
Abstract
Vibrio parahaemolyticus, an emerging food and waterborne pathogen, is a leading cause of seafood poisoning worldwide. Surface proteins can directly participate in microbial virulence by facilitating pathogen dissemination via interactions with host factors. Screening and identification of protective antigens is important for developing therapies against V. parahaemolyticus infections. Here, we systematically characterized a novel immunogenic enolase of V. parahaemolyticus. The enolase gene of V. parahaemolyticus ATCC33847 was cloned, sequenced, and expressed in Escherichia coli BL21. Enzymatic assays revealed that the purified recombinant V. parahaemolyticus enolase protein catalyzes the dehydration of 2-phospho-d-glycerate to phosphoenolpyruvate. Western blot analysis showed that V. parahaemolyticus enolase was detectable in the extracellular, outer membrane (OM) and cytoplasmic protein fractions using antibodies against the recombinant enolase. Surface expression of enolase was further confirmed by immunogold staining and mass spectrometry (liquid chromatography–tandem mass spectrometry) analysis of OM protein profiles. Notably, V. parahaemolyticus enolase was identified as a human plasminogen-binding protein with the enzyme-linked immunosorbent assay. The values obtained for adherence and inhibition suggest a role of surface-exposed enolase in epithelial adherence of V. parahaemolyticus. We further showed that enolase confers efficient immunity against challenge with a lethal dose of V. parahaemolyticus in a mouse model. To our knowledge, this is the first study to demonstrate the plasminogen-binding activity of enolase that is an adhesion-related factor of V. parahaemolyticus. Our findings collectively imply that enolase plays important roles in pathogenicity, supporting its utility as a novel vaccine candidate against V. parahaemolyticus infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vibrio parahaemolyticus, a Gram-negative motile bacterium inhabiting marine and estuarine environments, is commonly found free swimming, attached to underwater surfaces or commensally associated with different shellfish species (McCarter 1999; Shinoda 2011). The microbe is a leading cause of acute gastroenteritis after consumption of contaminated raw or undercooked seafood and occasionally causes wound infections in humans worldwide (Shimohata and Takahashi 2010; Su and Liu 2007). In approximately 5 % of cases, V. parahaemolyticus gastrointestinal infection progresses to septicemia and may be fatal for immunocompromised patients, including those with leukemia, liver disease, and individuals infected with human immunodeficiency virus (Baker-Austin et al. 2010; Qadri et al. 2003; Yeung and Boor 2004).
V. parahaemolyticus produces an array of factors associated with bacterial virulence, including thermostable direct hemolysin (TDH), TDH-related hemolysin (TRH), and type III secretion system (T3SS) (Broberg et al. 2011; Park et al. 2004; Yeung and Boor 2004; Nishibuchi et al. 1986). The global dissemination of this pathogen underscores the importance of understanding its numerous virulence factors and their effects on the human host. Microbial adhesion to host tissues is the initial event of most infectious process (Navarre and Schneewind 1999). Recent studies demonstrated that enolases from several pathogenic species, such as Aeromonas hydrophila (Sha et al. 2009), Streptococcus pneumoniae (Whiting et al. 2002), Mycoplasma suis (Schreiner et al. 2012) and Borrelia burgdorferi (Toledo et al. 2012), possess fibronectin-binding activity and affect bacterial virulence through different mechanism(s), playing critical roles in bacterial colonization, invasion, and adhesion to epithelial cells, which constitute significant early steps of infection.
Previous reports have shown that several pathogens capture plasminogen (Plg), which allows bacteria to acquire surface-associated proteolytic activity that facilitates invasion and dissemination in the infected host (Marcos et al. 2012; Kolberg et al. 2006). In particular, enolase plays a major role in microbial recruitment of Plg, a single-chain glycoprotein with a molecular mass of 92 kDa that is converted to plasmin in vivo (Song et al. 2012). Enolase is a ubiquitous enzyme that catalyzes the dehydration of 2-phosphoglycerate (2-PGE) to phosphoenolpyruvate (PEP) in the final steps of the catabolic glycolytic pathway (Han et al. 2012). Enolase is considered a multifunctional protein, in view of its contribution to several biological and pathophysiological processes in addition to its innate glycolytic and metabolic activities (Díaz-Ramos et al. 2012; Ghosh and Jacobs-Lorena 2011; Pancholi 2001). Considerable evidence has shown that enolase present on the surface of Gram-positive (Zhang et al. 2009; Ehinger et al. 2004) and Gram-negative (Han et al. 2012; Sha et al. 2009) bacteria, fungi (Marcos et al. 2012), and protozoa (Holmes et al. 2010; Mundodi et al. 2008) mainly acts as a receptor for plasminogen and functions as a mediator of microbial virulence (Avilán et al. 2011; Pancholi 2001; Pancholi and Chhatwal 2003).
Elucidation of the factors associated with virulence is a fundamental prerequisite for the search for new strategies targeting V. parahaemolyticus challenge. Enolase is found in both the cytosol and cell membrane in Paracoccidioides brasiliensis, and the presence of surface enolase is closely related to bacterial adherence to the host cell, where it interacts with Plg and facilitates microbial dissemination within hosts (Nogueira et al. 2010). Moreover, enolases of some bacteria (e.g., Streptococci and Aeromonas) have been characterized as protective immunogenic proteins (Feng et al. 2009; Sha et al. 2009; Zhang et al. 2009). However, Plg-binding activity has not been reported for V. parahaemolyticus enolase, and its role in cell adhesion is yet to be established. Therefore, it is important to examine enolase from the pathogenic and immunological points of view.
In the present study, we identified and characterized a functional V. parahaemolyticus enolase, with the aim of understanding its role in bacterial metabolism, pathogenesis, immunity, and infection. The potential of enolase for development as a novel vaccine antigen against V. parahaemolyticus was also investigated.
