
Abstract
Lysine acetylation is a dynamic, reversible posttranslational modification that is known to play an important role in regulating the activity of many key enzymes in bacteria. Acetylproteome studies have been performed on some bacteria. However, until now, there have been no data on Actinomycetes, which are the major producers of therapeutic antibiotics. In this study, we investigated the first acetylproteome of the erythromycin-producing actinomycete Saccharopolyspora erythraea using a high-resolution mass spectrometry-based proteomics approach. Using immune-affinity isolation of acetyl-peptides with an anti-acetyllysine antibody followed by nano ultra performance liquid chromatography tandem mass spectroscopy (nanoUPLC-MS/MS) analysis, we identified 664 unique lysine-acetylated sites on 363 proteins. Acetylated proteins are involved in many biological processes such as protein synthesis, glycolysis/gluconeogenesis, citric acid (TCA) cycle, fatty acid metabolism, secondary metabolism, and the feeder metabolic pathways of erythromycin synthesis. We characterized the acetylproteome and analyzed in detail the impact of acetylation on diverse cellular functions according to Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Four motif sequences surrounding the acetylation sites (KACH, KACY, KACXXXXR, and KACXXXXK) were found in the S. erythraea acetylproteome. These findings suggest that abundant lysine acetylation occurs in Actinomycetes, expand our current knowledge of the bacterial acetylproteome, and provide insight into the regulatory function of acetylation in primary and secondary metabolism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The dynamic and reversible Nε-lysine (Nε-Lys) acetylation (LysAc or Kac) of proteins is now recognized as a ubiquitous posttranslational modification (PTM) in prokaryotes and eukaryotes. Since the discovery of acetylation of the Salmonella enterica acetyl-CoA synthetase in 2002 (Starai et al. 2002), studies have shown that this PTM plays important regulatory roles in transcription, translation, and metabolism in bacteria. Recently, advancements in mass spectrometry (MS) and high-affinity purification of acetylated peptides have made it possible to identify thousands of lysine acetylation sites (the acetylproteome) in prokaryotic and eukaryotic cells. Acetylproteome studies have been conducted in some microorganisms, including Escherichia coli (Zhang et al. 2009, 2013; Yu et al. 2008; Colak et al. 2013; Weinert et al. 2013; Kuhn et al. 2014), Salmonella enterica (Wang et al. 2010), Bacillus subtilis (Kim et al. 2013), Geobacillus kaustophilus (Lee et al. 2013), Erwinia amylovora (Wu et al. 2013), and Thermus thermophilus (Okanishi et al. 2013). It was found that abundant lysine acetylation occurs in many bacterial proteins that are involved in protein synthesis, central metabolism, the stress response, and detoxification metabolism. Yu et al. (2008) identified 125 lysine acetylation sites in 85 proteins from E. coli strain W3110 grown in LB medium in both log and stationary phase cells, and 24 (28 %) of these are involved in protein biosynthesis and 16 (19 %) are involved in carbohydrate metabolism. Zhang et al. (2009) also reported that affinity enrichment of acetylated peptides followed by nanoscale liquid chromatograph conjugated tandem mass analysis (nanoLC-MS/MS) revealed 138 lysine acetylation sites in 91 proteins in E. coli MG1655. More than 70 % of these acetylated proteins are metabolic enzymes (53 %) and translation regulators (22 %). More recently, two large-scale studies of acetylated proteins in E. coli were performed by combining immunoaffinity enrichment with high-sensitivity mass spectrometry. More than 1070 lysine acetylation sites in 349 proteins were found in E. coli K12 DH10 (Zhang et al. 2013). Colak et al. (2013) reported the identification of 2803 lysine acetylation sites in 782 proteins, representing the largest LysAc dataset in wild-type E. coli. Wang et al. (2010) reported that 235 acetylated sites on 191 proteins were under LysAc modification in S. enterica grown in glucose-rich medium and found that enzymes involved the central metabolism are extensively acetylated and that their acetylation profiles change in response to different carbon sources, concomitant with changes in cell growth and metabolic flux. In a model gram-positive bacterium, B. subtilis, 332 unique lysine acetylation sites in 185 proteins were identified through MS-based proteomics (Kim et al. 2013). A similar proteomic analysis of LysAc has been performed in the gram-positive thermophilic bacterium G. kaustophilus, and the results revealed 253 LysAc sites in 114 proteins that function in a wide range of biological pathways (Lee et al. 2013). Wu et al. (2013) identified 141 LysAc sites in 96 proteins in E. amylovora, an enterobacterium that causes a serious fire blight disease of apples and pears, when grown in minimal medium at stationary phase and 20 LysAc sites in 17 proteins were common to both Ea273 and Ea1189 strains. Acetylproteome analysis with structural mapping revealed 197 lysine acetylation sites in 128 proteins in T. thermophilus HB8, an extremely thermophilic eubacterium (Okanishi et al. 2013). These results demonstrate that lysine acetylation occurs in several hundred proteins involved in a wide range of cellular functions, implying that acetylation as a PTM plays an extensive and well-conserved role in the regulation of transcription, translation, and metabolism in prokaryotic cells. Acetylproteome studies have greatly expanded our understanding of this PTM in various cellular processes. However, until now, no acetylproteome data were available in any high G + C gram-positive actinomycete, which are the main producers of therapeutic antibiotics and anticancer drugs. Knowledge on lysine acetylation and its functions in Actinomycetes is sparse. In some pathogenic Actinomycetes, e.g., Mycobacterium tuberculosis and Mycobacterium smegmatis, both Ac-CoA synthetase and a universal stress protein were found to be acetylated by two protein acetyltransferases, MtPatA and MsPat (Nambi et al. 2010; Xu et al. 2011).
For Streptomyces species, a SePat homologue (SlPatA) GNAT in Streptomyces lividans has been shown to have protein acetyltransferase activity and can acetylate acetoacetyl-CoA synthetase (Tucker et al. 2013). Recently, it was found that Ac-CoA synthetase was regulated in vivo by acetylation in Streptomyces coelicolor; however, the specific acetyltransferase responsible for this acetylation remained unidentified (Mikulik et al. 2012). These results suggest that reversible acetylation may also be a conserved regulatory PTM strategy in Actinomycetes. Further understanding of the role of lysine acetylation in the regulation of metabolism in Actinomycetes is of interest due to the diversity of the natural products synthesized by these organisms.
In this study, we conducted the first investigation of the acetylproteome of the actinomycete Saccharopolyspora erythraea using a high-resolution mass spectrometry-based proteomics approach. We identified 664 unique lysine acetylation sites in 363 proteins. It was found that the acetylated proteins are involved in many different metabolic processes, such as protein synthesis, glycolysis/gluconeogenesis, the TCA cycle, fatty acid metabolism, secondary metabolism, and the feeder metabolic pathways of erythromycin synthesis. We characterized the acetylproteome and analyzed in detail the impact of acetylation on diverse cellular functions according to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The four motif sequences surrounding acetylation sites, KACH, KACXXXXK, KACXXXXR, and KACY, were found in the S. erythraea acetylproteome. To the best of our knowledge, this is the first report that comprehensively discusses the acetylproteome of the actinomycete.
