
Abstract
Rhodococcus erythropolis CCM2595 is able to efficiently utilize phenol and other aromatic compounds. We cloned and sequenced its complete gene cluster — catA, catB, catC, catR, pheR, pheA2, pheA1 — involved in the ortho-cleavage pathway of phenol. The activity of the key enzyme of the phenol degradation pathway, two-component phenol hydroxylase, was found to be induced by phenol. When both phenol and succinate were present in the medium, phenol hydroxylase activity decreased substantially. To analyze the regulation of phenol degradation at the transcriptional level, the transcriptional fusions of the divergently oriented promoters PpheA2 and PpheR with the gfpuv reporter gene were constructed. The promoters driving expression of the genes of the pheR–pheA2pheA1 cluster were localized by determining the respective transcriptional start points. Measurements of GFP fluorescence as well as quantitative RT-PCR revealed that expression of the phe genes is induced by phenol at the transcriptional level. The transcription of pheA2A1 and pheR was repressed by succinate, whereas no repression by glucose or glycerol was observed. Activation of the R. erythropolis CCM2595 pheA2 promoter by PheR, an AraC-type transcriptional regulator, was demonstrated by overexpression of the pheR gene. Analysis of the transcriptional regulation of two similar phe clusters from R. jostii RHA1 by various substrates showed that the type of carbon catabolite repression and the temporal transcriptional pattern during cultivation are different in each of the three phe clusters analyzed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Efficient aerobic biodegradation of phenolic compounds has become an attractive alternative to physical and chemical methods to remove these toxic pollutants from the environment (Martínková et al. 2009). A number of phenol-utilizing bacterial strains were isolated and enzymes involved in the respective catabolic pathways were characterized. In the first step of phenol degradation, the aromatic ring is hydroxylated by phenol hydroxylase (PHH, phenol 2-monooxygenase EC 1.14.13.7). The catechol formed in this reaction is further degraded via an ortho-cleavage or meta-cleavage pathway to central metabolism intermediates. In many bacteria, phenol hydroxylases are the rate-limiting enzymes of phenol catabolism (Shingler 2003). Three different types of PHHs were described in terms of a number of protein subunits. Most phenol-degrading strains of the genera Pseudomonas, Acinetobacter, Comamonas and Burkholderia contain multicomponent PHHs (mPHs) encoded by six genes (Shingler 2003). Various Pseudomonas strains (Putrinš et al. 2007) and some Gram-positive bacteria (e.g., Bacillus stearothermophillus; Kim and Oriel 1995) contain a single-component PHH. Two-component flavin-dependent monooxygenases were reported in two Bacillus strains (Duffner and Müller 1998; Kirchner et al. 2003). A similar two-component PHH was described in R. erythropolis UVP-1 (Saa et al. 2009). Despite the significance of Rhodococcus strains as efficient phenol degraders in biotechnological processes (Paisio et al. 2012; Prieto et al. 2002), no detailed analysis of phenol catabolism and its genetic control has been performed in rhodococci so far.
Expression of the phenol degradation genes was found to be controlled in bacteria by a complex system including specific and global regulatory mechanisms (Shingler 2003). The effector compound, which is either phenol or an intermediate of its catabolic pathway (e.g., cis,cis-muconate) provides the specificity of gene expression induction. The respective protein regulators, which mediate the activation or repression of the genes, are usually encoded by genes adjacent to the catabolic genes. Expression of the dmpKLMNOP operon coding for mPH in Pseudomonas sp. CF600 is strictly controlled by the transcriptional activator DmpR (NtrC family) (Shingler et al. 1993). Genes encoding mPH in Comamonas testosteroni R5 were repressed by the PhcS regulator (GntR family) only in the absence of phenol, whereas this repressor did not affect the gene expression when the cells were grown on phenol. The PhcR activator (AraC/XylS family) was found to be responsible for the gratuitous expression of phenol-metabolizing genes in this strain (Teramoto et al. 2001). The first transcriptional regulator of the phenol metabolism in gram-positive bacteria was found in Geobacillus stearothermophilus, which degrades phenol via the meta-pathway; however, the mechanism of its action is still unknown (Omokoko et al. 2008). No data on the regulation of genes encoding two-component PHHs in rhodococci are available.
Expression of most of the genes involved in the catabolism of aromatic compounds including phenol is affected by the presence of alternative substrates in the growth media. Glucose, acetate or tricarboxylic acid cycle intermediates can inhibit the induction of the respective enzymes. A number of operons involved in the pathways for the catabolism of various aromatic compounds which are funneled into the β-ketoadipate pathway in Acinetobacter baylyi are controlled by carbon catabolite repression (CCR) mediated by succinate plus acetate. The global regulator Crc is involved in the repression of these operons (Bleichrodt et al. 2010). A negative control of phenol degradation genes by glucose, pyruvate, citrate and succinate was proved in P. putida. Inhibition of the activating function of the PhlR protein is involved in this regulation (Müller et al. 1996). The utilization hierarchy of carbon and energy sources may be generally considered to be a consequence of CCR although the underlying mechanisms, which are mostly unknown, may differ considerably in various bacteria.
Diverse strains of the genus Rhodococcus are able to degrade a wide range of aromatic compounds and persistent xenobiotics (Larkin et al. 2005; Martínková et al. 2009). These bacteria are able to survive in unfavorable conditions and possess various biotechnologically important capacities (Hernandez et al. 2008). The developed tools for gene manipulations in rhodococci currently allow bioengineers to construct new degrader strains suitable for use in biotechnological processes (van der Geize and Dijkhuizen 2004; Veselý et al. 2003).
We cloned and sequenced the gene cluster catABC–catR–pheR–pheA2A1 involved in phenol degradation in R. erythropolis CCM2595. We proved that the catABC operon is repressed by CatR, a transcriptional regulator of the IclR family. Expression of catR was found to be constitutive (Veselý et al. 2007). The R. erythropolis CCM2595 genes of the phenol degradation gene cluster were used for the development of recombinant strains efficiently degrading phenol in phenol-containing wastewater (Zídková et al. 2013). In the present work, we analyzed expression of the genes pheA2A1, encoding a two-component PHH, and pheR, encoding an AraC-type transcriptional regulator, that are involved in the first step of phenol degradation in R. erythropolis CCM2595 and R. jostii RHA1.To our knowledge, this is the first analysis of the transcriptional regulation of genes encoding two-component phenol hydroxylase.