Materials and methods
Bacterial strains and growth conditions
The V. parahaemolyticus strain ATCC33847, obtained from the Chinese Veterinary Culture Collection Center (CVCC), was grown on thiosulfate–citrate–bile salt–sucrose (TCBS) medium (Oxoid Ltd., Hampshire, UK) and incubated for 24 h at 37 °C. For polymerase chain reaction (PCR), bacterial adhesiveness, and virulence assays in mice, a single bacterial colony from TCBS agar was inoculated into brain heart infusion broth (BHI, Oxoid, UK) supplemented with 2 % NaCl and grown overnight at 37 °C with constant shaking. Luria-Bertani (LB) agar supplemented with 3 % NaCl was used for bacterial counting. The expression vector pET-28a (+) was purchased from Novagen (Madison, WI, USA). Prestained protein marker was obtained from MBI (Fermentas Inc., USA). All chemicals were of analytical grade and purchased from Sigma (St. Louis, MO, USA). Laboratory Escherichia coli strains, DH5α and BL21, were used as nonadherent and noninvasive recipients of pET-28a (+) plasmids expressing recombinant protein.
Cloning, expression, and purification of V. parahaemolyticus enolase
The enolase gene was amplified with PCR using the primers Eno-F (5′CCAGGATCCATGTCTAAGATCGTTAAAGTT3′) and Eno-R 5′GCAGAGCTCTTAAGCTTGGCCTTTAACT3′). Primers were designed according to the enolase gene sequence of V. parahaemolyticus RIMD 2210633 (NC_004603) and contained BamHI and SacI sites (underlined) at the 5′ and 3′ ends, respectively. PCR was performed in a thermal cycler using the genomic DNA of V. parahaemolyticus ATCC33847 obtained using a Genomic DNA isolation kit (Tiangen, Beijing, China) as the template under the following conditions: denaturation at 95 °C for 30 s, annealing at 56 °C for 30 s, and extension for 1.5 min at 72 °C for a total of 30 cycles. The 1,302-bp PCR product corresponding to enolase obtained was digested with BamHI/SacI and cloned into the corresponding sites of pET28a (+). The resulting plasmid, pET28a-eno, was used to transform E. coli DH5α (Invitrogen, Carlsbad, CA, USA). Sequencing was performed on a 3070xl DNA analyzer (Applied Biosystems, Foster City, CA, USA), and DNA analysis was performed with DNASTAR software (DNASTAR Inc., Madison, WI, USA). Protein was induced in E. coli BL21 (DE3, Stratagene, La Jolla, CA, USA) transformed with pET28-eno at 37 °C for 4 h with 0.1 mmol/L isopropyl b-d-1-thiogalactopyranoside (IPTG). After sonication on ice (99 times for 5 s at 400 W with 10-s intervals between repeats, Scientz, JY92-IIN), bacterial lysates were subjected to centrifugation at 10,000 g for 30 min for the removal of insoluble pellets. The acquired supernatant was purified using a His-Bind Purification Kit (Novagen), according to the manufacturer’s instructions. Fractions of recombinant protein were filtered through a 0.22-μm membrane (Millipore) and stored at −80 °C. Protein quantitation was performed with a BCA Protein Assay Kit (Thermo Scientific Pierce, Rockford, IL, USA).
Enolase activity and kinetics assay of V. parahaemolyticus enolase
Enzymatic assays for V. parahaemolyticus enolase were performed using standard procedures (Han et al. 2012; Feng et al. 2009; Esgleas et al. 2008) with modifications. Enolase activity was determined by measuring the conversion of 2-PGE to PEP. To address the capability of V. parahaemolyticus enolase to convert 2-PGE to PEP, 1 mM 2-PGE was added to a reaction buffer (100 mM HEPES buffer, pH 8.5, 7.7 mM KCl, 10 mM MgSO4, prewarmed to 25 °C); 10 μg of purified enolase protein was then added to initiate the reaction. PEP release was measured at 240 nm on a spectrophotometer at 1-min intervals for 10 min. Rabbit muscle enolase (Sigma) was used as the positive control. The kinetics of enolase activity was expressed as PEP production. To study V. parahaemolyticus enolase enzyme kinetics, different amounts of 2-PGE (0.25, 0.5, 1.0, 1.5, and 2.0 mM) were used for assay. Michaelis constant (K m) and maximum reaction velocity (V max) for enolase were determined from double-reciprocal Lineweaver–Burk plots.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot
Recombinant enolase (10 μg) was resolved on a 12 % (v/v) polyacrylamide vertical slab gel with a 5 % (v/v) stacking gel. After electrophoresis, one gel was stained with CBB R-250, and the other was applied for electrotransfer to a PVDF membrane (Amersham Pharmacia Biotech) using a semidry transfer cell (Bio-Rad) with a blotting buffer (39 mM glycine, 48 mM Tris, 0.037 % (w/v) SDS, 20 % methanol) at 15 V for 15 min. The membrane was blocked with a blocking buffer [5 % skimmed milk, 0.05 % Tween-20 in phosphate-buffered saline (PBS)] for 2 h at room temperature, washed with PBS containing 0.05 % Tween-20 (PBST), and incubated with convalescent-phase serum from a mouse clinically infected with the V. parahaemolyticus strain ATCC33847 (1:300) for 1 h, followed by incubation with anti-mouse IgG (whole-molecule) peroxidase conjugate (Sigma). Cross-reacting protein bands were visualized using a DAB Substrate Kit (Thermo). All experiments were performed in triplicate.
Antibody production
One milliliter (1 mL) of purified recombinant enolase (1 mg/mL) emulsified in Montanide ISA 50 V (SEPPIC, France) at a ratio of 1:1 was employed for subcutaneous immunization of two New Zealand rabbits (Slack Shanghai Laboratory Animal Co., Ltd., China), two times at 2-week intervals. Blood samples were collected when the second booster injection was administered 10 days later. The titers of rabbit antiserum against V. parahaemolyticus enolase were evaluated using enzyme-linked immunosorbent assay (ELISA). ELISA plate wells were coated with purified enolase (1 μg/well). Well contents were reacted with serial dilutions (1/2,000 to 1/32,000) of rabbit antienolase antibody, followed by HRP-conjugated anti-rabbit IgG antisera (1:4,000, Sigma). Reactions were developed with tetramethylbenzidine substrate solution (Sigma) and terminated with 2 M H2SO4, and OD450 of each well was read on an ELISA reader (BioTek, USA). The highest dilution of sera with OD450 > 2.1 times that of negative control wells was valued as ELISA titers. All samples were examined in triplicate.