Materials and methods
Materials
Water and acetonitrile were from Fisher Scientific (Pittsburgh, PA). Trifluoroacetic acid (TFA) was from Sigma-Aldrich (St. Louis, MO). Tryptone soya broth and yeast extract were from Oxoid (Basingstoke, UK). Protease inhibitor cocktail was from Calbiochem (Darmstadt, Germany). Nicotinamide, trichostatin A, and the ECL kit were from Beyotime (Beijing, China). Sequencing-grade trypsin was from Promega (Madison, WI). C18 ZipTips and iodoacetamide were from Millipore (Bedford, MA). Protein A-conjugated agarose beads were from Amersham Biosciences (Fairfield, CT). Luna C18 resin was from Phenomenex (Torrance, CA). Anti-acetyllysine pan antibodies were from PTM Biolabs, Inc. (Chicago, IL).
Preparation of crude extract
S. erythraea NRRL23338 was grown in tryptone soya broth with yeast (TSBY) medium (30 g of tryptone soya broth and 5 g of yeast extract per liter of distilled H2O) at 30 °C and 200 rpm. The cultured cells were harvested during the exponential growth phase by centrifugation at 4500×g for 10 min and washed three times by resuspending the pellet in ice-cold phosphate-buffered saline buffer (0.1 M Na2HPO4, 0.15 M NaCl, pH 7.2). The cells were broken by liquid nitrogen grinding and then resuspended in chilled lysis buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5 mM dithiothreitol, protease inhibitor cocktail [Protease Inhibitor Cocktail Set III; Calbiochem] and HDAC inhibitor [50 mM sodium butyrate, 30 mM nicotinamide, 3 μM trichostatin A]). Unbroken cells and cell debris were removed by centrifugation at 4 °C for 10 min at 20,000×g. The supernatant was divided into aliquots and stored at −80 °C until use.
Western blotting analysis
S. erythraea NRRL23338 was grown, harvested, and lysed as described in the “Preparation of crude extract” section. Protein concentration was determined using the Bradford assay (Bio-Rad, Hercules, CA). Proteins (150 μg) were resolved by 10 % SDS-PAGE and transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 3 % BSA at room temperature for 1 h. Then, the membrane was incubated with anti-acetyllysine antibody (1:1000, catalog no. PTM-101) in TBST (25 mM Tris-HCl, pH 8.0, 125 mM NaCl, 0.1 % Tween 20) with 3 % BSA. After the membrane was washed with TBST four times for 5 min each, it was incubated with horseradish peroxidase-conjugated anti-mouse IgG (1 μg/mL in TBST with 3 % BSA) at room temperature for 1 h. After the membrane was washed with TBST three times for 10 min each, an ECL kit was used for signal detection.
In-solution tryptic digestion
Five milligrams of protein was precipitated with 20 % trichloroacetic acid overnight at 4 °C. The resulting precipitate was washed three times with ice-cold acetone. The air-dried precipitate was resuspended in 100 mM NH4HCO3 and then digested with trypsin at an enzyme-to-substrate ratio of 1:50 for 12 h at 37 °C. The tryptic peptides were reduced with 5 mM dithiothreitol at 56 °C for 45 min and then alkylated with 15 mM iodoacetamide for 30 min at room temperature in the dark. The reaction was terminated with 30 mM cysteine at room temperature for 20 min. To ensure complete digestion, additional trypsin at an enzyme-to-substrate ratio of 1:100 was added, and the mixture was incubated for an additional 4 h. The digested peptides were dried in a SpeedVac (Thermo Scientific, Pittsburgh, PA).
Enrichment of lysine-acetylated peptides
The tryptic peptides obtained from in-solution digestion were dissolved in NETN buffer (100 mM NaCl, 1 mM EDTA, 50 mM Tris, 0.5 % Nonidet P-40, pH 8.0) and incubated with anti-acetyllysine agarose beads (catalog no. PTM-104) at 4 °C overnight with gentle shaking. After incubation, the beads were carefully washed three times with NETN buffer, twice with ETN buffer (100 mM NaCl, 1 mM EDTA, 50 mM Tris, pH 8.0), and once with water. The bound peptides were eluted from the beads with 1 % trifluoroacetic acid and dried in a SpeedVac. The resulting peptides were cleaned with C18 ZipTips according to the manufacturer’s instructions, prior to nano ultra performance liquid chromatography tandem mass spectroscopy (nanoUPLC-MS/MS) analysis.
NanoUPLC-MS/MS and protein sequence database search
The peptides were resuspended in buffer A (2 % ACN, 0.1 % FA) and centrifuged at 20,000×g for 2 min. The supernatant was transferred to a sample tube and loaded onto an Acclaim PepMap 100 C18 trap column (75 μm × 2 cm; Dionex, Sunnyvale, CA) on an EASY nLC1000 nanoUPLC (Thermo Scientific, Pittsburgh, PA), and then the peptides were eluted onto an Acclaim PepMap RSLC C18 analytical column (50 μm × 15 cm; Dionex). A 34-min gradient was run at a rate of 300 nL/min, from 5 to 30 % B (80 % ACN, 0.1 % FA), followed by a 2-min linear gradient from 30 to 40 % B, and a 2-min gradient from 40 to 80 % B, and then 80 % B was maintained for 4 min.
The peptides were subjected to nanoelectrospray ionization (NSI) followed by tandem mass spectrometry (MS/MS) using the Q Exactive, a High-Performance Benchtop Quadrupole Orbitrap Mass Spectrometer (Thermo Scientific, Pittsburgh, PA) coupled to the nanoUPLC. Intact peptides were detected in the Orbitrap at a resolution of 70,000. Peptides were selected for MS/MS using 25 % normalized collision energy (NCE) with 4 % stepped NCE, and ion fragments were detected in the Orbitrap at a resolution of 17,500. A data-dependent procedure that alternated between one MS scan followed by 15 MS/MS scans was used for the top 15 precursor ions above a threshold ion count of 40,000 in the MS survey scan with 2.5-s dynamic exclusion. The electrospray voltage was 1.8 kV. Automatic gain control (AGC) was used to prevent overfilling of the ion trap; the ion count is 200,000 and they were accumulated to generate the MS/MS spectra. For MS scans, the m/z scan range was 350 m/z to 1800 m/z.
Protein and acetylation site identification was performed by MaxQuant (http://www.maxquant.org/) with an integrated Andromeda search engine (v. 1.3.0.5). Tandem mass spectra were searched against the UniProt (http://www.uniprot.org/) S. erythraea NRRL23338 protein database (7165 sequences) concatenated with a reverse decoy database and protein sequences of common contaminants such as keratin, hemoglobin, lactoglobulin, etc. Trypsin/P was specified as cleavage enzyme, and up to three missed cleavages were allowed, as well as four modifications per peptide, and five charges. Mass error was set to 6 ppm for precursor ions and 0.02 m/z for fragment ions. Carbamidomethylation on Cys was specified as a fixed modification, whereas oxidation on Met, acetylation on Lys, and acetylation on protein N-terminal were specified as variable modifications. False discovery rate (FDR) thresholds for protein, peptide, and modification sites were specified at 0.01. The minimum peptide length was set at 7. All other parameters in MaxQuant were set to the default values. Lys acetylation site identifications with a localization probability less than 0.75 or from reverse or contaminant protein sequences were removed. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (Vizcaíno et al. 2014) via the PRIDE partner repository with the dataset identifier PXD001238.