Materials and methods
Strains, plasmids and growth conditions
The strains Rhodococcus erythropolis CCM2595 (Veselý et al. 2003) and R. jostii RHA1 (McLeod et al. 2006) were the sources of phenol degradation genes and regulatory pheR–pheA2 sequences. Corynebacterium glutamicum RES167 (Dusch et al. 1999) was used as a heterologous host lacking the PheR regulator. Escherichia coli DH5α (Hanahan 1985) was used for cloning. The plasmids used are listed in Table 1. Rhodococcus strains were cultivated at 25 °C in complete 2xYT medium (Sambrook and Russel 2001) or in minimal BSM basal salt medium (Veselý et al. 2007) with various substrates: phenol (3.5 mM), protocatechuic acid (3.5 mM), benzoate (3.5 mM), p-hydroxybenzoate (3.5 mM), glucose (10 mM), succinate (12 mM) and glycerol (20 mM). E. coli was grown in LB medium at 37 °C. When appropriate, kanamycin was added to the media for E. coli (30 μg/ml), C. glutamicum (30 μg/ml) and Rhodococcus (200 μg/ml) and tetracycline to the media for C. glutamicum (10 μg/ml).
DNA techniques
DNA isolation from E. coli, transformation of E. coli, polymerase chain reaction (PCR), DNA cloning and DNA analysis were performed following standard procedures (Sambrook and Russel 2001). Plasmid DNA from Rhodococcus strains was isolated as described previously (Veselý et al. 2007) and transformation of R. erythropolis, R. jostii and C. glutamicum was done by electroporation (Veselý et al. 2003).
RNA isolation and primer extension analysis
R. erythropolis CCM2595 and R. jostii RHA1 cells were cultivated in the BSM medium with phenol (3.5 mM) at 25 °C, harvested at OD600 = 1 (16 to 18 h), washed with PBS buffer and frozen at −70 °C. The cells were disintegrated in FastPrep FP120 (BIO101) (6 × 20 s, speed 6.0) using glass beads (size 10). The cell debris was removed by centrifugation and total RNA was isolated from 4 ml of extract using the High Pure RNA Isolation Kit (Roche). Primer extension analysis was done as described previously (Pátek et al. 2003). Reverse transcription was carried out using 50–60 μg of RNA, SuperScript III transcriptase (Invitrogen) and the fluorescein-labeled primer GFP1 (CTAATTCAACAAGAATTGGGAC) or GFP5 (GTTCTTCTCCTTTACTCATTT) complementary to the gfpuv sequence in the vector pEPR1. The reverse transcript was run on polyacrylamide gel electrophoresis in an automatic ALF DNA Sequencer (Pharmacia Biotech) with the DNA sequencing reactions generated using the same labeled primer.
Quantitative real-time RT-PCR
Total RNA was treated with TURBO DNase (Ambion) to remove DNA contamination and purified with the RNeasy kit (Qiagen). Reverse transcription was performed with SuperScript III Reverse Transcriptase (Invitrogen) in a 20-μl reaction mixture containing 2 μg of total RNA, 4 μl 5× First-Strand buffer, 1 μl dNTP mix (10 mM), 1 μl random hexamer primers (100 ng/μl) and 1 μl RNase OUT Recombinant Ribonuclease Inhibitor (40 U/μl). The reaction was run for 1 h at 50 °C and was stopped by heat inactivation of the enzyme (15 min at 70 °C).
Real-time PCR was carried out in a 25-μl reaction volume containing 2 μl cDNA,1 μl of each of the forward and reverse primers (400 nM), 12.5 μl 2× SYBR Green Supermix (Bio-Rad) in iQ5 Thermocycler (Bio-Rad). Each reaction was done in triplicate. The primers used are listed in Table 2. Cycling conditions: predenaturation at 95 °C for 5 min, 40 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 20 s and elongation at 72 °C for 20 s. After qPCR completion, melting curves were generated between 55 °C and 95 °C with 0.5 °C increments.
The ∆∆C t method was used for calculating the amount of transcripts with dinB (coding for DNA polymerase IV in R. erythropolis CCM2595) as a reference gene. The relative abundance of transcripts was calculated by normalization to dinB mRNA. Relative gene expression was reported as the change (n-fold) determined from the mean normalized expression relative to the mean normalized expression of the reference gene (∆∆C t method).
Phenol hydroxylase assay and phenol concentration measurements
R. erythropolis cells grown in minimal BSM medium with various substrates were disrupted with the FastPrep FP120 homogenizer (BIO101) (6 × 20 s, speed 6.0). Phenol hydroxylase activity (phenol monooxygenase, EC 1.14.13.7) was assayed spectrophotometrically at 340 nm in the 1.5-ml reaction mixture containing cell extract (0.05–0.1 mg protein), potassium phosphate buffer (0.1 mM, pH 7.6), 50 μl NADPH (5 mM), and 50 μl phenol (5 mM). One enzyme unit was defined as the amount of enzyme which causes the oxidation of 1 μmol NADPH/min in the presence of phenol (Neujahr and Gaal 1973). Specific activity was calculated as enzyme units per mg protein. Protein concentration was determined using the method of Bradford (1972).
Phenol concentration in the medium was measured using the colorimetric method (Martin 1949). Phenol reacts with 4-aminoantipyrine under alkaline conditions to form a red indophenol dye, which is assayed spectrophotometrically at 502 nm.