Preparation of V. parahaemolyticus protein fractions and localization of enolase
To determine the distribution of V. parahaemolyticus enolase, extracellular proteins, cytoplasm proteins, and outer membrane proteins (OMPs) from V. parahaemolyticus strains ATCC 33847 were obtained as previously described (Ni et al. 2010; Sha et al. 2003), with modifications. Briefly, after centrifugation at 10,000 g for 20 min, the supernatant of V. parahaemolyticus overnight cultures was collected and filtered through a 0.22-μm-pore-size filter to remove residual bacteria. Then, 10 μg/mL of a cocktail of protease inhibitors was added (Merck, USA) per milliliter of supernatant. The extracellular proteins were kept in the supernatant. The cell pellets were washed three times, resuspended in Tris–HCl (0.02 mo1/L, pH 7.5), and sonicated in the ultrasonic disintegrator (600 W). After sonication of the cells, debris was removed by centrifugation at 10,000 g for 20 min. The supernatant was ultracentrifuged at 150,000 g for 1 h, and then, the supernatant obtained contained cytoplasm proteins. The pellet was resuspended into 0.5 % sodium lauroyl sarcosine and incubated overnight at 4 °C. After ultracentrifugation at 150,000 g for 45 min, the pellet (OMPs) was resuspended in distilled water with a proteinase inhibitor. The extracellular and cytoplasm protein fractions in the supernatant were precipitated using trichloroacetic acid (TCA) and acetone, respectively. Protein samples or bovine serum albumin (BSA, negative control) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). For Western blotting, rabbit antiserum against V. parahaemolyticus enolase (1:600) was used as the primary antibody and goat anti-rabbit IgG (whole-molecule) peroxidase conjugate (Sigma) as the secondary antibody. Blots were developed with a DAB Substrate Kit. Experiments were repeated three times.
One-dimensional SDS-PAGE of OMPs and protein identification
In order to determine whether enolase is localized in the outer membrane of V. parahaemolyticus, OMPs of strain ATCC33847 were separated on a 1D SDS-PAGE using an acryl amide concentration of 5 % for the stacking gel and 12 % for the running gel. Separated protein bands in the SDS-PAGE gel were visualized with Coomassie Brilliant Blue. The protein band of about 48 kDa was excised and sent to Shanghai Applied Protein Technology Co. Ltd. (Shanghai, China) for tryptic in-gel digestion and liquid chromatography–tandem mass spectrometry (LC-MS/MS). LC-MS/MS profiles of the peptides from OMPs of the V. parahaemolyticus strain ATCC33847 were used for searching the sequence similarities to data from V. parahaemolyticus or closely related Gram-negative bacteria available in the National Center for Biotechnology Information (NCBI) database. MS/MS spectra were automatically searched against the NCBI database using the BioWorks Browser rev.3.1 (Thermo Electron, San Jose, CA, USA). Protein identification results were extracted from SEQUEST out files with BuildSummary. The peptides were constrained to be tryptic, and up to two missed cleavages were allowed. Carbamidomethylation of cysteine was treated as a fixed modification, whereas oxidation of methionine was considered as a variable modification. The mass tolerance allowed for the precursor ions and fragment ions was 0.8 and 2.0 Da, respectively. The protein identification criteria were based on Delta CN (≥0.1) and cross-correlation scores (Xcorr, one charge ≥ 1.9, two charges ≥ 2.2, three charges ≥ 3.75). Protein identifications were accepted if they could be established at >95 % probability and contained at least two identified peptides.
Immunogold staining
The surface exposure of enolase on V. parahaemolyticus was examined using immunelectron microscopy as previously described (Sha et al. 2003), with modifications. Briefly, after several washes with PBS, log-phase-grown cultures of the V. parahaemolyticus strain ATCC33847 were fixed with a mixture of 4 % paraformaldehyde and 0.1 % glutaraldehyde in 0.05 M cacodylate buffer (pH 7.3) and embedded in LR White resin (London Resin Company Limited, England), according to the manufacturer’s instructions. The ultrathin-sectioned LR White-embedded cells were reacted with mouse antiserum against V. parahaemolyticus enolase (prepared in our laboratory using the purified recombinant enolase from this study) diluted to 1/800, followed by colloidal gold-labeled (9–11-nm particles) anti-mouse IgG (Sigma) at a dilution of 1:20. After several washes with PBS, bacteria were finally stained with aqueous 3 % uranyl acetate and examined with an electron microscope (TEM, Philips CM120) at an accelerating voltage of 80 kV. Bacteria coated with normal mouse antibodies served as a negative control.
Plasminogen-binding assays
An ELISA was performed to determine the plasminogen-binding ability of V. parahaemolyticus enolase. Wells of multiwell plates (Nunc) were coated with 1 μg purified enolase diluted in carbonate buffer, pH 9.6. In a parallel experiment, ELISA wells were coated with OMPs from V. parahaemolyticus. Plates were incubated at 4 °C overnight. After subsequent blocking with 1 % (w/v) BSA in PBS, different concentrations (0.0156–2 μg/well) of human Plg (R&D Systems) or BSA were added to enolase-coated wells. Reactions proceeded at 37 °C for 1 h, and wells were subsequently washed. Bound protein was detected using anti-Plg monoclonal (R&D Systems) and horseradish peroxidase (HRP)-conjugated secondary antibodies. The competition experiment was performed by adding rabbit antienolase antibody (serial dilutions from 1/500 to 1/64,000), prior to the addition of Plg. The reaction was visualized with 3,3,5,5-tetramethylbenzidine (TMB) solution for 10 min and terminated with 2 M H2SO4. OD450 was measured using a microplate reader (BioTek Instruments Inc., Winooski, VT, USA) to determine binding activity. The assay was performed in triplicate, and data were reproducible, since the values did not vary by more than 5 % of the mean.