Bioinformatic analysis
Protein functional annotation and enrichment analysis
A GO annotation proteome was derived from the UniProt-GOA Database (http://www.ebi.ac.uk/GOA/). Proteins were classified by GO annotation based on three categories: biological process, cellular compartment, and molecular function. In addition, we used the KEGG (http://www.genome.jp/kegg/) to annotate pathways. First, we annotated proteins using the KEGG online service tool KEGG Automatic Annotation Server (KAAS) (http://www.genome.jp/tools/kaas/). Then, we mapped the annotation results to the KEGG pathway database (http://www.kegg.jp/kegg/pathway.html) using the KEGG online service tool KEGG mapper. Fisher’s exact test was used to test for GO and KEGG pathway enrichment or depletion (two-tailed test) of specific annotation terms among members of the resulting protein clusters. Derived p values were further adjusted to address multiple hypotheses testing by the method proposed by Benjamini and Hochberg (1995). Any terms with adjusted p values less than 0.05 in any of the clusters were treated as significant. For further hierarchical clustering based on GO terms, we first collated all the categories obtained after enrichment along with their p values, and then filtered for those categories, which were enriched in at least one of the clusters with a p value less than 0.05. This filtered p value matrix was transformed by the function x = −log10 (p value). Finally, these x values were z-transformed for each category. These z scores were then clustered by one-way hierarchical clustering (Euclidean distance, average linkage clustering) in Genesis. Cluster membership was visualized by using a heat map with the “heatmap.2” function from the “gplots” R-package.
Modeling of the sequences around the acetylation site
Soft motif-x (http://motif-x.med.harvard.edu/motif-x.html), a software tool designed to extract overrepresented patterns from any sequence dataset, was used to analyze the model of sequences with amino acids in specific positions of acety-21-mers (ten amino acids upstream and downstream of the acetylation site) in all protein sequences. The algorithm is an iterative strategy that builds successive motifs through comparison to a dynamic statistical background. All the database protein sequences were used as background database parameters and the other parameters were set at the default.
Interaction networks and domain architecture
We analyzed protein-protein interactions by using Cytoscape software (the Institute of Systems Biology, Seattle, WA) and direct physical interactions obtained from the STRING database (http://string.embl.de/). The InterPro database and InterProScan (a sequence analysis application) were used to annotate protein domains.
Conservation of acetylated lysines
The lysine conservation in each species was determined by counting the total number of conserved acetylated lysines, and then the total number of conserved nonacetylated lysines was considered conserved if both the S. erythraea protein and the query protein in the multiple sequence alignment have lysine residues at the aligned position. All the lysine residues of the proteins identified in this study were considered as controls. Mean conservation of the acetylated and nonacetylated lysines between the S. erythraea sequences and orthologous protein sequences from other actinomycete species were plotted separately. The p values were calculated for each comparison using Fisher’s exact test.
Results
Identification and analysis of lysine-acetylated proteins in S. erythraea
S. erythraea, a gram-positive filamentous bacterium, has been used for industrial-scale production of erythromycin A, a broad spectrum macrolide antibiotic against pathogenic gram-positive bacteria. The approximate 8.2-Mb genome of S. erythraea strain NRRL23338 was sequenced in 2007, and it encodes 7198 proteins (Oliynyk et al. 2007). To evaluate the extent of acetylation in the S. erythraea proteome, a cell-free protein extract derived from S. erythraea NRRL23338 grown in TSBY medium at the exponential growth phase was subjected to SDS-PAGE and Western blotting analysis with an anti-acetyllysine antibody. As shown in Fig. 1a, many bands with masses spanning a wide range visualized by Coomassie brilliant blue staining were found to exhibit strong reactivity with the anti-acetyllysine antibody, indicating abundant lysine acetylation in diverse S. erythraea proteins.

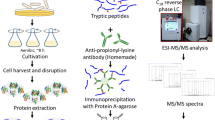
Outline of the experiments. a Western blotting analysis of lysine-acetylated proteins (150 μg total protein extracted from S. erythraea). The Western blotting result is shown on the right, and the Coomassie brilliant blue-stained SDS-PAGE is shown on the left. b Schematic showing an outline of the procedure to identify lysine-acetylated peptides through immune-affinity enrichment and high-sensitivity mass spectrometry
Global analysis of the acetylproteome was performed by selective enrichment of acetylated peptides, combined with highly sensitive Orbitrap mass spectrometry and bioinformatic analyses to systematically identify lysine-acetylated proteins in S. erythraea. In essence, the proteome-wide analysis of lysine acetylation consisted of three steps (as shown in Fig. 1b): (i) proteins from a whole cell lysate were digested with trypsin, affinity captured, and isolated with an anti-acetyllysine antibody; (ii) isolated lysine-acetylated peptides were analyzed by using high-sensitivity nanoUPLC-MS/MS; and (iii) lysine acetylation sites were identified by sequence alignment and characterization of protein lysine acetylation according to protein function, pathway, and interaction networks. Using the strategy described above, we identified 664 acetylated sites that matched 363 unique proteins from S. erythraea (see Table S1 in the Supplementary Material), demonstrating that 5 % of the proteins in S. erythraea are acetylated. The results were consistent with the ratios of acetylproteins to nonacetylated proteins in E. coli (7.8 %), T. thermophilus (5.7 %), B. subtilis (4.5 %), S. enterica (4.2 %), G. kaustophilus (3.1 %), and E. amylovora (2.6 %).
In this study, the S. erythraea acetylproteome showed about 1.8 lysine acetylation sites per protein. To investigate the distribution of the identified modification sites in the S. erythraea acetylproteome, we calculated the number of lysine acetylation sites per protein (Fig. 2a). More than 62 % of the proteins contained only one acetylation site, whereas approximately 38 % of the proteins were acetylated at multiple lysines. Our dataset contained 18 highly lysine-acetylated proteins with more than five LysAc sites, which mainly included metabolic enzymes: Ppa (SACE_0391, inorganic pyrophosphatase), FtsH (SACE_0396, ATP-dependent zinc metalloprotease) (Fig. 2b), ClpC (SACE_0414, putative ATP-dependent Clp protease), DldH2 (SACE_1639, dihydrolipoyl dehydrogenase), SACE_1764 (ribonucleoside diphosphate reductase) (Fig. 2d), TpiA (SACE_2145, triosephosphate isomerase), SahH (SACE_3897, adenosylhomocysteinase) (Fig. 2c), Kgd (SACE_6385, 2-oxoglutarate dehydrogenase E1 component), Icd (SACE_6636, isocitrate dehydrogenase), and SucC (SACE_6669, succinyl-CoA ligase subunit beta) and translation proteins: InfB (SACE_5926, translation initiation factor IF-2), RplF (SACE_6821, 50S ribosomal protein L6), RplC (SACE_6836, 50S ribosomal protein L3), Tsf (SACE_6037, elongation factor Ts), Tufa (SACE_6838, elongation factor Tu), FusA (SACE_6839, elongation factor G), and RplL (SACE_6867, 50S ribosomal protein L7/L12). Among the 18 proteins, Kgd had 14 lysine acetylation sites and GroEL (SACE_0527, 60 kDa chaperonin 1) was acetylated at 13 lysine residues, which is similar to the highly acetylated GroEL (TTHA0271, 12 lysine acetylation sites) reported in T. thermophilus. The acetylation was identified at 12 lysines in RpoC (SACE_6853, DNA-directed RNA polymerase subunit beta) and at 11 lysines in FusA elongation factor.