Construction and use of the two-plasmid system in C. glutamicum
The R. erythropolis CCM2595 pheR gene was amplified by PCR and cloned in the expression vector pEC-XT99A. The promoters PpheA2 Re and PpheA2 Re M8 (carrying 8 nt alterations upstream of the core promoter sequence as shown in Fig. 3) were cloned in parallel in promoter-test vector pEPR1 as 100-bp fragments (10 nt downstream and 89 nt upstream of TSP) synthetized by Sigma-Aldrich. The two-plasmid strains, C. glutamicum (pEC-XT99ApheR + pEPRPpheA2WT) and C. glutamicum (pEC-XT99ApheR + pEPRPpheA2M8), were cultivated in 2xYT medium at 30 °C with shaking, isopropyl-β-thiogalactoside (IPTG) was added (0.5 mM) at OD600 = 0.5 and cultivation continued at 25 °C for 2 h.
Promoter activity measurements
Activity of the promoters cloned in the promoter-test vector pEPR1 was quantified as the fluorescence of the cell suspension with the OD600 = 0.2 in arbitrary units (AU) measured with a Saphire2 microplate spectrophotometer (Tecan; excitation wavelength, 395 nm; emission wavelength, 509 nm) as described previously (Knoppová et al. 2007).
Results
Cloning and sequencing of the phe–cat gene cluster from R. erythropolis CCM2595
Analysis of the catR–catABC gene cluster from R. erythropolis CCM2595 showed that the genes encode the enzymes of a part of the phenol degradation pathway, which successively convert catechol into muconolactone via an ortho-cleavage pathway (Veselý et al. 2007). To find if the flanking regions encode the enzymes of the upstream or downstream part of the degradative pathway, we cloned these regions using the plasmid rescue technique (Veselý et al. 2007). Fragments flanking the catRABC cluster were sequenced. A cluster of three genes was detected downstream of the catR gene (Fig. 1). Two genes were found to encode proteins homologous to the subunits of two-component PHH and were therefore designated pheA1 and pheA2. The third gene coding for a transcriptional regulator homologous to AraC-type activators was designated pheR. The deduced amino acid sequences of PheA1 (large subunit of PHH, monooxygenase component) and PheA2 (small subunit of PHH, flavin reductase component) gene products exhibited a high degree of identity with the homologous potential proteins from R. erythropolis PR4 (99 % and 100 %, respectively) and lower similarity to the deduced PheA1 and PheA2 proteins from a few other Rhodococcus strains. Two similar clusters pheR–pheA2A1 (I and II) were found in the genome of R. jostii RHA1. These genes are not adjacent to the cat cluster as in R. erythropolis. The deduced R. jostii RHA1 PheA1 and PheA2 proteins exhibited 76 % to 89 % amino acid identity to their R. erythropolis CCM2595 homologs. The deduced amino acid sequence of the pheR product from R. erythropolis CCM2595 exhibited 99 % sequence identity with the putative PheR protein of R. erythropolis PR4. The two putative PheR proteins of R. jostii RHA1 showed lower similarities (54 % and 62 % identical amino acid residues, respectively) to their R. erythropolis CCM2595 homolog.

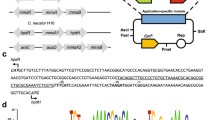
Scheme of phe–cat gene cluster of R. erythropolis CCM2595 and sequence of the pheR–pheA2 intergenic region with proposed regulatory elements. Experimentally determined transcriptional start points (TSP) and the proposed –10 and –35 promoter regions and ribosome-binding sites (RBS) are in bold and underlined. The proposed motifs of the binding sites for the CRP-type protein and the tandem binding sites for the AraC-type regulator are shaded, imperfect direct repeats are indicated by arrows. The organization of the catR–catA intergenic region responsible for the regulation of catABC expression has been described previously (Veselý et al. 2007)
The structure of the complete cluster of phenol degradation genes from R. erythropolis CCM2595 is shown in the upper part of Fig. 1. The respective DNA sequence (6,967 bp) was deposited in the GenBank under Acc. No. FM995530.
Activity of phenol hydroxylase
To prove that the pheA2 and pheA1 genes are responsible for the PHH activity of R. erythropolis CCM2595, we constructed a knock-out mutant by insertional inactivation. The pheA2A1 internal fragment (832 bp) was cloned in the EcoRI site of the E. coli vector pKSAC45 and the resulting construct was transferred to R. erythropolis CCM2595 by electrotransformation. Clones with the plasmid inserted into the chromosome by homologous recombination were selected on plates with kanamycin. The disruption of the pheA2A1 genes was confirmed by PCR. The pheA1A2 knock-out strain grew in BSM with succinate or glucose but not in the medium with phenol as the only carbon and energy source. This result indicated that the pheA1A2 genes encode the only phenol-degrading enzyme in R. erythropolis CCM2595. PHH activity was determined in the extract of the wild-type cells growing on phenol and on the mixture of phenol + protocatechuate. The activity levels were almost identical (0.55 ± 0.13 and 0.56 ± 0.08 U/mg protein). In contrast, PHH activity in extracts of the R. erythropolis pheA2A1 knock-out mutant cultivated on phenol + protocatechuate were lower than 0.01 U/mg protein. These results confirmed that the pheA1 and pheA2 genes encode components of PHH. To overexpress these genes in the natural host cells, a 3.5-kb fragment carrying pheA2A1 and pheR was cloned in the SmaI site of the multicopy E. coli–Rhodococcus vector pSRK21. PHH activity in the extracts of R. erythropolis cells carrying the resulting construct, pSRKpheRA2A1, cultivated on phenol reached 0.86 ± 0.2 U/mg protein.
Likewise, the activity of catechol 1,2-dioxygenase encoded by the catA gene in R. erythropolis CCM2595 (Veselý et al. 2007), PHH activity was induced by phenol in this strain. Specific PHH activity in the extracts of cells cultivated on glucose or succinate was very low, whereas the activity increased 14- to 28-fold on phenol (Table 3). To find out if some utilized substrates repress the induction of PHH, we cultivated R. erythropolis CCM2595 in the presence of phenol and an additional substrate (glucose, succinate or protocatechuate). The presence of protocatechuate did not affect the induction of PHH by phenol. When both phenol and glucose were present in the medium, PHH activity was 64 % of that with phenol alone and the addition of succinate to the medium with phenol resulted in much larger decrease of activity (18 % of that with phenol alone) (Table 3). These data indicate that some type of partial CCR controls the utilization of phenol in R. erythropolis CCM2595.
