
Abstract
The hypocrealean fungus Claviceps paspali is a parasite of wild grasses. This fungus is widely utilized in the pharmaceutical industry for the manufacture of ergot alkaloids, but also produces tremorgenic and neurotoxic indole-diterpene (IDT) secondary metabolites such as paspalitrems A and B. IDTs cause significant losses in agriculture and represent health hazards that threaten food security. Conversely, IDTs may also be utilized as lead compounds for pharmaceutical drug discovery. Current protoplast-mediated transformation protocols of C. paspali are inadequate as they suffer from inefficiencies in protoplast regeneration, a low frequency of DNA integration, and a low mitotic stability of the nascent transformants. We adapted and optimized Agrobacterium tumefaciens-mediated transformation (ATMT) for C. paspali and validated this method with the straightforward creation of a mutant strain of this fungus featuring a targeted replacement of key genes in the putative IDT biosynthetic gene cluster. Complete abrogation of IDT production in isolates of the mutant strain proved the predicted involvement of the target genes in the biosynthesis of IDTs. The mutant isolates continued to produce ergot alkaloids undisturbed, indicating that equivalent mutants generated in industrial ergot producers may have a better safety profile as they are devoid of IDT-type mycotoxins. Meanwhile, ATMT optimized for Claviceps spp. may open the door for the facile genetic engineering of these industrially and ecologically important organisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The indole-diterpenes (IDTs) are a group of chemically diverse mycotoxins that cause large economic losses in the livestock industry, especially in the Southern hemisphere (Bennett and Klich 2003; Cawdell-Smith et al. 2010). The IDTs are agonists of potassium ion channels in the nervous systems of insects and mammals and cause potent neurotoxic and tremorgenic effects (McMillan et al. 2003; Imlach et al. 2011; Uhlig et al. 2009). These secondary metabolites are produced by a number of ascomycetous fungi including Penicillium, Aspergillus, Claviceps, Epichloë, and Neotyphodium species (Parker and Scott 2004). In their native ecological contexts, these metabolites defend the overwintering structures of the fungus, and/or protect the host plant of the producing fungi against grazing by large animals and insects, thereby offering evolutionary advantages for the producing organism (di Menna et al. 2012; Thom et al. 2014; Panaccione et al. 2006). This antifeedant activity of the IDTs can be exploited in various plant protection strategies in agriculture as a part of integrated pest management systems (Panaccione et al. 2014; Saikkonen et al. 2016). In addition, IDTs are also investigated as lead compounds for potential drugs, for example in breast cancer therapies (Sallam et al. 2013).
During IDT biosynthesis, the diterpene moiety derives from geranylgeranyl diphosphate (GGPP), while the indole moiety originates from tryptophan or a tryptophan precursor (Laws and Mantle 1989; Byrne et al. 2002). The first IDT biosynthetic gene cluster to be genetically characterized was the paxilline cluster from Penicillium paxilli (Young et al. 2001), followed by additional gene clusters for various IDTs from diverse filamentous fungi (Nicholson et al. 2015; Young et al. 2006; Zhang et al. 2004). All these clusters encompass a conserved core set of genes that are responsible for the early steps of IDT biosynthesis, and a set of variable tailoring genes that ensure the remarkable chemical diversity of the final products (Zhang et al. 2004; Young et al. 2005, 2006; Nicholson et al. 2009). For example, paxG, paxM, paxB, and paxC from the paxilline cluster encode enzymes that are sufficient for the assembly of paspaline, the first stable cyclic IDT intermediate (Saikia et al. 2006). Not surprisingly, orthologues of these genes are present in all IDT gene clusters elucidated so far (Nicholson et al. 2015). The tailoring genes that are responsible for the chemical diversity of these metabolites may encode prenyl transferases, and cytochrome P450 monooxygenases and FAD-dependent monooxygenases that catalyze regiospecific and stereospecific oxidations of the IDT skeleton (Young et al. 2001, 2005; Nicholson et al. 2015; Liu et al. 2014).
The biosynthetic pathways of IDTs and the regulatory circuits governing the production of these metabolites have been studied in a number of model organisms such as Neotyphodium lolii and P. paxilli (Young et al. 2001, 2006). However, N. lolii is not well suited for large-scale production of secondary metabolites in the pharmaceutical industry due to its fastidious growth habits and genetic instability (Wiewióra et al. 2015). While P. paxilli has been extremely useful for the study of the biosynthesis of the IDT congener paxilline in the laboratory (Young et al. 2001), this organism has not been adopted for industrial strain development and fermentation process optimization. In contrast, the IDT producer Claviceps paspali has earned a good reputation in the fermentation industry for its relatively easy handling, and industrial scale fermentation processes for ergot alkaloid production have been developed and implemented with this fungus (Arcamone et al. 1960; Tudzynski et al. 2001). Unlike other IDT producers such as N. lolii (Young et al. 2006), C. paspali does not require the presence of any host plant to produce IDTs, thus its axenic submerged cultures may prove to be useful for the economical production of these metabolites. Conversely, IDT production represents an impediment for the industrial manufacture of ergot alkaloids with this species since precursors and cofactors that may be utilized for ergot production are depleted by IDT biosynthesis. In addition, IDTs represent hazardous impurities that must be excluded from the ergot alkaloid products and have to be safely disposed. Despite the potential benefits and the extant disadvantages of IDT production by C. paspali, the genetic basis of IDT biosynthesis has only been inadequately characterized in this strain. Thus, although putative IDT gene clusters have been identified in the genome sequences of Claviceps spp. including C. paspali RRC-1481 (Schardl et al. 2013), these bioinformatics studies have not been followed up by functional investigations of the constituent biosynthetic genes.
Agrobacterium tumefaciens-mediated gene transfer (ATMT) has long been used in plant molecular biology (Păcurar et al. 2011). This transformation technique was first adapted to the baker’s yeast Saccharomyces cerevisiae in 1995 (Bundock et al. 1995), and to the filamentous fungus Aspergillus awamori in 1998 (de Groot et al. 1998). This technique does not require laborious and often inefficient protoplast preparation and regeneration, and the success of the transformation reaction does not depend on the effectiveness of cell wall hydrolyzing enzyme preparations that often show significant batch-to-batch variability. In addition, the transferred DNA readily integrates into the genome of the host during ATMT. This allows this transformation method to be used for random insertional mutagenesis (Zhong et al. 2007; Kunitake et al. 2013) and for the targeted genetic modification of various filamentous fungi by gene deletion or replacement. In particular, ATMT has been used to validate the predicted roles of secondary metabolite biosynthetic genes, including core genes responsible for the assembly of polyketide or nonribosomal peptide metabolite skeletons in various fungi (Zhang et al. 2003; Xu et al. 2008, 2009). In spite of the advantages of this transformation method, there has been no ATMT protocol published so far, to the best of our knowledge, for any Claviceps species.