Acetylation sites of acetylproteome in S. erythraea. a Number of lysine acetylation sites identified per protein; annotation of lysine acetylation sites in three newly identified highly acetylated proteins: ATP-dependent zinc metalloprotease (b), adenosylhomocysteinase (c), and B12-dependent ribonucleoside diphosphate reductase (d)
Several representative MS/MS spectra of the newly identified lysine-acetylated peptides from S. erythraea proteins are presented in Fig. 3a (high-resolution spectra are presented in Fig. S1 in the Supplementary Material), including TFMKacWGAEIPAR from acetyl-coenzyme A synthetase (Sace_2375), FPLMPSGKacVDR from a putative nonribosomal peptide synthetase (Sace_2696), GPELEKacR from antibiotic biosynthesis monooxygenase (Sace_0428), and LAAGTAVKacSAQGR from TDP-4-keto-6-deoxyhexose 2,3-reductase (Sace_0727). Each acetylpeptide in our dataset was identified by MaxQuant at a FDR threshold of 0.01. Minimum peptide length was set at 7. Lysine acetylation site identifications with localization probabilities less than 0.75 or from reverse or contaminant protein sequences were removed.

Identification of acetylated proteins. a Representative MS/MS spectra of acetyl-peptides from proteins in S. erythraea; high-resolution MS/MS spectra are presented in Fig. S1 in the Supplementary Material. b Western analysis of immunoprecipitated acetyl-coenzyme A synthetase (AcsA) blotted with an anti-acetylated lysine antibody. Acetylated-BSA (Ac-BSA) was used as a positive control, and purified AcsA (AcsA) was used as a negative control
Acetyl-coenzyme A synthetase (AcsA) was selected for verification of acetylation in S. erythraea. To investigate whether AcsA is acetylated in vivo, the acetylation of acetyl-CoA synthetases was visualized by using immunoprecipitation (IP) and immunoblotting (IB) analyses. Proteins from S. erythraea cells were purified and immunoprecipitated with an antibody against AcsA (encoded by SACE_2375). Acetyl-lysines were detected in the AcsA immunoprecipitates with an anti-acetylated lysine antibody. As expected, the results demonstrated that acetyl-CoA synthetases in S. erythraea are acetylated in vivo (Fig. 3b).
Functional enrichment analysis of acetylproteome
To further characterize the acetylated proteins in S. erythraea, we classified the acetylated proteins into groups according to cell component, molecular function, and biological process. The cell component of the acetylated proteins was assigned based on GO annotation. Our results showed that the acetylated proteins were mainly distributed in the organelle (16.1 %, FDR 9.8 × 10−25) with 4.9-fold enrichment, cell part (39.1 %, FDR 2.4 × 10−24) with 1.6-fold enrichment, and macromolecular complex (12.2 %, FDR 4.9 × 10−13) with 3.3-fold enrichment, as shown in Fig. 4. Based on GO molecular functions, we categorized all 363 acetylated proteins with respect to their functions. Most acetylated proteins were assigned to three groups: 65.3 % of the proteins were associated with catalytic activity, 57.9 % of the proteins were involved in binding, and 12.1 % of the proteins were related with structural molecule activity. It was found that acetylated proteins were enriched 9.8-fold in structural molecule activity (FDR 1.6 × 10−34), which was the most significantly enriched gene ontology molecular function (GOMF) term (Table S2 in the Supplementary Material). The acetylated proteins were further characterized according to GO biological process terms (Table S2 in the Supplementary Material). The classification analysis revealed that 43 % of the lysine-acetylated proteins in S. erythraea participated in metabolic processes (FDR 9.6 × 10−10) and 36 % of the proteins were categorized as cellular processes (FDR 5.7 × 10−15). These results are agreement with previous reports in E. coli, B. subtilis, and S. enterica. The GO enrichment analysis of the acetylome demonstrated that acetylated proteins are relatively broadly distributed among cell components, biological processes, and molecular functions in S. erythraea, implying that this PTM has diverse regulatory roles in Actinomycetes, which is similar to other microorganisms.

The cellular components, molecular functions, and biological processes of the acetylated proteins in S. erythraea by gene ontology analysis. The horizontal axis is −log10AdjP, right of column number a(b) means acetylated proteins (the ratio of acetylated proteins)
To better understand the general function of lysine acetylation in S. erythraea, we analyzed the acetylproteome of S. erythraea by the function of acetylated proteins in the KEGG pathway. Among all identified acetylated proteins, 47.6 % were involved in metabolism, 35.1 % were linked to genetic information processing, and 6.9 % were assigned to environmental information processing (Fig. 5a). The acetylated proteins involved in metabolism were further categorized into several subclasses as shown in Fig. 5b, including biosynthesis of secondary metabolism (28.8 %), microbial metabolism in diverse environments (23.8 %), biosynthesis of amino acids (16 %), carbon metabolism (15.8), and nucleotide metabolism (9 %). The results showed that acetylated proteins were enriched by 6.2-, 4.6-, 2.8-, 2.1-, and 2.1-fold in the ribosome (FDR 3.6 × 10−27), aminoacyl-transfer RNA (tRNA) biosynthesis (6.8 × 10−6), citrate cycle (6.1 × 10−3), biosynthesis of amino acids (2.4 × 10−4), and carbon metabolism (3.3 × 10−4), respectively (Table S3 in the Supplementary Material).

KEGG pathway analysis of identified acetylated proteins in S. erythraea. a The KEGG pathway enrichment of all acetylated proteins; b acetylated proteins involved in metabolism were categorized
Acetylation of enzymes involved in metabolism
Many enzymes involved in central metabolic pathways and amino acid metabolism pathways are acetylated in both prokaryotes and eukaryotes. Five enzymes associated with glycolysis were acetylated in S. erythraea: fructose-bisphosphate aldolase (fba), glyceraldehyde-3-phosphate dehydrogenase (gap), 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase (gpmA), enolase (eno), and pyruvate kinase (pyk3). These enzymes were also found to be acetylated in more than two other prokaryotes. Lysine acetylation commonly occurs in citrate acid cycle enzymes. As expected, we identified nine S. erythraea-acetylated enzymes in this pathway (Fig. 6). For example, a total of nine lysine acetylation sites were identified in succinyl-CoA synthetase (SucC and SucD). Among the nine enzymes, GltA-2 (SACE_0649, citrate synthase) was reported to be acetylated in all previously reported bacterial acetylomes, whereas KorA (2-oxoglutarate ferredoxin oxidoreductase subunit alpha) was not reported to be acetylated in any previously reported bacterial acetylome.