Activity of the PpheA2 and PpheR promoters from R. erythropolis and R. jostii
To study the transcription of the R. erythropolis CCM2595 pheA2A1 and pheR genes, transcriptional fusions of the respective promoters (PpheA2 Re and PpheR Re ) with the gfpuv reporter gene were constructed. The 434-bp DNA fragment of the intergenic region pheR–pheA2 (including 62 and 72 bp of the pheR and pheA2 5′-ends, respectively) amplified by PCR was cloned in both orientations in the promoter-test vector pEPR1 carrying the gfpuv reporter gene. The green fluorescence of the R. erythropolis colonies carrying the resulting constructs pEPRPpheA2 Re and pEPRPpheR Re on the plates with phenol indicated that divergent promoters resided on the fragment. To localize the promoters precisely, the transcriptional start points (TSPs) of pheA2 and pheR were determined by primer extension (Fig. 2). The TSPs were detected 86 and 55 nt upstream of the translation initiation codons of pheA2 and pheR, respectively (Fig. 1, lower part). Potential –35 and –10 promoter hexamers were recognized closely upstream of the TSPs (Fig. 1).

Determination of R. erythropolis CCM2595 pheA2 and pheR transcription start points by non-radioactive primer extension (PEX) analysis. a pheA2; b pheR. The bottom peaks (PEX) represent cDNAs synthesized in reverse transcription (primer extension) with RNA from R. erythropolis CCM2595. The peaks in lanes A, C, G, T represent the products of the sequencing reactions carried out with the same fluorescein-labeled primer as that used for primer extension. TSP within the relevant part of the non-coding strand sequence shown below is in bold and underlined
To compare the transcriptional control of the phe genes from R. erythropolis with the control of the similar phe genes from R. jostii, we also cloned the pheR–pheA2 intergenic regions of the two phe clusters (I and II) from R. jostii RHA1. The resulting promoter-carrying plasmid constructs pEPRPpheA2 RjI, pEPRPpheR RjI, pEPRPpheA2 RjII, and pEPRPpheR RjII were used for the same analyses as those with R. erythropolis promoters. The TSPs of three genes (both pheA2 and pheR of cluster II and pheA2 of cluster I) were determined by primer extension (data not shown), whereas the TSP of pheR of cluster I could not be detected. The TSPs are shown in Fig. 3. The high level of similarity between deduced –35 and –10 motifs of R. erythropolis and R. jostii PpheA2 promoters is evident in Fig. 3.

Alignment of putative regulatory sequences within homologous pheR–pheA2 intergenic regions of R. erythropolis CCM2595 and related strains. Experimentally determined transcriptional start points (TSP) and proposed –35 and –10 promoter motifs are in bold and underlined, the proposed motifs of the binding sites for the CRP-type protein and the tandem binding sites for the AraC-type regulator are shaded, imperfect direct repeats are indicated by arrows. The 5′ end of the 100-bp fragment used for testing the activation of the pheA2 Re promoter by PheR is marked by vertical arrow. Nucleotide alterations introduced into the R. erythropolis CCM2595 PpheA2 promoter (resulting in PpheA2M8) are shown below the sequences. GenBank accession numbers of the sequences: R. erythropolis CCM2595 (FM995530), R. jostii RHA1 (NC_008268), R. opacus B4 (NC_012522), R. ruber BKS 20-38 (AOEX01000015), Nocardia sp. C-14-1 (DQ267826)
To test whether the induction of PHH is controlled at the transcriptional level, the activity of the promoters PpheA2 Re and PpheR Re in transcriptional fusion with gfpuv during growth on various substrates was determined by measuring the green fluorescence of the R. erythropolis cells. The fluorescence levels measured for the cells with empty vector pEPR1 (negative control) vary between 0.2 and 0.3 AU. Levels of transcription from PpheA2 Re close to the negative control were detected in cells grown on BSM with glucose, glycerol or succinate as the sole carbon source (Fig. 4a). In contrast, cells with PpheR Re exhibited 2- to 3-fold higher fluorescence when grown on succinate or glycerol (Fig. 4b). This indicated that there was a certain level of constitutive activity of PpheR Re . In agreement with this finding, R. erythropolis colonies carrying pEPRPpheR Re exhibited green fluorescence when grown on complete medium without phenol whereas the colonies with pEPRPpheA2 Re did not. The activity of both promoters was strongly induced by phenol (Fig. 4a, b).

Activity of PpheA2 and PpheR promoters from R. erythropolis CCM2595 and R. jostii RHA1 (promoters of clusters pheRA2A1 I and II) during growth on minimal medium BSM with various substrates. Activity was determined by measuring the intensity of fluorescence of the GFPuv reporter. AU arbitrary units. The values for the negative control (empty vector pEPR1) were 0.25 ± 0.05. The values shown are averages of three independent biological replicates; standard deviations were within 15 %
To examine the possible repression of phenol induction by other carbon sources, promoter activities were measured in cells grown in media containing phenol and an additional substrate. Transcription from both promoters was not decreased by the presence of glucose and glycerol in the medium with phenol. In contrast, the activity of PpheR Re was significantly decreased on phenol + succinate and was close to the activity of the promoter on succinate alone. The activity of PpheA2 Re on phenol + succinate was approximately 60 % of the activity on phenol alone for the first 18 h of cultivation (Fig. 4a). The transcription from both PpheA2 Re and PpheR Re increased in the later stages of cultivation (after 24 h), when the succinate was exhausted. We can conclude that there is a low-level constitutive transcription from PpheR Re , which is strongly induced by phenol. PpheA2 Re activity is completely induced by phenol.