Hereby, we describe the development of a facile and effective ATMT protocol for generating mitotically stable C. paspali transformants. We validate the suitability of this protocol for the genetic manipulation of this industrially important ergot producer by replacing a part of the predicted IDT biosynthetic gene cluster with a selectable marker gene. We show that this targeted gene replacement of the idtCBGF genes leads to the complete elimination of the biosynthesis of the whole spectrum of IDT secondary metabolites in C. paspali without adversely affecting the production of ergot alkaloids. This work thus provides a hitherto missing functional proof for the involvement of these genes in IDT production in C. paspali, and opens the way for the use of molecular genetics for the optimization of this important fungus for the production of useful bioactive metabolites on an industrial scale.
Materials and methods
Fungal strains and growth conditions
Although the genome sequence of C. paspali RRC-1481 was deposited in GenBank (Schardl et al. 2013), the strain itself is not available to the public from any mainstream strain collections. Thus, the IDT-producing C. paspali DSM-833 strain [equivalent to the now-discontinued ATCC 13893 (Chain et al. 1962)] was obtained from the DSMZ culture collection (Braunschweig, Germany) and was used throughout this study in lieu of C. paspali RRC-1481. C. paspali DSM-833 was maintained on potato dextrose agar (PDA, Sigma-Aldrich, St. Louis, MO). For the production of IDTs in axenic culture, C. paspali mycelium was collected from an agar slant (3.9% PDA, 0.05% yeast extract, pH 5.2) and homogenized with a Dounce Homogenizer in 5 mL distilled water. A 1 mL aliquot of the homogenized mycelium was used to inoculate 60 mL inoculum medium (5% mannitol, 1% succinic acid, 0.5% soy flour, 0.2% KH2PO4, 0.03% MgSO4 × 7H2O, pH 5.2), and the culture was incubated for 5 days at 28 °C with shaking at 240 rpm. A 5 mL aliquot of this preculture was inoculated into 60 mL production medium (10% sorbitol, 3.5% succinic acid, 1.5% corn steep liquor, 0.05% yeast extract, 1.5% NH4NO3, 0.07% MgSO4 x 7H2O, 0.0022% FeSO4 x 7H2O, 0.001% ZnSO4 x 7H2O, pH 5.2) in a 500 mL Erlenmeyer flask, and the resulting main culture was incubated for a further 12 days at 28 °C with shaking at 240 rpm. Mycelia were collected by centrifugation (5 min, 2000 g), freeze-dried, and homogenized under liquid nitrogen in a porcelain mortar. 0.5 g homogenate was extracted with 10 mL acetonitrile/water (4:1, v/v) overnight with shaking at 240 rpm. The supernatant was filtered through a 0.22 μm MultiScreen filter plate (Merck Millipore, Burlington, MA) before direct injection for LC-MS analysis.
Genomic DNA isolation and PCR reactions
The nucleotide sequence of the putative IDT biosynthetic gene cluster of C. paspali RRC-1481 (AFRC01000001-AFRC01002304) was obtained from the National Center for Biotechnology Information (NCBI) under the GenBank accession numbers JN613321.1 and JN613322.1 and was used to design primers for the PCR amplification of sequences from the genome of C. paspali DSM-833. For genomic DNA isolation, 0.1 g wet C. paspali DSM-833 mycelium was ground under liquid nitrogen in a mortar with a pestle. The homogenate was re-suspended in 400 μl sulfite buffer (0.7 M NaCl, 0.1 M Na2SO3, 0.1 M Tris-Cl pH 7.5, 0.05 M EDTA, 1% SDS) and digested with 3 U proteinase K (ThermoFisher) at 55 °C for 90 min. After inactivation of proteinase K at 95 °C for 5 min, cell debris was removed by centrifugation at 4000 g for 5 min. A 250 μl aliquot of the supernatant was further digested with 50 U RNase A (Thermo Fisher Scientific, Waltham, MA) at 37 °C for 12 h, and the RNA-free solution was used for the isolation of DNA with the MagNa Pure LC DNA Isolation Kit III (Roche Diagnostics, Indianapolis, IN) according to the manufacturer’s protocol. PCR reactions were carried out with 20 ng genomic DNA or 1 ng plasmid DNA as templates, respectively, in 50 μl reaction mixtures containing 0.2 mM of each dNTP, 1 pM of each primer, 1 μl Phusion HF DNA polymerase and 10 μl HF buffer (New England Biolabs, Ipswich, MA). Thermal cycling conditions for the PCR reactions were 180 s at 98 °C for the initial denaturation, followed by 31 cycles of amplification (98 °C for 10 s, 55 °C for 15 s, 72 °C for 30 s/kbp), and a final extension step of 60 s/kbp at 72 °C.
Assembly of the idtCBGF deletion construct
Construction of the idtCBGF deletion vector pAG-LTA-hph-RTA was carried out in two consecutive steps using Gibson assembly [Fig. 1; hph is the Escherichia coli gene coding for hygromycin B phosphotransferase (Gritz and Davies 1983)]. In the first step, the linearized pAg-H3 vector (Zhang et al. 2003) was PCR amplified with primers pAg-F1 and pAg-R1 and fused using the Gibson Assembly Master Mix (New England Biolabs, Ipswich, MA) with the left targeting arm (LTA, the 1.5 kb region of the C. paspali DSM-833 genomic DNA located upstream of the idtC gene, amplified with primers idtCBGF-LA-F and idtCBGF-LA-R). Primer sequences are displayed in Supplementary Table S1. After transformation of the assembly mixture into E. coli XL1Blue (New England Biolabs, Ipswich, MA), the pAg-LTA-hph plasmid was selected for the presence of the LTA by PCR. In the second step, pAg-LTA-hph was linearized by PCR with primers pAg-F2 and pAg-R2, and fused using the Gibson Assembly Master Mix with the right targeting arm (RTA, the 1.9-kb genomic region downstream from the idtF gene, amplified using primers idtCBGF-RA-F and idtCBGF-RA-R). The resulting pAG-LTA-hph-RTA plasmid was selected by PCR after transformation of the assembly mixture into E. coli XL1Blue, and its proper construction was verified by sequencing. The successfully assembled pAG-LTA-hph-RTA plasmid was then transformed into A. tumefaciens LBA4404 electrocompetent cells (Takara Bio Inc., Kusatsu, Japan) and the transformants were selected on LB agar (Formedium, Hunstanton, UK) medium supplemented with 25 μg/mL kanamycin (final concentration).