Acetylation of metabolic enzymes identified by acetylproteomics in the citric acid (TCA) cycle and glycolysis/gluconeogenesis in S. erythraea. The identified lysine-acetylated proteins are shown in red
Many enzymes involved in the biosynthesis of amino acids were also lysine acetylated (Fig. 7). Some acetylated enzymes identified in this study were not reported in previous bacteria acetylomes, including prephenate dehydrogenase (tyrA), chorismate synthase (aroF), glutamate synthase (gltB), cystathionine gamma-synthase (metB), histidinol dehydrogenase (hisD), diaminopimelate decarboxylase (lysA), adenosylhomocysteinase (sahH), and phosphoadenosine phosphosulfate reductase (cysH).

Acetylation of metabolic enzymes involved in amino acid biosynthesis identified by acetylproteomics of S. erythraea. The identified lysine-acetylated proteins are shown in red
Most importantly, some acetylated proteins associated with the biosynthesis of erythromycin and the precursor-supplied pathway were identified as acetylated proteins (Fig. 8). Understanding the mechanisms underlying the protein acetylation of the enzymes involved in the secondary metabolism and biosynthesis of erythromycin precursors is very important for improving the productivity of erythromycin in S. erythraea. Propionyl-CoA and methylmalonyl-CoA are the precursor metabolites of erythromycin production, which function as the starter unit and extender unit, respectively. Methylmalonyl-CoA can be derived from the methylmalonyl-CoA mutase (MCM) pathway, which catalyzes the reversible isomerization of succinyl-CoA and methylmalonyl-CoA by methylmalonyl-CoA epimerase (SACE_6238). Sace_1824 is an acetyl-CoA acetyltransferase that can transfer 2-methyl-acetoacetyl-CoA to propionyl-CoA. It was found that these two enzymes were acetylated in S. erythraea. In addition, TDP-4-keto-6-deoxyhexose 2,3-reductase (ery BII) is also an acetylated protein, and it is an important enzyme that irreversibly catalyzes the biosynthesis of erythromycin. These results indicate that reversible lysine acetylation may affect the relative activities of metabolic enzymes and modulate the metabolic flux for the biosynthesis of erythromycin. The S. erythraea genome houses seven gene clusters encoding nonribosomal peptide synthetases (NRPSs) for the biosynthesis of secondary metabolites, such as iron-scavenging compounds (siderophores) or peptide antibiotics. We also found that the nonribosomal peptide synthetase (Nrps3-3, SACE_2696; FPLMPSGK(2018)VDR) of the nrps3 cluster for the biosynthesis of siderophore in S. erythraea was acetylated. The effects of acetylation on the functions of these enzymes are being investigated.

Acetylation of metabolic enzymes in fatty acid metabolism and secondary metabolism precursor synthesis as identified by acetylproteomics in S. erythraea. The identified lysine-acetylated proteins are shown in red
Identification and analysis of lysine acetylation motifs in S. erythraea
The linear amino acid sequence motifs for protein phosphorylation are known; however, the motifs for lysine acetylation in E. coli, B. subtilis, S. enterica, G. kaustophilus, E. amylovora, and T. thermophilus have not yet been well defined. Recently, an acetylation motif has been proposed (PXXXXGK) that was found in AMP-forming acyl-CoA synthetases in Rhodopseudomonas palustris (Crosby et al. 2012). GNAT protein acetyltransferases recognize this motif and acetylate the last lysine residue. We also found that the S. erythraea acetyl-CoA synthetase AcsA (Sace_2375) contained a PXXXXGK motif and was acetylated on the last lysine residue (You et al. 2014). To further elucidate the acetylation motifs in S. erythraea, we analyzed the context of the amino acid sequence surrounding the acetylated lysines from the −10 to +10 position in 664 lysine-acetylated peptides identified using the motif-x program. Sequence windows centered on the acetylated lysine were compared with nonacetylated sequences to remove unwanted bias due to a general predisposition of certain amino acids close to lysine residues (Schwartz et al. 2009). Four conserved sequences around acetylation sites (KACH, KACY, KACXXXXR, and KACXXXXK) were found in the S. erythraea acetylproteome (Fig. 9a). In motif 1a, a histidine was located at the +1 position of the acetylation site (KACH). In motif 1b, a tyrosine was located at the +1 position amino acid (KACY). In motif 2a, an arginine at the +5 position was conserved (KACXXXXR). In motif 2b, a lysine was conserved at the +5 position (KACXXXXK). The four conserved sequences can be grouped into two types of motifs for lysine modifications (motif 1: KH/Y and motif 2: KXXXXR/K). Remarkably, the motif 1 sequence (with a histidine and tyrosine as the preferred amino acid residues at the +1 position) has also been reported in E. coli (Choudhary et al. 2009). In addition, motif 1 and motif 2 constituted 23.5 % (156/664) and 26.3 % (174/664) of the total lysine acetylation sites identified in this study (Fig. 9b). Approximately 16.1 and 16.3 % of the lysine-acetylated peptides in S. erythraea were categorized as motif 1a and motif 2a, respectively. All acetylated peptides with conserved motifs are listed in Table S4 in the Supplementary Material.

Identification of acetylation motifs. a Acetylation motifs were screened using the motif-x program; b the pie chart shows the number of acetylated peptides with different motifs. c Lysine conservation of S. erythraea acetylated lysines in other actinomycete species. Sco, Streptomyces coelicolor; Rxy, Rubrobacter xylanophilus; Nfa, Nocardia farcinica; Blj, Bifidobacterium longum; Srt, Segniliparus rotundus; Gpo, Gordonia polyisoprenivorans; Asd, Amycolicicoccus subflavus; Ams, Actinoplanes missouriensis; Tpr, Tsukamurella paurometabola; Pac, Propionibacterium acnes; Ami, Actinosynnema mirum; Mtu, Mycobacterium tuberculosis; Sro, Streptosporangium roseum; Rer, Rhodococcus erythropolis; Cgl, Corynebacterium glutamicum
To evaluate the degree of evolutionary conservation of the acetylated lysines identified in this study, we first used BLASTP to compare the acetylated protein sequences of S. erythraea NRRL23338 against the orthologous protein sequences in UniProtKB (http://www.uniprot.org/), which includes 15 actinomycete species: M. tuberculosis, Amycolicicoccus subflavus, Corynebacterium glutamicum, Nocardia farcinica, Rhodococcus erythropolis, Gordonia polyisoprenivorans, Tsukamurella paurometabola, Segniliparus rotundus, S. coelicolor, Propionibacterium acnes, Streptosporangium roseum, Actinosynnema mirum, Actinoplanes missouriensis, Bifidobacterium longum, and Rubrobacter xylanophilus. BLAST parameters are shown in Table S5 in the Supplementary Material. The orthologous proteins in these genomes were retrieved by applying a reciprocal best LAST hit approach and were aligned using MUSCLE v3.8.31 software (Edgar 2004). The degree of conservation in acetylated versus nonacetylated lysine residues across 15 actinomycete species and S. erythraea is shown in Fig. 9c. The results showed that the acetylated lysines in S. erythraea are more conserved than the nonacetylated lysines.