Promoter activity measurements using gfpuv reporter assay with promoters of the genes pheR I and II and pheA2 I and II from R. jostii showed that the trends of activity of pheA2 promoters during 24-h cultivation were similar to those of the respective pheR promoters (Fig. 4c–f). No transcription from the PpheA2 RjI and PpheR RjI promoters was detected on glucose, glycerol or succinate, whereas activity was moderately induced by phenol. Phenol-induced activity was low but not repressed by any additional substrate tested (Fig. 4c, d). The transcriptional pattern found for the promoters of the phe II R. jostii cluster was different from those of cluster I promoters. Phenol induction of both PpheA2 RjII and PpheR RjII was partially repressed by both glucose and glycerol, whereas strong repression of both promoters was observed when succinate was added to the medium with phenol (Fig. 4e, f).
Taken together, these results indicate that although the principle of the transcriptional regulation of the three analyzed phe gene clusters (one from R. erythropolis CCM 2595 and two from R. jostii RHA1) is the same (phenol induction), the phenotypic effects of various substrates differ.
In some previously described phenol degradation operons, the presence of other aromatic substrates in the growth medium affected the induction of catabolic genes (Putrinš et al. 2007). We therefore tested the influence of benzoate, p-hydroxybenzoate and protocatechuate on the activity of the PpheA2 Re and PpheR Re promoters. The activity of neither of the two promoters was induced when any of these compounds was used as the sole carbon source. None of these aromatic substrates affected transcription from either of the two promoters when present in the medium with phenol (data not shown).
Induction of R. erythropolis CCM2595 phe genes by low phenol concentration
To find the lowest concentration of phenol that can induce the transcription of phe genes, we determined the activity of the PpheA2 Re and PpheR Re promoters using R. erythropolis cultures grown in BSM with various phenol concentrations (1.2, 0.6, 0.12, 0.06, 0.012 mM). The lowest phenol concentration at which a promoter activity was detected was 0.12 mM (data not shown). Cultivation on 1.2 mM phenol resulted in lower maximal PpheA2 Re and PpheR Re activities (52 % and 63 %, respectively) than on 3.5 mM phenol (Fig. 5). The activity of both promoters quantitatively decreased with decreasing phenol concentration (data not shown). This indicates that expression of phe genes is induced by phenol in a dose-dependent manner rather than by an on/off mechanism. The maximum response of PpheR Re activity was achieved after 4 h of cultivation, whereas the maximum of PpheA2 Re activity was detected 6 h later (Fig. 5). The observed delayed activation of PpheA2 Re transcription in comparison with that of PpheR Re transcription is in agreement with the assumption that PheR acts as an activator of pheA2 transcription.

Activity of PpheA2 Re and PpheR Re promoters during growth on low phenol concentration (1.2 mM). Protocatechuate (3.5 mM) was used as an additional substrate in minimal medium BSM. AU arbitrary units. Standard deviations of three measurements are depicted by error bars
Quantification of the phe and cat transcripts by real-time RT-PCR
To confirm the results of promoter activity measurements using transcriptional fusion with gfpuv, we quantified the levels of cat and phe transcripts from R. erythropolis CCM2595 using quantitative real-time RT-PCR (qPCR). The lowest levels of the transcripts were found in cells grown on succinate, and these values were used as a basal level for comparison with the quantities determined on phenol. The levels of catA, catB and catC transcripts were approximately 60-fold higher on phenol than on succinate, whereas catR transcript was not induced by phenol and its level was similar on phenol and succinate (Fig. 6). These values are in good agreement with catA and catR promoter activities determined previously (Veselý et al. 2007). The pheA1 and pheA2 transcripts were upregulated to the same level in cells grown on phenol, which indicates that these genes form an operon. The pheR transcript increased much less, which probably reflects its partial constitutive expression.

Relative abundance of cat and phe transcripts in R. erythropolis CCM2595 cells grown on phenol and succinate determined by qPCR. The values obtained for the cultures grown on succinate were used as references and were set to 1.0 on the logarithmic scale. Averages of two independent biological replicates measured in triplicate are shown. Standard deviations are shown as error bars
PheR activates the pheA2 promoter
The pheR gene encodes an AraC-type transcriptional regulator and transcriptional patterns of pheA2 and pheR suggested that PheR acts as an activator of the pheA2A1 genes. The pheA2 promoter region of R. erythropolis CCM2595 carries a typical tandem target sequence (González-Pérez et al. 1999) for AraC/XylS-type activators (Fig. 1), which we also found by genome searching in several pheR–pheA2A1 clusters in various Rhodococcus strains (Fig. 3; see Discussion). However, all efforts to delete the R. erythropolis CCM2595 pheR gene using standard techniques of gene replacement failed (data not shown).
To examine whether the R. erythropolis CCM2595 PheR protein acts as an activator, we constructed the two-plasmid system in the heterologous host, C. glutamicum. C. glutamicum is a related bacterium in which promoters function similarly as in R. erythropolis (Knoppová et al. 2007) and its genome does not code for a PheR regulator. To test if the assumed binding site for PheR upstream of the PpheA2 Re promoter (carried at the 100-bp DNA fragment; Fig. 3) is involved in the promoter activation, we constructed a mutant promoter PpheA2M8 carrying 8 nt alteration within the supposed tandem PheR binding site upstream of the –35 region of PpheA2 Re promoter (Fig. 3). Both pEC-XT99ApheR (expression vector with the pheR gene) and pEPRPpheA2WT (or pEPRPpheA2M8) were introduced into the C. glutamicum and the effect of pheR overexpression on PpheA2 activity was measured by the reporter GFP fluorescence after IPTG addition. No significant promoter activity was observed for PpheA2 Re or for PpheA2M8 in comparison with the control (empty pEPR1) in absence of pheR (Fig. 7). The PpheA2 Re activity increased markedly after the induction of pheR expression, whereas no increase in PpheA2M8 activity was observed under the same conditions. These results indicate that PheR is necessary for PpheA2 function, and the altered nucleotides within the supposed PheR binding site are essential for promoter activation.