Construction of the pAg-LTA-hph-RTA deletion vector. Left and right targeting arms (LTA and RTA, respectively) were generated by PCR using C. paspali DSM-833 genomic DNA as the template, and fused with the vector using in vitro isothermal Gibson assembly. Gene symbols: hph, hygromycin phosphotransferase; nptIII, neomycin phosphotransferase III (kanamycin resistance); trfA, replication initiation gene; oriV, vegetative replication origin; LB and RB, left and right T-DNA border, respectively
Agrobacterium tumefaciens-mediated transformation of C. paspali DSM-833
A. tumefaciens-mediated transformation of C. paspali was carried out according to Yamada et al. (2009) with modifications (Supplementary Fig. S1). C. paspali vegetative mycelium was collected from the surface of PDA agar slants and homogenized with a Dounce Homogenizer. The homogenized mycelium was suspended in 5 mL distilled water, inoculated into 50 mL potato dextrose broth (PDB, Sigma-Aldrich, St. Louis, MO) in a 500 mL Erlenmeyer flask and was cultivated for 48 h at 28 °C with shaking at 240 rpm. A 5 mL aliquot of this preculture was inoculated into 50 mL fresh PDB medium in a 500 mL Erlenmeyer flask and was cultured for an additional 24 h at 28 °C with shaking at 240 rpm. Mycelia were collected by centrifugation for 5 min at 2000 g, washed in 40 mL of distilled water, suspended at 100 mg/mL in IM broth (Induction Medium, Michielse et al. 2005), supplemented with 200 μM acetosyringone, and incubated for 8 h at 28 °C with shaking at 240 rpm.
A. tumefaciens LBA4404 cells carrying the pAG-LTA-hph-RTA plasmid were inoculated into 50 mL of LB medium (Sigma-Aldrich, St. Louis, MO) supplemented with kanamycin (25 μg/mL) and streptomycin (50 μg/mL) in a 500 mL Erlenmeyer flask, and cultivated at 30 °C for 48 h with shaking at 120 rpm. A 1 mL aliquot of this preculture was used to inoculate 50 mL of LB medium supplemented with 25 μg/mL kanamycin and 50 μg/mL streptomycin in a 500 mL Erlenmeyer flask, and was cultured at 30 °C for 12–16 h with shaking at 120 rpm. The cells were collected by centrifugation for 5 min at 2000 g, washed twice with distilled water, suspended in 50 mL IM broth containing 200 μM acetosyringone, and incubated at 30 °C for 8 h with shaking at 120 rpm.
One hundred microliter aliquots of the C. paspali mycelia and the A. tumefaciens cells were mixed, spread onto IM agar plates containing 200 μM acetosyringone (Michielse et al. 2005), and the co-culture was incubated for 2–6 days at 28 °C. To select transformants and inhibit the further growth of the A. tumefaciens cells, the IM agar plates (20 mL) were overlaid with 10 mL of top agar (PDA, 14.5 g/L) supplemented with 600 μg/mL hygromycin and 600 μg/mL cefotaxime (final concentrations calculated for the full plate: 200 μg/mL each), and the plates were incubated at 28 °C for an additional 10 days. Transformation efficiency was estimated by calculating the average number of hygromycin-resistant colonies per IM plates (i.e., per 10 mg wet C. paspali mycelium).
Co-cultivation was also attempted on IM agar plates covered with cellulose acetate ester membranes (0.45 μm, Whatman PLC, Maidstone, UK). After incubation for 4 days at 28 °C, the membrane with the cells was lifted to a fresh PDA agar plate supplemented with 200 μg/mL hygromycin and 200 μg/mL cefotaxime and the incubation was continued for a further 10 days at 28 °C.
Enrichment to obtain homokaryotic strains was achieved by macrodissection of hyphae from the leading edges of the hygromycin-resistant colonies with a sterile toothpick, and inoculation of the cells onto PDA plates containing 200 μg/mL hygromycin. The resulting colonies were incubated at 28 °C for 7 days, and re-isolation was repeated at least four times to facilitate homogenotization of the multinucleate cells under prolonged antibiotic selection.
Liquid chromatography—tandem mass spectrometry (LC-MS/MS) analysis
LC-MS/MS analysis was performed with an Agilent (Santa Clara, CA) 1290 Infinity LC System connected to an Agilent 6550 iFunnel Q-TOF mass spectrometer equipped with an electrospray (Dual AJS ESI) source, operated in positive ion mode. Liquid chromatographic separation was performed on a Kinetex XB-C18 analytical column (150 × 4.6 mm, 2.6 μm, Phenomenex, Torrance, CA) at 1.0 mL/min flow rate. A linear gradient profile was used with 5 mM ammonium acetate in water (A) and acetonitrile (B) as follows: 0–20 min, 30% B to 95% B; hold 95% B for 5 min, then return to 30% B over 0.2 min and hold for 4.8 min (total run time was 30 min). The column temperature was maintained at 30 °C, and UV absorption was recorded at λ = 230 and 280 nm (see detailed conditions for mass spectrometry in the Supplementary Table S2).
HPLC analysis of ergot alkaloids
Five grams of the fermentation broth of the appropriate C. paspali strain was collected in a 50 mL volumetric flask, the flask was filled up to the mark with acetonitrile:water (15:85), and the resulting solution was filtered using a 0.22 um membrane (Merck Millipore, Burlington, MA) before injection (10 μL) into the HPLC system. A Waters (Milford, MA) XBridge C18 (100 × 4.6 mm; 3.5 μm) analytical HPLC column was used for the analyses with the eluent system of (A) 10 mM monobasic sodium phosphate (pH 7.0, adjusted with 25% sodium hydroxide solution):acetonitrile = 90:10 (v/v); and (B) the same components in a 70:30 (v/v) ratio. The ratio of eluent B was changed during the gradient run according to the following profile: 0–16 min: 0 to 50%; 16–24 min: 50 to 100%; 24–24.1 min: 100 to 0%; 24.1–28 min: constant 0%. The flow rate was maintained at 1.0 mL/min. UV detection was performed at 310 nm, and the column and the sample compartment temperatures were kept at 25 and 5 °C, respectively. Ergonovine maleate (Sigma-Aldrich, St. Louis, MO) solution (100 μg/mL) was used as the external standard for quantitation. Main ergotamide compounds, such as ergonovine, lysergic acid methyl carbonyl amide, isolysergic acid methyl carbonyl amide, ergine, and erginine were identified by comparing their chromatographic mobility and UV absorption spectra with those of in-house reference materials.
Since lysergic acid and paspalic acid co-eluted under these conditions the separation of these compounds was achieved by a different HPLC method with UV detection at λ = 230 nm. In this assay, the samples were diluted twofold with acetonitrile:water = 5:95 (v/v) and filtered through a 0.22 μm membrane (Merck Millipore, Burlington, MA) prior to analysis. A reversed phase column (Zorbax Extend C18, 100 × 4.6 mm, 3.5 μm, Agilent Technologies, Santa Clara, CA) was used with an eluent system that consists of 2 g/L aqueous solution of ammonium carbamate (A) and acetonitrile (B), and the total flow rate was 1.7 mL/min. The ratio of eluent B was changed during the gradient run according to the following profile: 0–5 min: 10 to 15%; 5–7.5 min: 15 to 35%; 7.5–10 min: 35 to 80%; 10–12 min: constant 80%; 12–13 min: 80 to 10%; the total run time was 15 min. Typically, 20 μL aliquots of the specimens were injected onto the column, which was kept at 30 °C during the analyses. Product yields per 1 g C. paspali fermentation broths were determined and are presented as mean ± SD values, calculated from three independent experiments.