Motif 1a, motif 1b, motif 2a, and motif 2b are present in 107, 49, 108, and 66 acetylated peptides representing 101, 44, 97, and 50 S. erythraea proteins, respectively. GO analysis was performed on the four classes of proteins to investigate the cellular component (GOCC), molecular function (GOMF), and biological processes (GOBP) they are involved in (Table S6-8 in the Supplementary Material). Acetylated proteins with motif 1a, motif 1b, and motif 2a were more frequently present in the cytoplasm (p value < 1.96 × 10−8). Three GOCC terms (ribosome, ribonucleoprotein complex, and intracellular) were significantly overrepresented for acetylated proteins with motif 2 (p value < 2.1 × 10−5). Large ribosomal subunit and small ribosomal subunit proteins were conserved with the motif 2a acetylation site sequence (p value < 0.01). Based on the p value of <0.001, motif 1a proteins were enriched in several groups based on GOMF terms, including nucleotide binding, ligase activity, ATP binding, aminoacyl-tRNA ligase activity, and RNA binding; motif 1b proteins were enriched in only two GOMF terms, nucleotide binding and ATP binding; motif 2a proteins were enriched in four GOMF terms, rRNA binding, structural constituent of ribosome, RNA binding, and translation elongation factor activity; and motif 2b proteins were enriched in two GOMF terms, structural constituent of ribosome and RNA binding. The functional characterization of acetylated proteins by motif was assigned according to the GOBP. The results showed that motif 1a proteins were mainly involved in translation (8.3 × 10−7), tRNA aminoacylation for protein translation (4.2 × 10−5), cellular amino acid biosynthetic processes (0.004), and regulation of translational fidelity (0.01). Motif 1b proteins were only enriched in glycolysis (0.005). Motif 2a proteins were enriched in translation (1.9 × 10−13), translational elongation (0.0006), and the tricarboxylic acid cycle (0.002). Motif 2b proteins were enriched in translation (2.9 × 10−9), protein folding (0.001), and the tricarboxylic acid cycle (0.008). We further mapped the acetylated proteins with motifs to KEGG metabolic pathways. Motif 1a proteins were mainly involved in aminoacyl-tRNA biosynthesis pathway (0.001). Motif 2a proteins were involved in ribosome (2.8 × 10−6) and biosynthesis of amino acids (0.0007). Motif 2b proteins were involved in four pathways, ribosome, citrate cycle, tuberculosis, and carbon metabolism, with p value less than 0.001 (Table S9 in the Supplementary Material).
Interaction networks of acetylated proteins
We also analyzed the protein-protein interaction network and domain architecture of the identified acetylproteome in S. erythraea according to a previously described protocol (Zhang et al. 2013). An S. erythraea protein-protein interaction network of all acetylated proteins was created from the STRING database using Cytoscape software. The analysis parameters and Cytoscape network are shown in Figs. S2 and S3, respectively, in the Supplementary Material. A complete interaction STRING network has 363 acetylated proteins as nodes, connected by 2357 identified direct physical interactions (Fig. S4 in the Supplementary Material). Four highly confident protein networks (p < 10 × 10−20) are shown in Figs. S5–S8 in the Supplementary Material, including aminoacyl-tRNA biosynthesis, citrate cycle, ribosome, and RNA degradation. Sixteen acetylated proteins were involved in aminoacyl-tRNA biosynthesis, and they were connected in a relatively high-density protein-protein interaction network, especially SACE_0409, tyr, alaS, and aspS, as shown in Fig. S5 in the Supplementary Material. Several enzymes assigned to the citrate cycle produced a high-density interaction network (Fig. S6 in the Supplementary Material). For instance, gltA-2, sucC, sucD, and icd-2, which are the key enzymes in the citrate cycle, are lysine acetylated. Other high-density protein networks are presented in Figs. S7 and S8 in the Supplementary Material, such as the ribosome network, which consisted of more than 40 acetylated proteins in ribosome and RNA degradation. The characteristics of the networks are listed in Figs. S9–S11 in the Supplementary Material.
Discussion
Conservation of acetylation in Actinomycetes
N-lysine acetylation is a dynamic, reversible, regulatory PTM in prokaryotes that modulates the activity of enzymes. Growing evidence suggests that metabolic pathways are coordinated through reversible acylation of metabolic enzymes in response to nutritional status in the cell to maintain homeostasis. Actinomycetes are filamentous bacteria that are the major producers of therapeutic antibiotics. However, very few proteins (acetyl-CoA synthetase and acetoacetyl-CoA synthetases) were previously reported to be acetylated in Actinomycetes. In this study, we identified 664 unique lysine-acetylated sites on 363 proteins in S. erythraea. The acetylated proteins are involved in many biological processes, such as protein synthesis, glycolysis/gluconeogenesis, the citric acid (TCA) cycle, fatty acid metabolism, secondary metabolism, and precursor synthesis. These findings suggest that abundant lysine acetylation occurs in Actinomycetes. In addition, the findings expand our current knowledge of the bacterial acetylproteome and demonstrate the conservation of acetylation in Actinomycetes.
The role of acetylation in secondary metabolism
Reversible lysine acylation was detected in numerous enzymes in central metabolic pathways providing building blocks, cofactors, and energy for cell growth and antibiotic biosynthesis. AMP-forming acyl-CoA synthetases, such as acetyl-CoA synthetase, propionyl-CoA synthetase, malonyl-CoA synthetase, and succinyl-CoA synthetase, are under the control of dynamic and reversible N-lysine acylation (acetylation, succinylation, propionylation, and malonylation) in response to intracellular acyl-CoA pools, while variants of acyl-CoA metabolites also are precursors for the biosynthesis of antibiotics and other secondary metabolites. Propionyl-CoA and methylmalonyl-CoA are the precursor metabolites of erythromycin production as the starter unit and extender unit, respectively. Methymalonyl-CoA can be derived from different pathways such as carboxylation of propionyl-CoA and rearrangement of succinyl-CoA. In many Streptomycetes, at least four pathways, which have already been characterized, are connected to the methylmalonyl-CoA pool: the MCM pathway, which catalyzes the reversible isomerization of succinyl-CoA and methylmalonyl-CoA; the CCR (crotonyl-CoA reductase) pathway, which utilizes crotonyl-CoA reductase or adenosylcobalamin-dependent isobutyryl-CoA mutase; the MeaA (meaA gene coding for MCM-like B12-Dependent Mutase) pathway from acetoacetyl-CoA; and the PCC (propionyl-CoA carboxylase) pathway through carboxylation of propionyl-CoA by propionyl-CoA carboxylase or methylmalonyl-CoA transcarboxylase. We found that methylmalonyl-CoA epimerase (SACE_6238) and acetyl-CoA acetyltransferase (SACE_1824), which are involved in the production of methymalonyl-CoA and propionyl-CoA, respectively, were acetylated in S. erythraea. It was known that methylmalonyl-CoA epimerases and acetyl-CoA acetyltransferases in mouse, rat, and human mitochondria were acetylated at multiple lysine sites for influencing metabolic pathways in response to environmental signals (Masri et al. 2013; Rardin et al. 2013; Still et al. 2013; Choudhary et al. 2009). Such observations may suggest that reversible lysine acetylation may affect the relative activities of metabolic enzymes in the precursor-supplied pathways and modulate metabolic flux for the biosynthesis of secondary metabolites.