Indirect immunofluorescence assays
To visualize whether V. parahaemolyticus enolase specifically adheres to the surface of Hep-2 cells, immunofluorescence assays were conducted (Chen et al. 2011; Zhang et al. 2009) with specific modifications. Hep-2 cells cultured in 96-well cell plates were washed twice with PBS, fixed with cold acetone/methanol (1:1) for 20 min at −20 °C, and allowed to air-dry. Purified enolase (10 μg) or bovine serum albumin (blank control) was incubated with fixed Hep-2 cells at 37 °C for 1 h and washed three times with PBS (100 μL/well). Rabbit antibodies against enolase preadsorbed with Hep-2 cells were added and incubated for 30 min at 37 °C. Hep-2 cells were incubated with recombinant enolase, washed with PBS, and incubated with preimmunization rabbit serum, which was tested as a negative control. After washing, cells were incubated with 100 μL FITC-conjugated goat anti-rabbit IgG (1:200, Sigma) at 37 °C for 1 h. Finally, samples were washed three times and visualized under a confocal laser scanning microscope (Nikon). To determine only enolase, but not the fused peptide (His6 tag) of the recombinant enolase that contributes to protein binding to the cells, the His6-tag was removed from the expressed target protein and followed by removal of thrombin using a Thrombin Cleavage Capture Kit (Novagen), according to the manufacturer’s instructions. Protein was applied to the His-Bind Purification Kit (Novagen) as above for a second time, and flow-through fractions were collected and concentrated by ultrafiltration (Millipore Amicon Ultra 10 K device). The resulting cleaved protein was verified by SDS-PAGE and Western blot with an anti-His-tag monoclonal antibody (Beyotime Institute of Biotechnology, China), while its adherence ability to host cells was confirmed as above by indirect immunofluorescence assays.
Inhibition assays
To evaluate the potential role of enolase in V. parahaemolyticus adhesion to host cells, the inhibition assays were performed. Hep-2 cells were grown to confluence in 24-well tissue culture plates in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10 % fetal calf serum at 37 °C in a 5 % CO2-humidified atmosphere without antibiotics. After washing with PBS three times, increasing amounts of purified recombinant enolase (10–20 μg/well) were added to cells and incubated at 37 °C for 2 h. Cells treated with 20 μg/well BSA served as the control. All cells were washed with PBS three times, infected with the ATCC33847 strain at a multiplicity of infection (MOI) of 1:50 in DMEM without fetal calf serum, and incubated for 2 h at 37 °C in 5 % CO2. Cells were washed with PBS to remove nonadherent bacteria and treated with a lysis buffer (PBS containing 0.1 % (v/v) trypsin and 1.0 % (v/v) Triton 100) at 37 °C for 5 min. Serial dilutions of this cell lysate were plated onto LB agar supplemented with 3 % NaCl. After incubation at 37 °C overnight, the numbers of colonies were determined. The percentage of inhibition of enolase to adherence was calculated as (number of CFU recovered in the enolase-treated cells / number of CFU recovered in the BSA-treated cells) × 100.
To investigate the effect of antienolase antibody to the adherence of the V. parahaemolyticus strain ATCC33847, bacteria were pretreated with either specific antienolase antibody or preimmume serum for 1 h at 37 °C. Pretreated bacteria were used to infect Hep-2 cells as described above. All experiments were performed in triplicate and repeated three times.
Animal challenge experiment
ICR mice (6 weeks old) were purchased from Slack Shanghai Laboratory Animal Co., Ltd., China. Before challenge, the median lethal dose (LD50) for the V. parahaemolyticus strain ATCC33847 was measured to determine virulence in a mouse model, as described previously (Baffone et al. 2001; Vongxay et al. 2008). Briefly, bacteria were grown to the exponential phase, collected, washed twice in PBS, and adjusted to the appropriate doses. ICR mice were inoculated intraperitoneally (i.p.) with 0.2 mL of each bacterial suspension containing different numbers of CFU. The numbers of bacterial CFU in injected inoculum were confirmed by plating on LB agar supplemented with 3 % NaCl. Negative controls were injected with PBS. Ten animals were used per dose, and mortality was monitored until 7 days postinfection. The experiment were repeated three times, and the results were averaged and calculated using the method of Reed and Muench (1938).
Forty-five female ICR mice were randomly assigned to three groups of 15 mice each. Mice in group 1 were immunized subcutaneously at multiple sites with 100 μL of purified recombinant enolase (500 μg/mL) emulsified in Montanide ISA 50 V (SEPPIC, France) at a ratio of 1:1. Mice received one booster injection after 14 days with the same concentration of antigen and were reimmunized after 7 days. Mice in group 2 immunized with the inactivated V. parahaemolyticus strain ATCC33847 served as the positive control. Specifically, bacterial strains were grown for 24 h in LB medium supplemented with 3 % NaCl with shaking (250 rpm) at 28 °C and inactivated with formaldehyde at a final solubility of 0.4 % for 24 h. The injection dose was 100 μL of inactivated whole-cell vaccine (1 × 1010 CFU/mL) diluted with ISA 50 V adjuvant (SEPPIC, France) at a ratio of 1:1. Mice in group 3 inoculated with PBS emulsified in the same adjuvant served as the negative control. After 1 week, each group was challenged with a lethal dose of 5 LD50 of the log-phase V. parahaemolyticus strain ATCC33847 in 0.5 mL PBS. Mice were monitored and scored for survival for 7 days. All animal infection experiments were approved by the Animal Ethics Committee of Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences (no. SYXK < HU > 2011-0116).
Statistical analysis
Antibody levels are expressed as geometric mean ELISA units of n independent observations ± standard deviations. Data were analyzed using Student’s t test, and statistical significance was accepted at P < 0.05. Statistical analyses for in vitro and in vivo experiments were determined using the chi-square test with SPSS 16.0, and significance was established at P values of <0.05.
Results
Expression and immunogenicity of V. parahaemolyticus enolase
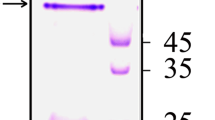
The enolase gene (eno) was amplified from the V. parahaemolyticus strain ATCC33847. The open reading frame of eno gene is 1,302 bp in length encoding an enzyme of 433 amino acids with a calculated molecular mass of 48 kDa. DNA sequence analysis showed that the amplified enolase gene displays 100 % identity with V. parahaemolyticus RIMD 2210633 (GenBank: BA000031). BLAST analyses indicated that the sequence is highly homologous to the protein sequences of enolase from a variety of other Vibrio species (85 to 98 %). Recombinant fusion protein V. parahaemolyticus enolase tagged with hexa histidine (His6) at its N-terminus was successfully expressed in E. coli BL21 and purified with the His-Bind Purification Kit (Novagen, Madison, WI, USA), which was observed as a 52-kDa band in SDS-PAGE analysis (Fig. 1). Western blot analysis showed that V. parahaemolyticus enolase has a good immunoreactive to convalescent phase serum (1:300) from a mice clinically infected with V. parahaemolyticus (Fig. 1), suggesting that enolase may be an infection-associated antigen.