Results
Agrobacterium tumefaciens-mediated transformation of C. paspali
Although putative genes for the biosynthesis of IDTs in C. paspali were annotated (Schardl et al. 2013), experimental confirmation of the involvement of these genes in IDT production is still necessary. To provide such a proof, we set out to disrupt IDT biosynthesis in C. paspali DSM-833. Unfortunately, we found that protoplast-mediated transformation protocols developed previously for Claviceps spp. (van Engelenburg et al. 1989), at least in our hands, suffer from low protoplast regeneration efficiency when applied to C. paspali DSM-833, yield very few antibiotic-resistant putative transformants and that these transformants display high mitotic instability. This latter obstacle is partially due to the multinucleate nature of C. paspali hyphae (Hareven and Koltin 1970), whereby wild type nuclei without the incoming DNA persist in transformants even under antibiotic selection and are responsible for mixed phenotypes displayed by heterokaryotic mycelia. Worse, in the absence of selection (such as during industrial scale fermentation), these wild type nuclei displace those that have undergone transformation, leading to the de facto reversion of the transformed strain to the wild type. Thus, effective and industrially useful molecular genetic strain improvement of C. paspali demands the development of a more convenient and more reliable transformation procedure.
To devise such a protocol, we turned to A. tumefaciens-mediated transformation (ATMT, Supplementary Fig. S1), and set out to replace the idtCBGF genes with a selectable resistance marker gene. We built a derivative of the pAg-H3 binary vector (Zhang et al. 2003), pAg-LTA-hph-RTA (Fig. 1). In this vector, the hph hygromycin resistance gene is bracketed by a 1.5-kb left targeting arm and a 1.9-kb right targeting arm, taken from the sequences bordering the area to be deleted on the C. paspali genome as depicted on Fig. 1. Next, we determined the minimum inhibitory concentration of hygromycin against wild type C. paspali mycelia as 200 μg/mL (Supplementary Fig. S2), and optimized the A. tumefaciens cell density and the length of the co-incubation time of the bacterium with C. paspali (Fig. 2). The highest transformation frequency, approximately 80 hygromycin-resistant colonies per 10 mg wet C. paspali mycelium, was recorded at an A. tumefaciens cell density of OD600 = 0.5, after 4 days of co-incubation. Longer co-incubation or higher A. tumefaciens cell densities reduced the apparent efficiency of the transformation, likely due to the reduced efficiency of antibiotic selection against denser A. tumefaciens cultures reaching stationary phase, and the inhibitory/competitive effects of the denser bacterial cultures on developing C. paspali mycelia. We also confirmed the necessity to supplement the IM agar with acetosyringone during co-incubation in order to induce T-DNA formation in A. tumefaciens (Michielse et al. 2005, Supplementary Fig. S3). Finally, we found no improvement in transformation frequency when co-cultivation was carried out on the surface of a cellulose acetate ester membrane covering the agar plate, as opposed to plating the two microorganisms directly on IM agar plates (compare Supplementary Fig. S4 to Supplementary Fig. S3).

Optimization of the co-incubation step during ATMT. Effects of the length of the co-incubation period on the number of hygromycin-resistant C. paspali transformants. Co-incubations were conducted at three different cell densities of the A. tumefaciens culture. Transformant numbers (per 10 mg wet C. paspali mycelium plated) are shown as averages ± SD over three independent experiments
To test the stability of the transformants, 50 primary hygromycin-resistant colonies were re-isolated onto PDA agar supplemented with 200 μg/mL hygromycin. All of these transformants retained hygromycin resistance and displayed vigorous growth. During subsequent re-isolations, putative C. paspali transformants tended to develop larger colonies within the same incubation timespans, likely due to the enrichment of the transformed nuclei at the expense of the wild type ones within the mycelia.
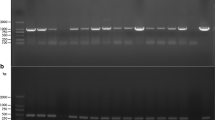
To check whether or not the hph gene integrated into the genomes of the transformants at the expected site and thus replaced the idtCBGF genes, genomic DNA was isolated from 12 independent hygromycin-resistant colonies after four rounds of re-isolation and were examined by a series of five PCR reactions (Fig. 3). All 12 strains were confirmed to carry the hph hygromycin resistance marker gene. Three out of the 12 transformants, CPIDT2, 8 and 9 were validated to have undergone site-specific integration of the hph gene with the concomitant loss of the genomic region encompassing the wild type idtCBGF genes. The absence of the idtC and idtF amplicons also demonstrated that these strains are homokaryotic for the gene replacement allele. One transformant, CPIDT5, yielded amplicons indicative of the presence of both the intact idtCBGF allele and the gene replacement allele, suggesting that this strain remained heterokaryotic even after repeated strain re-isolation. Finally, the rest of the transformants carried only the intact idtCBGF allele, indicating that the hph gene has undergone ectopic integration in these isolates.

PCR analysis of the genomic DNA of the transformants. a The hph gene is detected in C. paspali transformants CPIDT1–12, but not in the wild type control (WT), by amplifying a 415-bp internal fragment of the gene using the hph-F and hph-R primers. b PCR primers designed to detect the presence of the idtC (primer pair: idtC-F and idtC-R) and idtF genes (primer pair: idtF-F and idtF-R). The predicted sizes of the PCR amplicons are also shown. c PCR primers designed to detect the replacement of the idtCBGF genes by the hph gene (primer pairs: LOUT-F and hph-R; hph-F and LOUT-R) and the predicted sizes of the PCR amplicons. d PCR analyses of the CPIDT1-12 transformants and the wild type C. paspali for the presence of the idtC and idtF genes and for the correct site-specific integration of the hph gene
LC-MS/MS analysis of IDT production
Production of IDT congeners by the wild type C. paspali DSM-833 and its ∆idtCBGF replacement mutants CPIDT2 and CPIDT8 were investigated by liquid chromatography—high-resolution tandem mass spectrometry (LC-MS/MS) analysis of the extracts of 12 days old submerged liquid cultures (Fig. 4). We have tracked the production of five IDT congeners: paspaline, paxilline, paspalinine, and paspalitrems A and B (Fig. 4) by monitoring their retention times, corresponding [M + H]+ ions and their major fragment ions (Supplementary Table S3), and comparing these with literature examples (Uhlig et al. 2014), or with an available standard as in the case of paxilline. For paspalitrem A, comprehensive analysis of the mass spectrometric data (including fragment ions) was particularly important since the selected ion chromatogram of the extract from the wild type strain revealed several peaks with near-identical mass to charge ratios, whose fragmentation patterns nevertheless differed from that of paspalitrem A. Taken together, these analyses unequivocally showed that production of all monitored IDT congeners is completely abrogated in the ∆idtCBGF replacement mutants CPIDT2 and CPIDT8.