Effect of PheR overexpression on the activity of PpheA2 and PpheA2M8 promoters from R. erythropolis CCM2595. Promoter activity was determined by measuring the intensity of fluorescence of the GFP reporter. AU; arbitrary units. IPTG was added at time zero. Standard deviations of three measurements are depicted by error bars
Discussion
The gene cluster catABC–catR–pheR–pheA2A1 from R. erythropolis CCM2595 was found to encode four enzymes of the phenol degradation pathway and two regulatory proteins. Clusters with the same gene organization can be detected within the genome of R. erythropolis PR4 (NC_012490) and the genome shotgun sequences of R. erythropolis SK121 (ACNO01000001) and R. qingshengii BKS 20-40 (AODN01000022). The intergenic sequences pheR–pheA2 in these strains are very similar to that of R. erythropolis CCM2595 (92 % to 100 % identity). We also found the same organization of the cat–phe genes within the sequenced parts of the genomes of the related strains, R. pyridinivorans AK37 (AHBW01000047), R. ruber 20-38 (AOEX01000015) and Nocardia sp. C-14 (DQ267826). The intergenic regions pheR–pheA2 of these bacteria differ in terms of length and sequence distinctly from those of the first group. A different organization of the phe genes was found in R. jostii RHA1 and some other rhodococci. Two pheR–pheA2A1 clusters (I and II), not adjacent to catRABC, were found in the genomes of R. jostii RHA1 (NC_008268) and R. opacus B4 (NC_012522). Similar phe I and phe II clusters can be found in R. opacus M213, R. opacus 1CP, R. wratislaviensis IFP 2016 and R. imtechensis RKJ300. No studies concerning regulation of phe–cat gene expression in the mentioned bacteria have been reported.
We proved that there is a single PHH encoded by pheA2A1 in R. erythropolis CCM2595. Although a number of Rhodococcus strains were described as efficient phenol degraders useful in biotechnological processes (Paisio et al. 2012; Prieto et al. 2002), PHH, the key enzyme of phenol catabolism and expression of the pheA2A1 genes were mostly not studied. Activity of PHH from R. erythropolis UVP1 that is closely similar to PHH from R. erythropolis CCM2595 was analyzed after expression of the respective pheA2A1 genes into the two his-tagged PHH components in E. coli (Saa et al. 2009); however, expression control of these genes in Rhodococcus strains was not examined. We initiated the studies of regulatory mechanisms of the phenol degradation pathway in Rhodococcus by the analysis of expression of the catABC genes involved in the downstream part of the pathway (Veselý et al. 2007). We found that the expression of catABC is repressed by the IclR-type regulator CatR and induced by phenol in R. erythropolis CCM2595 (Veselý et al. 2007). Expression of the genes involved in the catabolism of aromatic compounds studied particularly in soil bacteria is controlled by a number of specific and global regulatory mechanisms which integrate diverse signals from the environment (Cases and de Lorenzo 2005). Most apparent regulatory phenomena are the induction of gene expression by the presence of the substrate and CCR of this induction exerted by more easily metabolizable substrates (Rojo 2010). We proved that the induction of phenol hydroxylase is triggered at the level of transcription of the pheR and pheA2A1 genes by the presence of phenol. The activity of the mPH was also found to be induced by phenol, e.g., in Pseudomonas strains (Shingler 2003) or in Acinetobacter calcoaceticus (Yu et al. 2011).However, no data on induction of the two-component PHH and modulation of expression of the respective genes mediated by a carbon catabolic repression or utilization hierarchy of aromatic substrates have been available until now.
Many soil bacteria carry sets of genes determining the catabolic activities which allow them to degrade and utilize a wide range of aromatic compounds, either of a natural origin (e.g., lignin metabolites) or xenobiotics (e.g., chlorophenols and polychlorinated biphenyls). Since mostly heterogenous mixtures of aromatic compounds of a natural or man-made origin are present in soil, a hierarchy of the substrate utilization evolved in most bacteria. Out of the aromatic compounds for which the substrate hierarchy was described (Nichols and Harwood 1995; Choi et al. 2007), we tested the effects of benzoate, p-hydroxybenzoate and protocatechuate on the phenol utilization. None of these aromatic substrates repressed the induction of PHH and utilization of phenol.
The presence of the simple, easily metabolizable carbon and energy sources, such as glucose or organic acids, in a mixture with aromatic compounds, generally results in the inhibition of the aromatic substrate catabolism by CCR. This phenomenon was observed in many bacterial species, but the mechanisms of CCR differ between bacterial species (Rojo 2010). We have already shown that glucose did not repress the catechol degradation pathway in R. erythropolis CCM2595 (Veselý et al. 2007). Similarly, expression of the pheA2A1 genes was repressed neither by glucose nor glycerol (Fig. 4a). In contrast, we observed partial pheA2A1 repression by succinate at the transcriptional level. The observed partial repression of the PHH activity by glucose (Table 3) which did not correlate with the results of the transcriptional analysis (Fig. 4a) may be mediated by an unknown posttranscriptional regulation. However, this partial PHH repression did not affect the flux through the phenol degradation pathway since phenol was degraded completely after 26 h independently of the presence of glucose (data not shown).