Two identified acetylated enzymes in the S. erythraea acetylproteome are directly involved in the biosynthesis of secondary metabolites, TDP-4-keto-6-deoxyhexose 2,3-reductase (EryBII, SACE_0727, LAAGTAVK(237)SAQGR), which is involved in the biosynthesis of erythromycin, and nonribosomal peptide synthetase (Nrps3-3, SACE_2696, FPLMPSGK(2018)VDR), which is involved in siderophore biosynthesis. Such observations suggested that reversible acetylation may have a direct regulatory role in antibiotic biosynthesis in Actinomycetes. It was indeed proposed that some enzymes involved in the biosynthesis of secondary metabolites could be regulated by acetylation modification, such as gramicidin synthetase I (GrsA, Brevibacillus brevis), chloroeremomycin synthetases (CepA, Amycolotopsis orientalis), and calcium-dependent antibiotic peptide synthetase I (cda PSI, SCO3230, S. coelicolor) (Starai et al. 2002).
Actinomycetes, such as S. coelicolor, S. avermitilis, S. lividans, and S. erythraea, have been genetically engineered for the overproduction of therapeutic antibiotics. Such strain improvement typically increases the activities of specific regulator proteins and/or overexpresses key enzymes in precursor pathways and biosynthetic pathways. Nevertheless, these genetically engineered strains have not taken full advantage of the global regulatory mechanism of reversible lysine acylation. Therefore, a deeper understanding and optimization of the acylation modification landscape of metabolism enzymes involved in secondary metabolism could provide a novel approach for highly efficient engineering of industrial overproducing strains.
Protein acetyltransferases and deacetylases
Advancements in high-resolution MS and high-affinity purification of acetylated peptides allow the identification of thousands of lysine acetylation sites in cells. However, it remains unclear how many protein acetyltransferases and deacetylases are involved in this PTM. Generally, 40–80 protein acetyltransferases, one to two NAD-dependent sirtuin deacetylases, or one NAD-independent protein deacetylase are encoded in actinomycete genomes. The S. coelicolor genome encodes 77 putative GNAT acetyltransferases (Pfam00583), two sirtuin-type deacetylases (SCO0452, SCO6464), and an AcuC-like NAD-independent deacetylase (SCO3330). The S. avermitilis genome encodes 43 putative GNAT acetyltransferases, one sirtuin-type deacetylases (SAV_537), and one AcuC-like deacetylase (SAV_4729). S. lividans genome encodes 72 putative GNAT acetyltransferases, two sirtuin-type protein deacetylases (EFD65580, EFD71509), and one NAD-independent deacetylase (EFD68590). S. erythraea encodes 42 putative GNAT acetyltransferases, one NAD-dependent deacetylase (SACE_3798), and one NAD-independent deacetylase (SACE_1779). Therefore, the number of GNAT acetyltransferases in a bacterial species appears to reflect its metabolic complexity. These enzymes control the acetylation/deacetylation of hundreds of proteins. Each of GNAT acetyltransferases controls the specific acetylation of different proteins under various physiological conditions or in response to changes in the levels of intracellular signals (Xu et al. 2014). Although an individual GNAT acetyltransferase modifies multiple substrates, one protein can be acetylated by multiple acetyltransferases. Which protein acetyltransferases modify the various proteins needs to be elucidated in future studies.
In addition to the established enzyme-catalyzed acetylation, it is worth noting that recent and historical evidence demonstrates that nonenzymatic acetylation and acylation can also lead to protein lysine modification (Delpech et al. 1983; Weinert et al. 2013; Kuhn et al. 2014). Recently, it was found that most E. coli acetylation occurs at a low level and is independent on the only known protein lysine Gcn5-like acetyltransferase Pat/YfiQ in E. coli. The results suggested that acetyl-phosphate (AcP) can chemically acetylate lysine residues in vitro and that AcP levels are correlated with acetylation levels in vivo, indicating that AcP may acetylate proteins nonenzymatically in cells (Weinert et al. 2013). Further study showed that the acP-dependent protein acetylation appears to be quite extensive and specific, with specificity determined by the local three-dimensional environment that surrounds the substrate lysine residue, as well as by its solvent accessibility and pKa (Kuhn et al. 2014). In bacteria, the cellular levels of acetyl-phosphate, as an intermediate of acetate dissimilation Pta-AckA pathway, depend upon the rate of its formation from acetyl-CoA by the phosphotransacetylase (Pta) and the rate of its degradation into acetate by the acetate kinase (AckA). The Pta-AckA pathway and AcP metabolism in Actinomycetes remain poorly understood. S. erythraea possesses one gene coding for acetate kinase, namely, ackA (SACE_1922). However, no phosphotransacetylase-encoding gene was found in the genome of S. erythraea. It is possible that the nonenzymatic acetylation also plays a role to S. erythraea acetylproteome identified.
Lysine acetylation motifs in S. erythraea
Schwartz et al. (2009) employed the motif-x program to extract lysine acetylation motifs from human acetylproteomes, including KK, KR, KF, KY, KXF, GK, KXXXK, and KXXK. This analysis found a preference for glycine and lysine in the residues immediately surrounding the acetylation site as well as a preference for aromatic residues at the +1 position. These motifs may be specially recognized by different acetyltransferase enzymes in different subcellular compartments. GO analysis of proteins containing the KY acetylation motif revealed a significant overenrichment of mitochondrial proteins, suggesting that a unique acetyltransferase with a preference for tyrosine at the +1 position is active in the mitochondrial compartment. In rat acetylomes, there is a strong preference for glycine in position −1 and proline in position +1 in nuclear proteins, whereas cytoplasmic proteins are enriched with glutamate in the vicinity of the acetylation site (Lundby et al. 2012). In Plasmodium falciparum, the dataset of acetylation sites in histones showed glycine as the preferred amino acid residue at the −1 position (GK motif) (Miao et al. 2013). Acetylproteome studies have been performed in some prokaryotes, including E. coli, B. subtilis, S. enterica, G. kaustophilus, E. amylovora, and T. thermophilus. The acetylation motifs in most microorganisms studied thus far remain largely unknown. In this study, we identified four definitive acetylation motifs in the S. erythraea acetylproteome: motif 1a, with a histidine located at the +1 position of the acetylation site (KACH); motif 1b, with a tyrosine located at the +1 position (KACY); motif 2a, with an arginine conserved at the +5 position (KACXXXXR); and motif 2b, with a lysine conserved at the +5 position (KACXXXXK). Similar to the motif 1 sequence, at the +1 position, histidine and tyrosine are most commonly found in E. coli (Choudhary et al. 2009). Interestingly, when considering the mitochondrial subset of lysine acetylation substrates identified in the mammalian dataset, a preference for histidine and tyrosine at the +1 position was also observed (Kim et al. 2006). Two preferred residues (histidine and tyrosine) at the +1 position of acetylated lysine sites in S. erythraea, E. coli, and mammalian mitochondria suggest that this PTM might be catalyzed by conserved protein acetyltransferases with unique substrate preferences. Acetylation motif 2 differs greatly from motif 1, which indicates that other protein acetyltransferases with different substrate preferences exist in S. erythraea. Therefore, further studies are needed to identify the unique protein acetyltransferase that recognize these acetylation motifs.