Metabolic profiling of C. paspali strains. Extracted ion chromatograms are presented for the [M + H]+ ions of key IDT products in extracts of the submerged cultures of the wild type C. paspali DSM-833 and the strain CPIDT2, a representative ∆idtCBGF mutant. The structures, molecular formulas, and the mass to charge ratios (m/z) of the [M + H]+ ions of the IDT congeners are also shown
Analysis of ergot alkaloid production
In order to examine whether alkaloid biosynthesis in C. paspali DSM-833 is affected by the inactivation of the idtCBGF genes, we examined the ergot alkaloid productivity of the wild type, CPIDT2 and CPIDT8 strains. HPLC analysis of the extracts of the fermentation broths showed (Supplementary Table S4, Supplementary Figs. S5 and S6) that the total ergot alkaloid productivities of these strains were statistically identical. Wild type C. paspali DSM-833 produced 18.30 ± 1.05 μg/g total ergot alkaloids including 1.25±0.09 μg/g paspalic acid. The CPIDT2 and CPIDT8 knockout strains produced 19.09 ± 1.30 μg/g and 18.41 ± 0.50 μg/g total ergot alkaloids, including 1.17±0.15 μg/g and 1.28±0.11 μg/g paspalic acid, respectively. Lysergic acid was not detectable in the fermentation broths of the wild type or the CPIDT2 or CPIDT8 strains (Supplementary Fig. S6). Distribution of the detected ergot alkaloids such as ergine, lysergic acid methyl carbonyl amide, ergonovine, isolysergic acid methyl carbonyl amide, erginine, and paspalic acid (Supplementary Table S4) were similarly not affected by the disruption of IDT biosynthesis.
Discussion
C. paspali is an ergot fungus of great veterinary, food security, public health, and pharmaceutical importance (Cawdell-Smith et al. 2010; Keller and Tudzynski 2002; Flieger et al. 2003; Hulvová et al. 2013). The aim of this study was to establish an easy-to-use and reliable genetic modification system for the stable transformation of C. paspali, and to validate this system by generating a functional proof for the involvement of putative core genes identified in the genome sequence of this species in the biosynthesis of IDTs (Schardl et al. 2013). The resulting convenient ATMT protocol and the functional verification of the IDT biosynthetic gene cluster pave the way for the development of new C. paspali mutant strains with superior productivity of various secondary metabolites, including ergot alkaloids and IDTs of pharmaceutical interest.
C. paspali has long been used in the pharmaceutical industry to produce lysergic acid and similar ergot alkaloids in submerged liquid cultures (Ricicová et al. 1982; Socic et al. 1986). Nevertheless, the targeted molecular genetic modification of this fungus remained a major challenge due to the low efficiencies of most steps in the transformation workflow, including protoplast regeneration, DNA introduction, mitotically stable integration of the incoming DNA, and homogenization of the transformant allele. Here, we report that ATMT can be successfully applied to overcome these challenges to generate targeted, stable, and homokaryotic gene replacements in the genome of C. paspali, and by extension, in other Claviceps spp.
ATMT was developed as an effective tool for the genetic engineering of a number of industrially important fungi. This method has been used to increase the production of various hydrolytic and biosynthetic enzymes and also to modify secondary metabolite profiles, e.g., via the elimination of polyketide synthases (Zhang et al. 2003). ATMT has also been employed successfully to verify the roles of selected genes in some biosynthetic pathways (Xu et al. 2008, 2009).
The most important factors for the successful application of ATMT for C. paspali are the cell density of the A. tumefaciens donor cells and the co-incubation time. Optimization of the hygromycin concentration to block the outgrowth of untransformed wild type colonies and supplementation of the IM medium with acetosyringone to activate T-DNA generation in A. tumefaciens cells were also essential. ATMT can also be carried out with a similarly high efficiency on cellulose acetate ester membranes laid on agar plates. To eliminate heterokaryotic cells that are typical in Claviceps species (Amici et al. 1967; Esser and Tudzynski 1978, Tudzynski et al. 2001), hygromycin-resistant primary colonies had to be re-isolated by repeated macrodissection of the edge of the colonies and serial passage on hygromycin-containing plates. The final, optimized workflow of C. paspali transformation by A. tumefaciens provided us with mitotically highly stable, homokaryotic transformants. The observed proportion of homologous recombination (~ 33%) vs. ectopic integration was also encouraging in this experiment.
Functional identification of the IDT biosynthetic gene cluster also allowed us to reconstruct the biochemical pathway leading to the biosynthesis of the tremorgenic IDTs paspalitrems A and B in C. paspali (Fig. 5). For this, we considered previous genome annotations, and the results of published chemical and functional analyses in P. paxilli (Saikia et al. 2006), Aspergillus flavus (Zhang et al. 2004), Penicillium janthinellum (Nicholson et al. 2015), and C. paspali itself (Cole et al. 1977; Schardl et al. 2013; Uhlig et al. 2014). The putative IDT biosynthetic gene cluster spans two separate contigs on the genome sequence assembly of C. paspali RRC-4128 (Schardl et al. 2013). Contig JN613321 includes idtQ, idtP, idtF, idtG, idtB, and idtC, while contig JN613322 harbors the idtM gene. The translated protein products of the C. paspali idtG, idtM, idtB, and idtC genes show 53, 38, 56, and 45% identities with the protein products of the paxG, paxM, paxB, and paxC genes resident in the paxilline cluster of P. paxilli. These genes encode a GGPP synthase, a FAD-dependent monooxygenase, an integral membrane protein and a prenyl transferase, respectively (Saikia et al. 2006; Scott et al. 2013). Collectively, these enzymes are responsible for the assembly of paspaline, the first stable, cyclic IDT intermediate (Saikia et al. 2006; Scott et al. 2013). The gene idtP codes for a putative cytochrome P450 monooxygenase that shows 41% identity to PaxP of P. paxilli (Scott et al. 2013). We propose that like PaxP, IdtP catalyzes the conversion of paspaline to 13-desoxypaxilline via the intermediate β-PC-M6 by removing the C-30 methyl group and installing the carbonyl oxygen at C-10 (Scott et al. 2013). The deduced IdtQ is a P450 monooxygenase that is orthologous to PaxQ of P. paxilli (37% identity). Just as PaxQ, IdtQ may catalyze the C-13 oxidation of 13-desoxypaxilline to afford paxilline (Scott et al. 2013). However, orthologues of IdtQ also take part in the biosynthesis of aflatrems and sheraninine A in A. flavus and P. janthinellum, respectively (Zhang et al. 2004; Ehrlich and Mack 2014; Nicholson et al. 2015). Just as AtmQ of the aflatrem producer A. flavus NRRL6541 (Nicholson et al. 2009), IdtQ may alternatively catalyze the formation of paspalinine from 13-desoxypaxilline via paspalicine as an intermediate. Considering that both paspalicine and paxilline were detected in the C. paspali—Paspalum dilatatum (dallisgrass) association (Uhlig et al. 2014), it is reasonable to assume that IdtQ does in fact catalyze both the C-13 and the C-7 oxidations of 13-desoxypaxilline. Whether paxilline may be converted to paspalinine by IdtQ, or paxilline represents a shunt product of the pathway remains to be determined. Finally, the prenylation of the C ring of paspalinine to form paspalitrem A is catalyzed by the IdtF prenyl transferase (Schardl et al. 2013), followed by hydroxylation at C-32 by a still unknown oxidase to afford paspalitrem B.