SDS-PAGE and Western blot analysis of the recombinant V. parahaemolyticus enolase expressed in E. coli BL21. Lane M, MW marker (SM0671, Fermentas); lane 1, pET28a-enolase in E. coli BL21, induced; lane 2, purified recombinant enolase; lane 3, pET28a-enolase in E. coli BL21, uninduced; lane 4, Western blot of recombinant enolase with convalescent sera from mouse against V. parahaemolyticus ATCC33847; lane 5, Western blot of BSA as the negative control
Antiserum against V. parahaemolyticus enolase was obtained from rabbits on day 10 after the booster injection and tested for ELISA titers as 1:32,000, which indicated that V. parahaemolyticus enolase has strong immunogenicity.
Enzymatic activity and kinetics assay of V. parahaemolyticus enolase
V. parahaemolyticus enolase activity in catalyzing the conversion of 2-PGE to PEP was similar to that of rabbit muscle enolase, as determined with the spectrophotometric assay. The production of PEP increased with time for both V. parahaemolyticus enolase and rabbit muscle enolase, and V. parahaemolyticus enolase reached maximal activity at 5 min, which was earlier than that of rabbit muscle enolase (positive control) at 9 min (Fig. 2a). The Michaelis–Menten plot produced using V. parahaemolyticus enolase kinetics data with varying substrate concentrations showed that V. parahaemolyticus enolase is able to fully convert 2-PGE to PEP for all five substrate concentrations, clearly implying that recombinant enolase retains enzymatic activity. The K m and V max values were determined as 1.9 mM and 154 μM/min, respectively (Fig. 2b).

Enzymatic characterization of enolase. a The production of PEP increased with time for both recombinant V. parahaemolyticus enolase and rabbit muscle enolase (positive control). b Determination of Vmax and Km for V. parahaemolyticus enolase by means of a Lineweaver–Burk plot (double-reciprocal plot). Data from three independent assays were fitted to the Equation V = VmaxS / (S + Km). The values found were the Vmax = 154 μM/min and Km = 1.9 mM. Data shown here were the mean values ± standard deviations
Localization of V. parahaemolyticus enolase
To determine in which portion of the cell, the majority of the enolase was located, Western blot, LC-MS/MS, and immunoelectron microscopy were performed. The extracellular, outer membrane, and cytoplasmic protein fractions from V. parahaemolyticus culture were prepared and reacted with rabbit antiserum against recombinant V. parahaemolyticus enolase. Western blot analysis showed that a band corresponding to the size of enolase protein (~48 kDa) was detected in all the above fractions (Fig. 3). Further, OMP preparations from V. parahaemolyticus were separated by 1D SDS-PAGE (Fig. 4a), and the protein band of about 48 kDa was excised and digested with trypsin. The resultant peptides were then analyzed with the help of LC-MS/MS. A total of 68 proteins were predicted to be localized to the outer membrane of V. parahaemolyticus, including maltoporin, OmpTolC, long-chain fatty acid transport protein, OmpU, OmpA, OmpK, polar flagellar assembly protein FliH, and so on (date not shown). As expected, enolase was also identified in the OMP preparations. Simplified LC-MS/MS spectrum of a peptide of amino acid sequence of V. parahaemolyticus enolase, and the sequence coverage was shown in Fig. 4b, c. The eno gene sequence has been deposited in GenBank under the accession number KF835607.

Western blot analysis of enolase in different cell fractions from V. parahaemolyticus. Lane 1, cytoplasmic protein fractions; lane 2, outer membrane protein fractions; lane 3, extracellular protein fractions; lane 4, BSA, negative control; lane M prestained protein marker (SM1811, Fermentas)

The localization of enolase in outer membrane. a SDS-PAGE analysis of purified recombinant enolase (lane 1) and OMPs of V. parahaemolyticus strain ATCC33847 (lane 2). The band pointed by the arrow was cut from the gel and further analyzed with the help of LC-MS/MS. Numbers on the left refer to commercial molecular weight markers (SM1811, Fermentas). b Simplified LC-MS/MS spectrum of the DAGYTAVISHR peptide. c Amino acid sequence of V. parahaemolyticus enolase. Identified peptide sequences are highlighted in yellow, which cover 17.3 % of the full-length sequence of enolase
In order to further examine the distribution of enolase, the V. parahaemolyticus strain ATCC33847 was fixed and stained with primary antibodies against V. parahaemolyticus enolase followed by gold-labeled secondary antibodies. Immunoelectron microscopy confirmed the surface and intracellular locations of enolase on bacterial cells (Fig. 5a). In contrast, no labeling was observed for the negative control (Fig. 5b).

Surface localization of enolase on V. parahaemolyticus. a Immunogold electron microscopic detection of the location of enolase (original magnification ⋅58,000) on V. parahaemolyticus pretreated with mouse anti-enolase antibody followed by colloidal-gold-labeled anti-mouse IgG. Colloidal gold particles were localized at the surface of bacterial cells (indicated by thin arrows) and in the cytoplasm of bacterial cells (indicated by thick arrows). b Normal mouse serum used as a negative control. Bars, 500 nm
V. parahaemolyticus enolase binds plasminogen
ELISA analysis revealed specific concentration-dependent interactions between Plg and immobilized V. parahaemolyticus enolase. A similar pattern was observed in wells coated with V. parahaemolyticus outer membrane proteins. Binding was detected using anti-plasminogen and secondary antibodies. The differences between Plg (0.0156 to 2 μg/well) and control (BSA, negative control) were significant (P < 0.05) (Fig. 6a, b). Competition experiments were performed by adding V. parahaemolyticus enolase rabbit polyclonal antibodies to the system. The addition of increasing concentrations of anti-enolase antibody led to a dose-dependent decrease in Plg binding to V. parahaemolyticus (Fig. 6c).