Different expression patterns were found in the two R. jostii RHA1 phe clusters. The transcription from the PpheA2 RjII promoter was substantially repressed only by succinate (Fig. 4e), whereas PpheA2 RjI activity was not decreased by any of the substrates tested in the presence of phenol (Fig. 4c). The diverse transcription patterns of the three phe clusters from R. erythropolis CCM2595 and R. jostii RHA1 during growth suggest that some subtle regulatory mechanisms of the gene expression differ in the individual cases. Different regulation of two phe genes in R. jostii suggests that the phe clusters I and II are involved in redundant catabolic reactions that may be triggered under different growth conditions. The temporal transcriptional patterns of PpheA2 and PpheR were found to correlate in each of the PpheA2–PpheR pair (Fig. 4). Activity of PpheA2 Re was delayed during the cultivation in comparison with PpheR Re when a low phenol concentration was used for growth (Fig. 5). We found that overexpression of pheR resulted in a sharp increase of PpheA2 Re activity in the heterologous host, C. glutamicum (Fig. 7). A moderate increase of PpheA2 Re activity was also apparent in the presence of pEC-XT99ApheR without addition of IPTG (due to the leaky pheR expression from the trc promoter). None of these effects was observed when pheR was missing or the assumed PheR binding site was altered. These results suggest that PheR is an activator of the pheA2A1 genes. AraC-type regulator NphR was also found to activate transcription of the genes encoding two-component 4-nitrophenol hydroxylase in the Rhodococcus sp., strain PN1. However, only 4-nitrophenol induced nitrophenol degradation pathway and expression of the nphR gene was found to be constitutive, in the PN1 strain (Takeo et al. 2008). Inspection of the pheA2 promoter sequence of all three phe analyzed clusters showed that the tandem binding sites for the AraC-type regulator are present at the exactly same position overlapping two nucleotides of the putative –35 promoter hexamer of PpheA2 (Fig. 3). The proposed PheR Re binding site shares 15 nt out of 18 nt of the AraC/XylS-type activator binding site upstream of the Pm promoter which drives transcription of the genes involved in alkylbenzoates catabolic pathway in P. putida (González-Pérez et al. 1999). In both cases the downstream GGNTA/C motif overlaps by 2 nt with the –35 hexamer.
To isolate the PheR protein from R. erythropolis CCM2595 and use it for direct biochemical and biophysical analyses, e.g., the electrophoretic mobility shift assay, which may directly prove binding of the protein to the supposed target site, we have overexpressed the pheR gene in E. coli and purified the PheR protein. However, the procedure yielded mostly insoluble inclusion bodies (data not shown), which is a typical feature of the AraC-family proteins (Schleif 2010). Alignment of the pheA2–pheR promoter regions from R. erythropolis and R. jostii with the respective parts of the sequences of the homologous phe clusters from the related strains showed the similarity of the key regulatory sequences in these bacteria (Fig. 3). Nearly identical potential binding sites for a CRP-type regulator were detected within the pheR promoter regions of all aligned sequences (Fig. 3). Closely similar sequence CTGTGT–N6–TCACAG within the promoters of two genes involved in tetralin utilization was proved to be involved in binding of the CRP-like protein which exerted glucose-mediated CCR in Rhodococcus sp. TFB strain (Tomás-Gallardo et al. 2012). Potential function of the CRP-like protein from R. erythropolis CCM2595 and R. jostii RHA1 in CCR or its connection to the pheR gene expression in Rhodococcus strains remain to be elucidated.
References
Bleichrodt FS, Fischer R, Gerischer UC (2010) The β-ketoadipate pathway of Acinetobacter baylyi undergoes carbon catabolite repression, cross-regulation and vertical regulation, and is affected by Crc. Microbiology 156:1313–1322
Bradford MA (1972) A rapid and sensitive method for the quantitation of microgram quantities of protein using the principle of dye binding. Anal Biochem 72:248–252
Cases I, de Lorenzo V (2005) Promoters in the environment: transcriptional regulation in its natural context. Nat Rev Microbiol 3:105–118
Choi KY, Zylstra GJ, Kim E (2007) Benzoate catabolite repression of the phthalate degradation pathway in Rhodococcus sp. strain DK17. Appl Environ Microbiol 73:1370–1374
Duffner FM, Müller R (1998) A novel phenol hydroxylase and catechol 2,3-dioxygenase from the thermophilic Bacillus thermoleovorans strain A2: nucleotide sequence and analysis of the genes. FEMS Microbiol Lett 161:37–45
Dusch N, Pühler A, Kalinowski J (1999) Expression of the Corynebacterium glutamicum panD gene encoding L-aspartate-α-decarboxylase leads to pantothenate overproduction in Escherichia coli. Appl Environ Microbiol 65:1530–1539
González-Pérez MM, Ramos JL, Gallegos MT, Marqués S (1999) Critical nucleotides in the upstream region of the XylS-dependent TOL meta-cleavage pathway operon promoter as deduced from analysis of mutants. J Biol Chem 274:2286–2290
Hanahan E (1985) Techniques for transformation of E. coli. In: Glover DM (ed) DNA cloning. A practical approach, vol 1. IRL, Oxford, pp 109–135
Hernandez MA, Mohn WW, Martinez E, Rost E, Alvarez AF, Alvarez HM (2008) Biosynthesis of storage compounds by Rhodococcus jostii RHA1 and global identification of genes involved in their metabolism. BMC Genomics 9:600
Holátko J, Elišáková V, Prouza M, Sobotka M, Nešvera J, Pátek M (2009) Metabolic engineering of the L-valine biosynthesis pathway in Corynebacterium glutamicum using promoter activity modulation. J Biotechnol 139:203–210
Kim IC, Oriel PJ (1995) Characterization of the Bacillus stearothermophilus BR219 phenol hydroxylase gene. Appl Environ Microbiol 61:1252–1256
Kirchner O, Tauch A (2003) Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J Biotechnol 104:287–299
Kirchner U, Westphal AH, Müller R, van Berkel WJ (2003) Phenol hydroxylase from Bacillus thermoglucosidasius A7, a two-protein component monooxygenase with a dual role for FAD. J Biol Chem 278:47545–47553