Proposed biosynthetic pathway for IDTs in C. paspali. IPP, isopentenyl diphosphate; DMAPP, dimethylallyl diphosphate; GGPP, geranylgeranyl diphosphate
In this study, we successfully replaced the idtCBGF region of the IDT biosynthetic gene cluster of C. paspali with the hph hygromycin resistance marker gene. This genomic region encodes three (IdtC, B, and G) of the four core biosynthetic enzymes necessary for the assembly of the hexacyclic skeleton of IDTs. It also encodes the prenyl transferase IdtF that decorates paspalinine to yield paspalitrem A. Replacement of these genes eliminated the biosynthesis of the whole spectrum of IDTs that have been reported to be produced by this ergot fungus (Cole et al. 1977; Uhlig et al. 2014), including paspaline, paspalinine, paxilline, paspalitrem A, and paspalitrem B. Importantly, the ∆idtCBGF strains continued to produce ergot alkaloids undisturbed. Elimination of IDT production thus not only verifies the deduced role of the putative IDT biosynthetic gene cluster of C. paspali (Schardl et al. 2013) but also serves as a model for the generation of similar mutants in industrially relevant C. paspali strains. The blockade of IDT biosynthesis in such mutants may reduce heightened competition for precursors and cofactors during high-titer ergot alkaloid production, and thus may improve ergot productivity. Further, utilization of such mutants may also reduce the costs associated with the downstream processing of ergot alkaloid products by eliminating the co-production of IDT mycotoxins during industrial fermentations.
The optimized ATMT protocol described in this article may be easily adapted for other Claviceps spp., and can find utility in other strain improvement projects. For example, the copy numbers of selected biosynthetic genes may be increased, or promoters and other regulatory elements may be replaced to improve the yields of desired metabolites. Deeper insights may also be gained about the organization, regulation, and evolution of industrially important secondary metabolite biosynthetic gene clusters present in the family Clavicipitaceae (Panaccione and Schardl 2003; Haarmann et al. 2005; Schardl et al. 2006, 2013; Hulvová et al. 2013; Young et al. 2015; Kishimoto et al. 2016). In addition, synthetic biological platforms based on C. paspali strains adapted for industrial scale fermentation may provide us with novel metabolites with useful biological activities. Outside the pharmaceutical industry, facile genetic manipulation of clavicipitalean fungi may also help us to shed light on the complex ecological interplay that these fungi engage in during plant-fungus-herbivore multitrophic interactions (Panaccione et al. 2006).
References
Amici AM, Scotti T, Spalla C, Tognoli L (1967) Heterokaryosis and alkaloid production in Claviceps purpurea. Appl Microbiol 15(3):611–615
Arcamone F, Bonino C, Chain EB, Ferretti A, Pennella P, Tonolo A, Vero L (1960) Production of lysergic acid derivatives by a strain of Claviceps paspali Stevens and Hall in submerged culture. Nature 187(4733):238–239. https://doi.org/10.1038/187238a0
Bennett JW, Klich M (2003) Mycotoxins. Clin Microbiol Rev 16(3):497–516. https://doi.org/10.1128/CMR.16.3.497-516.2003
Bundock P, den Dulk-Ras A, Beijersbergen A, Hooykaas PJ (1995) Trans-kingdom T-DNA transfer from Agrobacterium tumefaciens to Saccharomyces cerevisiae. EMBO J 14:3206–3214
Byrne KM, Smith SK, Ondeyka JG (2002) Biosynthesis of nodulisporic acid A: precursor studies. J Am Chem Soc 124(24):7055–7060. https://doi.org/10.1021/ja017183p
Cawdell-Smith AJ, Scrivener CJ, Bryden WL (2010) Staggers in horses grazing paspalum infected with Claviceps paspali. Aust Vet J 88(10):393–395. https://doi.org/10.1111/j.1751-0813.2010.00624.x
Chain EB, Bonino C, Tonolo A (1962) Process for the production of alkaloid derivatives of lysergic acid. US Patent Office 3,038,840
Cole RJ, Dorner JW, Lansden JA, Cox RH, Pape C, Cunfer B, Nicholson SS, Bedell DM (1977) Paspalum staggers: isolation and identification of tremorgenic metabolites from sclerotia of Claviceps paspali. J Agric Food Chem 25(5):1197–1201. https://doi.org/10.1021/jf60213a061
de Groot MJ, Bundock P, Hooykaas PJ, Beijersbergen AG (1998) Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat Biotechnol 16(9):839–842. https://doi.org/10.1038/nbt0998-839
di Menna ME, Finch SC, Popay AJ, Smith BL (2012) A review of the Neotyphodium lolii/Lolium perenne symbiosis and its associated effects on animal and plant health, with particular emphasis on ryegrass staggers. N Z Vet J 60(6):315–328. https://doi.org/10.1080/00480169.2012.697429
Ehrlich KC, Mack BM (2014) Comparison of expression of secondary metabolite biosynthesis cluster genes in Aspergillus flavus, A. parasiticus, and A. oryzae. Toxins (Basel) 6(6):1916–1928. https://doi.org/10.3390/toxins6061916
Esser K, Tudzynski P (1978) Genetics of the ergot fungus Claviceps purpurea: I. Proof of a monoecious life cycle and segregation patterns for mycelial morphology and alkaloid production. Theor Appl Genet 53(4):145–149. https://doi.org/10.1007/BF00273574
Flieger M, Mehta P, Mehta A (2003) Biotechnological potential of ergot alkaloids. In: Arora DK (ed) Fungal biotechnology in agricultural, food, and environmental applications. Marcel Dekker, New York, pp 91–99
Gritz L, Davies J (1983) Plasmid-encoded hygromycin B resistance: the sequence of hygromycin B phosphotransferase gene and its expression in Escherichia coli and Saccharomyces cerevisiae. Gene 25(2-3):179–188. https://doi.org/10.1016/0378-1119(83)90223-8
Haarmann T, Machado C, Lübbe Y, Correia T, Schardl CL, Panaccione DG, Tudzynski P (2005) The ergot alkaloid gene cluster in Claviceps purpurea: extension of the cluster sequence and intra species evolution. Phytochemistry 66(11):1312–1320. https://doi.org/10.1016/j.phytochem.2005.04.011
Hareven D, Koltin Y (1970) Nuclear distribution in the mycelium of Claviceps and the problem of strain selection. Appl Microbiol 19(6):1005–1006