Plasminogen binding assays. ELISA results indicated that plasminogen (Plg) (0.0156 to 2 μg/well) binds to purified recombinant enolase (a) or OMPs from V. parahaemolyticus (b) in a concentration-dependent manner. Binding of plasminogen is inhibited by increasing concentrations of rabbit antienolase antibody (in serial dilutions from 1/500 to 1/64,000) in a competition assay (c). Three independent experiments were performed in triplicate
Inhibition of bacterial adherence
Immunofluorescence analyses were conducted to determine whether V. parahaemolyticus enolase contributes to bacterial adherence. Significant green fluorescence was detected from the surface of Hep-2 cells incubated with V. parahaemolyticus enolase (Fig. 7a), while no specific green fluorescence was observed from negative controls (Fig. 7b, c). To exclude the possible contribution of His6 tag in the adherence, we removed the tag from the recombinant enolase using the Thrombin Cleavage Capture Kit. SDS-PAGE and Western blot analyses showed that the cleaved protein was successfully obtained, and its purity was >95 % (Fig. S1). The immunofluorescence assay indicated that the resulting cleaved protein could still contribute to protein binding to Hep-2 cells (Fig. S2).

Immunofluorescence assay (IFA). Adherence of enolase to Hep-2 cells was confirmed by an indirect immunofluorescence assay, which indicates enolase bound to the surface of the Hep-2 cell. Hep-2 cells were incubated with recombinant V. parahaemolyticus enolase and then with rabbit anti-enolase antibody (a) or with preimmunization rabbit serum (b), or Hep-2 cells were incubated with BSA and then with rabbit anti-enolase antibody (c), stained with goat anti rabbit IgG-FITC before being examined with a confocal laser scanning microscope (original magnification × 400)
Further, Hep-2 cells were treated with increasing amounts of purified V. parahaemolyticus enolase before adherence of V. parahaemolyticus to cells. V. parahaemolyticus adherence to Hep-2 cells was inhibited by enolase in a dose-dependent manner. At 15 μg/well enolase, we observed ~50 % inhibition of V. parahaemolyticus adherence (Fig. 8a). To exclude the possibility that V. parahaemolyticus enolase prevents a bacterial adhesin from interacting with its receptor on the epithelial cell surface, bacterial enolase was blocked with a specific anti-enolase antibody. As shown in Fig. 8b, anti-enolase antibody treatment decreased the adhesion of V. parahaemolyticus to Hep-2 cells to ~63 % compared with the control. Our results collectively indicate that enolase is a V. parahaemolyticus adherence-related factor.

Inhibition assays. a The recombinant enolase inhibits binding capacity of V. parahaemolyticus to Hep-2 cells. Dose-dependent inhibition of V. parahaemolyticus adherence to Hep-2 cells was determined in the presence of increasing amounts of purified enolase. b The anti-enolase antibody inhibits binding capacity of V. parahaemolyticus to Hep-2 cells. The V. parahaemolyticus strain ATCC33847 was incubated with preimmune serum (control) and with anti-enolase antibody, respectively. *P < 0.05 in comparison with the level of adhesion with control (considered to be 100 %)
Vaccine protective efficacy
The virulence of V. parahaemolyticus ATCC33847 was tested with mouse models. Mortality of animals was observed for 7 days after the challenge. The LD50 value was 4.0 × 108 CFU/mouse with the strain ATCC33847. Three groups of 15 mice were injected with V. parahaemolyticus enolase, inactivated V. parahaemolyticus strain ATCC33847, and PBS, respectively. At 10 days after booster immunization, each group was challenged i.p. with 5 LD50 of V. parahaemolyticus ATCC33847. At 1 day after inoculation, all mice in the negative control group died, and most exhibited marked clinical signs, such as ruffled hair, difficulties in breathing, and poor appetite. At 2 days after challenge, two mice died in both the group immunized with the inactivated V. parahaemolyticus strain ATCC33847 and the enolase-vaccinated group. Some of the surviving mice showed clinical signs of V. parahaemolyticus infection, but these were gradually convalescing. In both groups, 86.7 % of mice (13/15) successfully survived the V. parahaemolyticus infection. From day 3, no deaths were observed until the end of the study, which was indicative of high protection from cause-specific mortality.
Discussion
Enolase is a glycolytic enzyme that participates in a variety of normal cellular activities (Miles et al. 1991). The role of enolase in the pathogenesis of Gram-negative bacteria is far from clear. Moreover, no reports to date have focused on V. parahaemolyticus enolase and its encoding gene, although those from several other pathogenic organisms, including bacterial enolases, have been investigated (Agarwal et al. 2008; Bergmann et al. 2003; Ehinger et al. 2004). In the present study, enolase from V. parahaemolyticus ATCC 33847 was cloned, expressed, and characterized. Our enzymatic test results revealed that in vitro overexpressed V. parahaemolyticus enolase, a functional protein, successfully catalyzes the conversion of 2-PGE to PEP, indicating that recombinant enolase retains enzymatic activity. The kinetic coefficients K m and V max values for V. parahaemolyticus enolase were comparable with those published for the enolases of several other pathogens such as Brucella abortus (Han et al. 2012) and Streptococcus suis (Feng et al. 2009).
In addition to its property as a glycolytic enzyme, the V. parahaemolyticus enolase sequence contains typical Plg-binding site motifs characteristic of other enolase proteins. The ability of V. parahaemolyticus enolase to bind Plg was examined using ELISA, and our results clearly suggest that V. parahaemolyticus enolase is a Plg-binding protein enolase and binds plasminogen in a dose-dependent manner. Several recent studies have demonstrated that enolase plays a major role in the microbial recruitment of Plg, and the Plg-binding property of bacteria, including pathogenic spirochetes, is suggested to be a contributory factor in tissue invasion and survival in hosts (Bergmann et al. 2003; Crowe et al. 2003; Ehinger et al. 2004; Nogueira et al. 2010).