In conclusion, lysine acetylation is a dynamic and reversible PTM in eukaryotes and prokaryotes that regulates enzyme activity and gene expression. Here, we report the first investigation of lysine acetylation from the perspective of a proteome-wide analysis of the actinomycete S. erythraea. We succeeded in identifying 664 unique lysine-acetylated sites on 363 proteins. We found that the acetylated proteins are involved in many biological processes, such as protein synthesis, glycolysis/gluconeogenesis, the TCA cycle, fatty acid metabolism, secondary metabolism, and the precursor synthesis. We characterized the acetylproteome and analyzed in detail the impact of acetylation on diverse cellular functions according to GO and KEGG pathways. Four acetylation motifs (KACH, KACY, KACXXXXR, and KACXXXXK) were identified in the S. erythraea acetylproteome. Our study was the first to present the catalog of lysine acetylation substrates and sites in Actinomycetes, which are the major producers of therapeutic antibiotics.
References
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 57(1):289–300
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325(5942):834–840
Colak G, Xie Z, Zhu AY, Dai L, Lu Z, Zhang Y, Wan X, Chen Y, Cha YH, Lin H, Zhao Y, Tan M (2013) Identification of lysine succinylation substrates and the succinylation regulatory enzyme CobB in Escherichia coli. Mol Cell Proteomics 12(12):3509–3520
Crosby HA, Pelletier DA, Hurst GB, Escalante-Semerena JC (2012) System-wide studies of N-lysine acetylation in Rhodopseudomonas palustris reveal substrate specificity of protein acetyltransferases. J Biol Chem 287(19):15590–15601
Delpech M, Levy-Favatier F, Kruh J (1983) In vitro, non enzymatic labelling of histone H1 with [14C] acetyl CoA. Biochimie 65:291–294
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797
Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23(4):607–618
Kim D, Yu BJM, Lee YJ, Choi SG, Kang S, Pan JG (2013) The acetylproteome of Gram-positive model bacterium Bacillus subtilis. Proteomics 13(10–11):1726–1736
Kuhn ML, Zemaitaitis B, Hu LI, Sahu A, Sorensen D, Minasov G, Lima BP, Scholle M, Mrksich M, Anderson WF, Gibson BW, Schilling B, Wolfe AJ (2014) Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS ONE 9(4):e94816
Lee DW, Kim D, Lee YJ, Kim JA, Choi JY, Kang S, Pan JG (2013) Proteomic analysis of acetylation in thermophilic Geobacillus kaustophilus. Proteomics 13(15):2278–2282
Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, Olsen JV (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep 2(2):419–431
Masri S, Patel VR, Eckel-Mahan KL, Peleg S, Forne I, Ladurner AG, Baldi P, Imhof A, Sassone-Corsi P (2013) Circadian acetylome reveals regulation of mitochondrial metabolic pathways. Proc Natl Acad Sci U S A 110(9):3339–3344
Miao J, Lawrence M, Jeffers V, Zhao FQ, Parker D, Ge Y, Sullivan EJ Jr, Cui L (2013) Extensive lysine acetylation occurs in evolutionarily conserved metabolic pathways and parasite-specific functions during Plasmodium falciparum intraerythrocytic development. Mol Microbiol 89(4):660–675
Mikulik K, Felsberg J, Kudrnacova E, Bezouskova S, Setinova D, Stodulkova E, Zidkova J, Zidek V (2012) CobB1 deacetylase activity in Streptomyces coelicolor. Biochem Cell Biol 90(2):179–187
Nambi S, Basu N, Visweswariah SS (2010) Cyclic AMP-regulated protein lysine acetylases in Mycobacteria. J Biol Chem 285(32):24313–24323
Okanishi H, Kim K, Masui R, Kuramitsu S (2013) Acetylome with structure mapping reveals the significance of lysine acetylation in Thermus thermophilus. J Proteome Res 12(9):3952–3968
Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF (2007) Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol 25(4):447–453
Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, Gibson BW (2013) Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A 110(16):6601–6606
Schwartz D, Chou MF, Church GM (2009) Predicting protein post-translational modifications using meta-analysis of proteome scale data sets. Mol Cell Proteomics 8(5):365–379
Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC (2002) Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298(5602):2390–2392
Still AJ, Floyd BJ, Hebert AS, Bingman CA, Carson JJ, Gunderson DR, Dolan BK, Grimsrud PA, Dittenhafer-Reed KE, Stapleton DS, Keller MP, Westphall MS, Denu JM, Attie AD, Coon JJ, Pagliarini DJ (2013) Quantification of mitochondrial acetylation dynamics highlights prominent sites of metabolic regulation. J Biol Chem 288(36):26209–26219
Tucker AC, Escalante-Semerena JC (2013) Acetoacetyl-CoA synthetase activity is controlled by a protein acetyltransferase with unique domain organization in Streptomyces lividans. Mol Microbiol 87(23):152–167
Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Ríos D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolomé S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H (2014) ProteomeXchange provides globally co-ordinated proteomics data submission and dissemination. Nat Biotechnol 30(3):223–226
Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327(5968):1004–1007
Weinert BT, Iesmantavicius V, Wagner SA, Schölz C, Gummesson B, Beli P, Nyström T, Choudhary C (2013) Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol Cell 51(2):265–272
Wu X, Vellaichamy A, Wang DP, Zamdborg L, Kelleher NL, Huber SC, Zhao Y (2013) Differential lysine acetylation profiles of Erwinia amylovora strains revealed by proteomics. J Proteomics 79:60–71
Xu H, Hegde SS, Blanchard JS (2011) Reversible acetylation and inactivation of Mycobacterium tuberculosis acetyl-CoA synthetase is dependent on cAMP. Biochemistry 50(26):5883–5892
Xu JY, You D, Leng PQ, Ye BC (2014) Allosteric regulation of a protein acetyltransferase in Micromonospora aurantiaca by the amino acids cysteine and arginine. J Biol Chem 289(39):27034–27045
You D, Yao LL, Huang D, Escalante-Semerena JC, Ye BC (2014) Acetyl-CoA synthetase is acetylated on multiple lysine residues by a protein acetyltransferase with single GNAT domain in Saccharopolyspora erythraea. J Bacteriol 196(17):3169–3178
Yu BJ, Kim JA, Moon JH, Ryu SE, Pan JG (2008) The diversity of lysine-acetylated proteins in Escherichia coli. J Microbiol Biotechnol 18(9):1529–1536
Zhang JM, Sprung R, Pei JM, Tan XH, Kim S, Zhu H, Liu CF, Grishin NV, Zhao YM (2009) Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol Cell Proteomics 8(2):215–225
Zhang K, Zheng S, Yang JS, Chen Y, Cheng Z (2013) Comprehensive profiling of protein lysine acetylation in Escherichia coli. J Proteome Res 12(2):844–851
Acknowledgments
This work was financially supported by grants from the National Key Technologies R&D Program (2014AA021502), the National Natural Science Foundation of China (21276079, 31401592) and SRFDP (No. 20120074110009). We thank Prof. Yingming Zhao at University of Chicago for critical reading of the manuscript and helpful comments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Huang, D., Li, ZH., You, D. et al. Lysine acetylproteome analysis suggests its roles in primary and secondary metabolism in Saccharopolyspora erythraea . Appl Microbiol Biotechnol 99, 1399–1413 (2015). https://doi.org/10.1007/s00253-014-6144-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6144-2