Hulvová H, Galuszka P, Frébortová J, Frébort I (2013) Parasitic fungus Claviceps as a source for biotechnological production of ergot alkaloids. Biotechnol Adv 31(1):79–89. https://doi.org/10.1016/j.biotechadv.2012.01.005
Imlach WL, Finch SC, Zhang Y, Dunlop J, Dalziel JE (2011) Mechanism of action of lolitrem B, a fungal endophyte derived toxin that inhibits BK large conductance Ca2+-activated K+ channels. Toxicon 57(5):686–694. https://doi.org/10.1016/j.toxicon.2011.01.013
Keller U, Tudzynski P (2002) Ergot Alkaloids. In: Osiewacz HD (ed) Industrial applications. The Mycota (a comprehensive treatise on fungi as experimental systems for basic and applied research), vol 10. Springer, Berlin, pp 157–181. https://doi.org/10.1007/978-3-662-10378-4_8
Kishimoto S, Sato M, Tsunematsu Y, Watanabe K (2016) Evaluation of biosynthetic pathway and engineered biosynthesis of alkaloids. Molecules 21(8):e1078. https://doi.org/10.3390/molecules21081078
Kunitake E, Tani S, Sumitani J, Kawaguchi T (2013) A novel transcriptional regulator, ClbR, controls the cellobiose- and cellulose-responsive induction of cellulase and xylanase genes regulated by two distinct signaling pathways in Aspergillus aculeatus. Appl Microbiol Biotechnol 97(5):2017–2028. https://doi.org/10.1007/s00253-012-4305-8
Laws I, Mantle PG (1989) Experimental constraints in the study of the biosynthesis of indole alkaloids in fungi. J Gen Microbiol 135(10):2679–2692. https://doi.org/10.1099/00221287-135-10-2679
Liu C, Noike M, Minami A, Oikawa H, Dairi T (2014) A fungal prenyltransferase catalyzes the regular di-prenylation at positions 20 and 21 of paxilline. Biosci Biotechnol Biochem 78(3):448–454. https://doi.org/10.1080/09168451.2014.882759
McMillan LK, Carr RL, Young CA, Astin JW, Lowe RG, Parker EJ, Jameson GB, Finch SC, Miles CO, McManus OB, Schmalhofer WA, Garcia ML, Kaczorowski GJ, Goetz M, Tkacz JS, Scott B (2003) Molecular analysis of two cytochrome P450 monooxygenase genes required for paxilline biosynthesis in Penicillium paxilli, and effects of paxilline intermediates on mammalian maxi-K ion channels. Mol Gen Genomics 270(1):9–23. https://doi.org/10.1007/s00438-003-0887-2
Michielse CB, Hooykaas PJ, van den Hondel CA, Ram AF (2005) Agrobacterium-mediated transformation as a tool for functional genomics in fungi. Curr Genet 48(1):1–17. https://doi.org/10.1007/s00294-005-0578-0
Nicholson MJ, Koulman A, Monahan BJ, Pritchard BL, Payne GA, Scott B (2009) Identification of two aflatrem biosynthesis gene loci in Aspergillus flavus and metabolic engineering of Penicillium paxilli to elucidate their function. Appl Environ Microbiol 75(23):7469–7481. https://doi.org/10.1128/AEM.02146-08
Nicholson MJ, Eaton CJ, Stärkel C, Tapper BA, Cox MP, Scott B (2015) Molecular cloning and functional analysis of gene clusters for the biosynthesis of indole-diterpenes in Penicillium crustosum and P. janthinellum. Toxins (Basel) 7(8):2701–2722. https://doi.org/10.3390/toxins7082701
Păcurar DI, Thordal-Christensen H, Păcurar ML, Pamfil D, Botez C, Bellini C (2011) Agrobacterium tumefaciens: from crown gall tumors to genetic transformation. Physiol Mol Plant Pathol 76(2):76–81. https://doi.org/10.1016/j.pmpp.2011.06.004
Panaccione DG, Schardl CL (2003) Molecular genetics of ergot alkaloid biosynthesis. In: White JF Jr, Bacon CW, Hywel-Jones NL, Spatafora JW (eds) The clavicipitalean fungi: evolutionary biology, chemistry, biocontrol, and cultural impacts. Marcel-Dekker, New York, pp 399–424. https://doi.org/10.1201/9780203912706.ch13
Panaccione DG, Cipoletti JR, Sedlock AB, Blemings KP, Schardl CL, Machado C, Seidel GE (2006) Effects of ergot alkaloids on food preference and satiety in rabbits, as assessed with gene-knockout endophytes in perennial ryegrass (Lolium perenne). J Agric Food Chem 54(13):4582–4587. https://doi.org/10.1021/jf060626u
Panaccione DG, Beaulieu WT, Cook D (2014) Bioactive alkaloids in vertically transmitted fungal endophytes. Funct Ecol 28(2):299–314. https://doi.org/10.1111/1365-2435.12076
Parker EJ, Scott DB (2004) Indole-diterpene biosynthesis in ascomycetous fungi. In: An Z (ed) Handbook of Industrial Mycology, Vol. 22. Marcel Dekker, New York, pp. 405–426
Ricicová A, Flieger M, Rehácek Z (1982) Quantitative changes of the alkaloid complex in a submerged culture of Claviceps paspali. Folia Microbiol (Praha) 27(6):433–445. https://doi.org/10.1007/BF02876456
Saikia S, Parker EJ, Koulman A, Scott B (2006) Four gene products are required for the fungal synthesis of the indole-diterpene, paspaline. FEBS Lett 580(6):1625–1630. https://doi.org/10.1016/j.febslet.2006.02.008
Saikkonen K, Young CA, Helander M, Schardl CL (2016) Endophytic Epichloë species and their grass hosts: from evolution to applications. Plant Mol Biol 90(6):665–675. https://doi.org/10.1007/s11103-015-0399-6
Sallam AA, Ayoub NM, Foudah AI, Gissendanner CR, Meyer SA, El Sayed KA (2013) Indole diterpene alkaloids as novel inhibitors of the Wnt/β-catenin pathway in breast cancer cells. Eur J Med Chem 70:594–606. https://doi.org/10.1016/j.ejmech.2013.09.045
Schardl CL, Panaccione DG, Tudzynski P (2006) Ergot alkaloids—biology and molecular biology. Alkaloids Chem Biol 63:45–86. https://doi.org/10.1016/S1099-4831(06)63002-2
Schardl CL, Young CA, Hesse U, Amyotte SG, Andreeva K, Calie PJ, Fleetwood DJ, Haws DC, Moore N, Oeser B, Panaccione DG, Schweri KK, Voisey CR, Farman ML, Jaromczyk JW, Roe BA, O’Sullivan DM, Scott B, Tudzynski P, An Z, Arnaoudova EG, Bullock CT, Charlton ND, Chen L, Cox M, Dinkins RD, Florea S, Glenn AE, Gordon A, Güldener U, Harris DR, Hollin W, Jaromczyk J, Johnson RD, Khan AK, Leistner E, Leuchtmann A, Li C, Liu J, Liu J, Liu M, Mace W, Machado C, Nagabhyru P, Pan J, Schmid J, Sugawara K, Steiner U, Takach JE, Tanaka E, Webb JS, Wilson EV, Wiseman JL, Yoshida R, Zeng Z (2013) Plant-symbiotic fungi as chemical engineers: multi-genome analysis of the Clavicipitaceae reveals dynamics of alkaloid loci. PLoS Genet 9(2):e1003323. https://doi.org/10.1371/journal.pgen.1003323