To function as a Plg-binding protein, enolase must localize to the microbial surface. Enolase had been shown to be present in both the soluble cytoplasmic and membrane fractions in several other bacterial species, such as A. hydrophila (Sha et al. 2009), Bacillus anthracis (Agarwal et al. 2008), B. burgdorferi (Toledo et al. 2012; Floden et al. 2011), S. pneumoniae (Kolberg et al. 2006), and S. suis (Esgleas et al. 2008). In the present study, we found that enolase was detectable in the extracellular, outer membrane, and soluble cytosolic fractions of V. parahaemolyticus by Western blotting. Further, we demonstrated its presence in the cytoplasma as well as localization on bacterial surface by LC-MS/MS analysis of OMP profiles and immunogold staining of V. parahaemolyticus. Diverse localization of enolase suggests its pivotal role as a glycolytic enzyme.
The exact mechanism by which enolase is secreted and translocated to the bacterial surface is still unknown. It is known that the amino acid sequence of enolase is absent for an N-terminal signal sequence. The findings from Yang et al. (2011) showed that protein structure, such as a hydrophobic α-helical domain of enolase, might be a contributing factor in enolase secretion of B. subtilis. A recent study from Leptospira interrogans (Nogueira et al. 2013) speculated that a secretion system might involve in enolase secretion, and once secreted by a yet-unknown mechanism, enolase probably localizes on the bacterial surface by reassociation. Nine protein secretion systems have been unravelled so far in Gram-negative bacteria (Chagnot et al. 2013). A secreted protein can be translocated across outer membrane through secretion systems. Besides, the contribution of the so-called nonclassical (NC) secretion and other protein trafficking mechanisms in bacterial colonization (e.g., allolysis, phage-mediated lysis, membrane budding) remains to be determined. Nevertheless, the molecular mechanism of how enolase is exported, secreted, or become bacterial cell-surface localization needs further investigation.
Although the mechanism underlying the translocation of protoplasmic enolase across cellular membranes remains unknown at present, in view of the collective data showing that V. parahaemolyticus enolase binds to human Plg, we propose that enolase on the cell surface of V. parahaemolyticus functions in the colonization and/or invasion of the pathogen. Accumulating data support the hypothesis that surface-related proteins are involved in bacterial pathogenesis via adhesion, invasion, and bacterial defense mechanisms (Boleij et al. 2011; Patti et al. 1994). The localization of enolase is consistent with the etiology of adherence to the host cells. In general, proteins localized on the cell surfaces of pathogenic bacteria may act as potential adhesins. Indeed, for many infectious agents, surface-displayed enolase is proposed to function as an adhesion (Marcos et al. 2012; Nogueira et al. 2012; Song et al. 2012). Undoubtedly, adherence to epithelial cells is important for V. parahaemolyticus to break through the first host barrier. To elucidate its possible role in the early events of V. parahaemolyticus infection, indirect immunofluorescence and inhibition assays were conducted in this study and V. parahaemolyticus enolase-mediated adherence to the Hep-2 cell surface was confirmed.
The virulence roles of microbial antigens can be effectively analyzed using molecular genetic approaches. However, our attempts to develop an enolase-deficient mutant of V. parahaemolyticus were unsuccessful (data not shown), possibly due to its indispensable involvement in glycolysis. It suggests that enolase is essential for bacterial life of V. parahaemolyticus and may thus be successfully employed as a diagnostic or protective antigen.
To investigate the immunological characteristics of V. parahaemolyticus enolase, an immunization assay and animal challenge experiment were performed. Our immunological data revealed strong antigenicity and immunogenicity of V. parahaemolyticus enolase. The recombinant protein enolase reacted strongly with convalescent phase serum from mouse clinically infected with V. parahaemolyticus, showing that enolase triggered an antibody response during the course of infection. In addition, immunoassay results showed that V. parahaemolyticus enolase is immunogenic in rabbits, and an ELISA titer of 1:32,000 could be achieved. Western blot analysis further revealed that the acquired anti-enolase antibody not only responds effectively to purified recombinant enolase but also specifically recognizes native enolase from V. parahaemolyticus cytosolic proteins or outer membrane proteins. Our data showing the generation of enolase-specific antibody responses in infected hosts as well as extracellular or microbial surface-associated localization of enolase suggest that the protein may facilitate the pathogen’s infection in the host.
The issue of whether V. parahaemolyticus enolase exhibits strong immunogenicity and confers protective efficacy is of significant interest. Enolases from several pathogens have been identified as an important immunogenic protein and a protective antigen (Feng et al. 2009; Pal-Bhowmick et al. 2007; Sha et al. 2009; Zhang et al. 2009). For instance, mice immunized with purified recombinant enolase subjected to a lethal challenge dose of wild-type (WT) A. hydrophila were protected against mortality. López-Villar et al. (2006) reported that enolase of the pathogenic yeast Candida albicans constitutes an immunodominant antigen during invasive candidiasis. Moreover, Zhang et al. (2009) demonstrated that enolase of S. suis can be used as an immunogen to protect mice from infection. V. parahaemolyticus enolase could effectively serve as a vaccine candidate, as confirmed with the challenge study. Among mice immunized with purified recombinant enolase in our study, 86.7 % were protected against mortality from a lethal challenge dose of the TDH-positive V. parahaemolyticus strain ATCC33847. This relative protective efficacy was similar to that with inactivated ATCC33847, confirming the suitability of V. parahaemolyticus enolase as a candidate subunit vaccine against bacterial infection. In addition, the enolase gene was detected in all 150 strains of V. parahaemolyticus isolated from seafood or diarrhea patients using PCR (data not shown), indicating that enolase is reasonably conserved among V. parahaemolyticus. BLAST analysis showed high similarity of V. parahaemolyticus enolase gene and amino acid sequences to homologs from other Vibrio species. The findings suggest that V. parahaemolyticus enolase may be a suitable candidate for developing vaccine against different Vibrio species.
In conclusion, our studies have clearly shown that V. parahaemolyticus enolase is a Plg-binding, surface-exposed protein facilitating bacterial adherence to Hep-2 cells, which possibly functions in colonization and/or invasion of bacteria into host epithelial cells and promotes pathogen–host interactions. Additionally, V. parahaemolyticus enolase plays an important role in pathogenicity and confers protective efficacy against V. parahaemolyticus infection, supporting its utility as an effective subunit vaccine for development.
