics A, Benedek T, Farkas M, Máthé I, Márialigeti K, Szoboszlay S, Kukolya J, Kriszt B (2014) Sequence analysis of 16S rRNA, gyrB and catA genes and DNA-DNA hybridization reveal that Rhodococcus jialingiae is a later synonym of Rhodococcus qingshengii. Int J Syst Evol Microbiol 64:298–301
Tao F, Zhao P, Li Q, Su F, Yu B, Ma C, Tang H, Tai C, Wu G, Xu P (2011) Genome sequence of Rhodococcus erythropolis XP, a biodesulfurizing bacterium with industrial potential. J Bacteriol 193:6422–6423
Van Beilen JB, Funhoff EG, Van Loon A, Just A, Kaysser L, Bouza M, Holtackers R, Röthlisberger M, Li Z, Witholt B (2006) Cytochrome P450 alkane hydroxylases of the CYP153 family are common in alkane-degrading eubacteria lacking integral membrane alkane hydroxylases. Appl Environ Microbiol 72:59–65
Van Der Geize R, Dijkhuizen L (2004) Harnessing the catabolic diversity of rhodococci for environmental and biotechnological applications. Curr Opin Microbiol 7:255–261
Van Hylckama Vlieg JET, Leemhuis H, LutjeSpelberg JH, Janssen DB (2000) Characterization of the gene cluster involved in isoprene metabolism in Rhodococcus sp. strain AD45. J Bacteriol 182:1956–1963
Vilchez-Vargas R, Junca H, Pieper DH (2010) Metabolic networks, microbial ecology and “omics” technologies: towards understanding in situ biodegradation processes. Environ Microbiol 12:3089–3104
Watcharakul S, Röther W, Birke J, Umsakul K, Hodgson B, Jendrossek D (2016) Biochemical and spectroscopic characterization of purified latex clearing protein (Lcp) from newly isolated rubber degrading Rhodococcus rhodochrous strain RPK1 reveals novel properties of Lcp. BMC Microbiol 16:92
Whyte LG, Smits THM, Labbé D, Witholt B, Greer CW, Van Beilen JB (2002) Gene cloning and characterization of multiple alkane hydroxylase systems in Rhodococcus strains Q15 and NRRL B-16531. Appl Environ Microbiol 68:5933–5942
Xu-Xiang Z, Shu-Pei C, Cheng-Jun Z, Shi-Lei S (2006) Microbial PAH-degradation in soil: degradation pathways and contributing factors. Pedosphere 16:555–565
Yoneda A, Henson WR, Goldner NK, Park KJ, Forsberg KJ, Kim SJ, Pesesky MW, Foston M, Dantas G, Moon TS (2016) Comparative transcriptomics elucidates adaptive phenol tolerance and utilization in lipid-accumulating Rhodococcus opacus PD630. Nucleic Acids Res 44:2240–2254
Yoo M, Kim D, Choi KY, Chae JC, Zylstra GJ, Kim E (2012) Draft genome sequence and comparative analysis of the superb aromatic-hydrocarbon degrader Rhodococcus sp. strain DK17. J Bacteriol 194(16):4440
Zampolli J, Collina E, Lasagni M, Di Gennaro P (2014) Biodegradation of variable-chain-length n-alkanes in Rhodococcus opacus R7 and the involvement of an alkane hydroxylase system in the metabolism. AMB Express 4:73
Zídková L, Szoköl J, Rucká L, Pátek M, Nešvera J (2013) Biodegradation of phenol using recombinant plasmid-carrying Rhodococcus erythropolis strains. Int Biodeterior Biodegrad 84:179–184
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in this study were in compliance with ethical standards. This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zampolli, J., Zeaiter, Z., Di Canito, A. et al. Genome analysis and -omics approaches provide new insights into the biodegradation potential of Rhodococcus. Appl Microbiol Biotechnol 103, 1069–1080 (2019). https://doi.org/10.1007/s00253-018-9539-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-9539-7