Scott B, Young CA, Saikia S, McMillan LK, Monahan BJ, Koulman A, Astin J, Eaton CJ, Bryant A, Wrenn RE, Finch SC, Tapper BA, Parker EJ, Jameson GB (2013) Deletion and gene expression analyses define the paxilline biosynthetic gene cluster in Penicillium paxilli. Toxins (Basel) 5(8):1422–1446. https://doi.org/10.3390/toxins5081422
Socic H, Gaberc-Porekar V, Pertot E, Puc A, Milicić S (1986) Developmental studies of Claviceps paspali seed cultures for the submerged production of lysergic acid derivatives. J Basic Microbiol 26(9):533–539. https://doi.org/10.1002/jobm.3620260906
Thom ER, Popay AJ, Waugh CD, Minne EMK (2014) Impact of novel endophytes in perennial ryegrass on herbage production and insect pests from pastures under dairy cow grazing in northern New Zealand. Grass Forage Sci 69(1):191–204. https://doi.org/10.1111/gfs.12040
Tudzynski P, Correia T, Keller U (2001) Biotechnology and genetics of ergot alkaloids. Appl Microbiol Biotechnol 57(5-6):593–605. https://doi.org/10.1007/s002530100801
Uhlig S, Botha CJ, Vrålstad T, Rolén E, Miles CO (2009) Indole-diterpenes and ergot alkaloids in Cynodon dactylon (Bermuda grass) infected with Claviceps cynodontis from an outbreak of tremors in cattle. J Agric Food Chem 57(23):11112–11119. https://doi.org/10.1021/jf902208w
Uhlig S, Egge-Jacobsen W, Vrålstad T, Miles CO (2014) Indole-diterpenoid profiles of Claviceps paspali and Claviceps purpurea from high-resolution Fourier transform Orbitrap mass spectrometry. Rapid Commun Mass Spectrom 28(14):1621–1634. https://doi.org/10.1002/rcm.6938
van Engelenburg F, Smit R, Goosen T, van den Broek H, Tudzynski P (1989) Transformation of Claviceps purpurea using a bleomycin resistance gene. Appl Microbiol Biotechnol 30(4):364–370. https://doi.org/10.1007/BF00296625
Wiewióra B, Żurek G, Pańka D (2015) Is vertical transmission of Neotyphodium lolli in perennial ryegrass the only possible way to the spread of endophytes? PLoS One 10(2):e0117231. https://doi.org/10.1371/journal.pone.0117231
Xu Y, Orozco R, Wijeratne KEM, Gunatilaka LAA, Stock SP, Molnár I (2008) Biosynthesis of the cyclooligomer depsipeptide beauvericin, a virulence factor of the entomopathogenic fungus Beauveria bassiana. Chem Biol 15(9):898–907. https://doi.org/10.1016/j.chembiol.2008.07.011
Xu Y, Orozco R, Wijeratne KEM, Espinosa-Artiles P, Gunatilaka LAA, Stock SP, Molnár I (2009) Biosynthesis of the cyclooligomer depsipeptide bassianolide, an insecticidal virulence factor of Beauveria bassiana. Fungal Genet Biol 46(5):353–364. https://doi.org/10.1016/j.fgb.2009.03.001
Yamada M, Yawata K, Orino Y, Ueda S, Isogai Y, Taguchi G, Shimosaka M, Hashimoto S (2009) Agrobacterium tumefaciens-mediated transformation of antifungal-lipopeptide-producing fungus Coleophoma empetri F-11899. Curr Genet 55(6):623–630. https://doi.org/10.1007/s00294-009-0275-5
Young C, McMillan L, Telfer E, Scott B (2001) Molecular cloning and genetic analysis of an indole-diterpene gene cluster from Penicillium paxilli. Mol Microbiol 39(3):754–764. https://doi.org/10.1046/j.1365-2958.2001.02265.x
Young CA, Bryant MK, Christensen MJ, Tapper BA, Bryan GT, Scott B (2005) Molecular cloning and genetic analysis of a symbiosis-expressed gene cluster for lolitrem biosynthesis from a mutualistic endophyte of perennial ryegrass. Mol Gen Genomics 274(1):13–29. https://doi.org/10.1007/s00438-005-1130-0
Young CA, Felitti S, Shields K, Spangenberg G, Johnson RD, Bryan GT, Saikia S, Scott B (2006) A complex gene cluster for indole-diterpene biosynthesis in the grass endophyte Neotyphodium lolii. Fungal Genet Biol 43(10):679–693. https://doi.org/10.1016/j.fgb.2006.04.004
Young C, Schardl CL, Panaccione DG, Florea S, Takach JE, Charlton ND, Moore N, Webb JS, Jaromczyk J (2015) Genetics, genomics and evolution of ergot alkaloid diversity. Toxins (Basel) 7(4):1273–1302. https://doi.org/10.3390/toxins7041273
Zhang A, Lu P, Dahl-Roshak AM, Paress PS, Kennedy S, Tkacz JS, An Z (2003) Efficient disruption of a polyketide synthase gene (pks1) required for melanin synthesis through Agrobacterium-mediated transformation of Glarea lozoyensis. Mol Gen Genomics 268(5):645–655. https://doi.org/10.1007/s00438-002-0780-4
Zhang S, Monahan BJ, Tkacz JS, Scott B (2004) Indole-diterpene gene cluster from Aspergillus flavus. Appl Environ Microbiol 70(11):6875–6883. https://doi.org/10.1128/AEM.70.11.6875-6883.2004
Zhong YH, Wang XL, Wang TH, Jiang Q (2007) Agrobacterium-mediated transformation (AMT) of Trichoderma reesei as an efficient tool for random insertional mutagenesis. Appl Microbiol Biotechnol 73(6):1348–1354. https://doi.org/10.1007/s00253-006-0603-3
Funding
This work was supported by the European Union and the European Social Fund through the project EFOP-3.6.1-16-2016-00022 (to IP) and by the National Institutes of Health project NIGMS R01GM114418-01A1 (to IM).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
IM has a disclosed financial interest in, and LK, ZS, BV, AK, and LT are employees of Teva Pharmaceutical Works Ltd., Hungary. Responsibility for the design of the experiments, the evaluation of the results, the conclusions drawn, and the opinions expressed in this article are solely those of the authors and are not shared by Teva Pharmaceutical Works Ltd. IP declares no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the Authors.
Electronic supplementary material
ESM 1
(PDF 737 kb)
Rights and permissions
About this article

Cite this article
Kozák, L., Szilágyi, Z., Vágó, B. et al. Inactivation of the indole-diterpene biosynthetic gene cluster of Claviceps paspali by Agrobacterium-mediated gene replacement. Appl Microbiol Biotechnol 102, 3255–3266 (2018). https://doi.org/10.1007/s00253-018-8807-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-8807-x