Knoppová M, Phensaijai M, Veselý M, Zemanová M, Nešvera J, Pátek M (2007) Plasmid vectors for testing in vivo promoter activities in Corynebacterium glutamicum and Rhodococcus erythropolis. Curr Microbiol 55:234–239
Larkin MJ, Kulakov LA, Allen CC (2005) Biodegradation and Rhodococcus—masters of catabolic versatility. Curr Opin Biotechnol 16:282–290
Martin RW (1949) Rapid colorimetric estimation of phenol. Nature 21:1419–1420
Martínková L, Uhnáková B, Pátek M, Nešvera J, Křen V (2009) Biodegradation potential of the genus Rhodococcus. Environ Int 35:162–177
McLeod MP, Warren RL, Hsiao WW, Araki N, Myhre M, Fernandes C, Miyazawa D, Wong W, Lillquist AL, Wang D, Dosanjh M, Hara H, Petrescu A, Morin RD, Yang G, Stott JM, Schein JE, Shin H, Smailus D, Siddiqui AS, Marra MA, Jones SJ, Holt R, Brinkman FS, Miyauchi K, Fukuda M, Davies JE, Mohn WW, Eltis LD (2006) The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc Natl Acad Sci U S A 103:15582–15587
Müller C, Petruschka L, Cuypers H, Burchhardt G, Herrmann H (1996) Carbon catabolite repression of phenol degradation in Pseudomonas putida is mediated by the inhibition of the activator protein PhlR. J Bacteriol 178:2030–2036
Neujahr HY, Gaal A (1973) Phenol hydroxylase from yeast. Purification and properties of the enzyme from Trichosporon cutaneum. Eur J Biochem 35:386–400
Nichols NN, Harwood CS (1995) Repression of 4-hydroxybenzoate transport and degradation by benzoate: a new layer of regulatory control in the Pseudomonas putida β-ketoadipate pathway. J Bacteriol 177:7033–7040
Omokoko B, Jäntges UK, Zimmermann M, Reiss M, Hartmeier W (2008) Isolation of the phe-operon from G. stearothermophilus comprising the phenol degradative meta-pathway genes and a novel transcriptional regulator. BMC Microbiol 8:197
Paisio CE, Talano MA, Gonzalez PS, Busto VD, Talou JR, Agostini E (2012) Isolation and characterization of a Rhodococcus strain with phenol-degrading ability and its potential use for tannery effluent biotreatment. Environ Sci Pollut Res Int 19:3430–3439
Pátek M, Muth G, Wohlleben W (2003) Function of Corynebacterium glutamicum promoters in Escherichia coli, Streptomyces lividans, and Bacillus subtilis. J Biotechnol 104:325–334
Prieto MB, Hidalgo A, Rodriguez-Fernandez C, Serra JL, Llama MJ (2002) Biodegradation of phenol in synthetic and industrial wastewater by Rhodococcus erythropolis UPV-1 immobilized in an air-stirred reactor with clarifier. Appl Microbiol Biotechnol 58:853–859
Putrinš M, Tover A, Tegova R, Saks Ü, Kivisaar M (2007) Study of factors which negatively affect expression of the phenol degradation operon pheBA in Pseudomonas putida. Microbiology 153:1860–1871
Rojo F (2010) Carbon catabolite repression in Pseudomonas: optimizing metabolic versatility and interactions with the environment. FEMS Microbiol Rev 34:658–684
Saa L, Jaureguibeitia A, Largo E, Llama MJ, Serra JL (2009) Cloning, purification and characterization of two components of phenol hydroxylase from Rhodococcus erythropolis UPV-1. Appl Microbiol Biotechnol 86:201–211
Sambrook J, Russel DV (2001) Molecular cloning. A laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Schleif R (2010) AraC protein, regulation of the L-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol Rev 34:779–796
Shingler V (2003) Integrated regulation in response to aromatic compounds: from signal sensing to attractive behaviour. Environ Microbiol 5:1226–1241
Shingler V, Bartilson M, Moore T (1993) Cloning and nucleotide sequence of the gene encoding the positive regulator (DmpR) of the phenol catabolic pathway encoded by pVI150 and identification of DmpR as a member of the NtrC family of transcriptional activators. J Bacteriol 175:1596–1604
Takeo M, Murakami M, Niihara S, Yamamoto K, Nishimura M, Kato D, Negoro S (2008) Mechanism of 4-nitrophenol oxidation in Rhodococcus sp. Strain PN1: characterization of the two-component 4-nitrophenol hydroxylase and regulation of its expression. J Bacteriol 190:7367–7374
Teramoto M, Harayama S, Watanabe K (2001) PhcS represses gratuitous expression of phenol-metabolizing enzymes in Comamonas testosteroni R5. J Bacteriol 183:4227–4234
Tomás-Gallardo L, Santero E, Floriano B (2012) Involvement of a putative cyclic AMP receptor protein (CRP)-like binding sequence and a CRP-like protein in glucose-mediated catabolite repression of thn genes in Rhodococcus sp. strain TFB. Appl Environ Microbiol 78:5460–5462
van der Geize R, Dijkhuizen L (2004) Harnessing the catabolic diversity of rhodococci for environmental and biotechnological applications. Curr Opin Microbiol 7:255–261
Veselý M, Pátek M, Nešvera J, Čejková A, Masák J, Jirků V (2003) Host–vector system for phenol-degrading Rhodococcus erythropolis based on Corynebacterium plasmids. Appl Microbiol Biotechnol 61:523–527
Veselý M, Knoppová M, Nešvera J, Pátek M (2007) Analysis of catRABC operon for catechol degradation from phenol-degrading Rhodococcus erythropolis. Appl Microbiol Biotechnol 76:159–168
Yu H, Peng Z, Zhan Y, Wang J, Yan Y, Chen M, Lu W, Ping S, Zhang W, Zhao Z, Li S, Takeo M, Lin M (2011) Novel regulator MphX represses activation of phenol hydroxylase genes caused by a XylR/DmpR-type regulator MphR in Acinetobacter calcoaceticus. PLoS ONE 6:e17350
Zídková L, Szököl J, Rucká l, Pátek M, Nešvera J (2013) Biodegradation of phenol using recombinant plasmid-carrying Rhodococcus erythropolis strains. Int Biodeter Biodegradation 84:179–184
Acknowledgments
This work was supported by a grant (2B08062 AROMAGEN) from the Czech Ministry of Education, Youth and Sports, grant P504/11/0394 from Czech Science Foundation and internal project RVO61388971 (Institute of Microbiology).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Szőköl, J., Rucká, L., Šimčíková, M. et al. Induction and carbon catabolite repression of phenol degradation genes in Rhodococcus erythropolis and Rhodococcus jostii . Appl Microbiol Biotechnol 98, 8267–8279 (2014). https://doi.org/10.1007/s00253-014-5881-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5881-6