References
Agarwal S, Kulshreshtha P, Bambah Mukku D, Bhatnagar R (2008) α-Enolase binds to human plasminogen on the surface of Bacillus anthracis. Biochim Biophys Acta 1784:986–994
Avilán L, Gualdrón-López M, Quiñones W, González-González L, Hannaert V, Michels PA, Concepción JL (2011) Enolase: a key player in the metabolism and a probable virulence factor of trypanosomatid parasites-perspectives for its use as a therapeutic target. Enzym Res 2011:932549
Baffone W, Citterio B, Vittoria E, Casaroli A, Pianetti A, Campana R, Bruscolini F (2001) Determination of several potential virulence factors in Vibrio spp. isolated from sea water. Food Microbiol 18:479–488
Baker-Austin C, Stockley L, Rangdale R, Martinez-Urtaza J (2010) Environmental occurrence and clinical impact of Vibrio vulnificus and Vibrio parahaemolyticus: a European perspective. Environ Microbiol Rep 2:7–18
Bergmann S, Wild D, Diekmann O, Frank R, Bracht D, Chhatwal GS, Hammerschmidt S (2003) Identification of a novel plasmin (ogen)-binding motif in surface displayed α-enolase of Streptococcus pneumoniae. Mol Microbiol 49:411–423
Boleij A, Laarakkers CM, Gloerich J, Swinkels DW, Tjalsma H (2011) Surface-affinity profiling to identify host-pathogen interactions. Infect Immun 79:4777–4783
Broberg CA, Calder TJ, Orth K (2011) Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes Infect 13:992–1001
Chagnot C, Zorgani MA, Astruc T, Desvaux M (2013) Proteinaceous determinants of surface colonization in bacteria: bacterial adhesion and biofilm formation from a protein secretion perspective. Front Microbiol 4:303
Chen H, Yu S, Shen X, Chen D, Qiu X, Song C, Ding C (2011) The Mycoplasma gallisepticum α-enolase is cell surface-exposed and mediates adherence by binding to chicken plasminogen. Microb Pathog 51:285–290
Crowe JD, Sievwright IK, Auld GC, Moore NR, Gow NA, Booth NA (2003) Candida albicans binds human plasminogen: identification of eight plasminogen-binding proteins. Mol Microbiol 47:1637–1651
Díaz-Ramos À, Roig-Borrellas A, García-Melero A, Lopez-Alemany R (2012) α-Enolase, a multifunctional protein: its role on pathophysiological situations. J Biomed Biotechnol 2012:156795
Ehinger S, Schubert WD, Bergmann S, Hammerschmidt S, Heinz DW (2004) Plasmin (ogen)-binding α-enolase from Streptococcus pneumoniae: crystal structure and evaluation of plasmin (ogen)-binding sites. J Mol Biol 343:997–1005
Esgleas M, Li Y, Hancock MA, Harel J, Dubreuil JD, Gottschalk M (2008) Isolation and characterization of α-enolase, a novel fibronectin-binding protein from Streptococcus suis. Microbiology 154:2668–2679
Feng Y, Pan X, Sun W, Wang C, Zhang H, Li X, Ma Y, Shao Z, Ge J, Zheng F (2009) Streptococcus suis enolase functions as a protective antigen displayed on the bacterial cell surface. J Infect Dis 200:1583–1592
Floden AM, Watt JA, Brissette CA (2011) Borrelia burgdorferi enolase is a surface-exposed plasminogen binding protein. PLoS One 6:e27502
Ghosh AK, Jacobs-Lorena M (2011) Surface-expressed enolases of Plasmodium and other pathogens. Mem Inst Oswaldo Cruz 106:85–90
Han X, Ding C, Chen H, Hu Q, Yu S (2012) Enzymatic and biological characteristics of enolase in Brucella abortus A19. Mol Biol Rep 39:2705–2711
Holmes M, Liwak U, Pricop I, Wang X, Tomavo S, Ananvoranich S (2010) Silencing of tachyzoite enolase 2 alters nuclear targeting of bradyzoite enolase 1 in Toxoplasma gondii. Microbes Infect 12:19–27
Kolberg J, Aase A, Bergmann S, Herstad TK, Rødal G, Frank R, Rohde M, Hammerschmidt S (2006) Streptococcus pneumoniae enolase is important for plasminogen binding despite low abundance of enolase protein on the bacterial cell surface. Microbiology 152:1307–1317
López-Villar E, Monteoliva L, Larsen MR, Sachon E, Shabaz M, Pardo M, Pla J, Gil C, Roepstorff P, Nombela C (2006) Genetic and proteomic evidences support the localization of yeast enolase in the cell surface. Proteomics 6:107–118
Marcos CM, Fátima da Silva J, Oliveira HC, Moraes da Silva RA, Mendes-Giannini MJ, Fusco-Almeida AM (2012) Surface-expressed enolase contributes to the adhesion of Paracoccidioides brasiliensis to host cells. FEMS Yeast Res 12:557–570
McCarter L (1999) The multiple identities of Vibrio parahaemolyticus. J Mol Microbiol Biotechnol 1:51–57
Miles LA, Dahlberg CM, Plescia J, Felez J, Kato K, Plow EF (1991) Role of cell-surface lysines in plasminogen binding to cells: identification of alpha-enolase as a candidate plasminogen receptor. Biochemistry 30:1682–1691
Mundodi V, Kucknoor A, Alderete J (2008) Immunogenic and plasminogen-binding surface-associated α-enolase of Trichomonas vaginalis. Infect Immun 76:523–531
Navarre WW, Schneewind O (1999) Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol Mol Biol Rev 63:174–229
Ni XD, Wang N, Liu YJ, Lu CP (2010) Immunoproteomics of extracellular proteins of the Aeromonas hydrophila China vaccine strain J-1 reveal a highly immunoreactive outer membrane protein. FEMS Immunol Med Microbiol 58:363–373
Nishibuchi M, Hill WE, Zon G, Payne WL, Kaper JB (1986) Synthetic oligodeoxyribonucleotide probes to detect Kanagawa phenomenon-positive Vibrio parahaemolyticus. J Clin Microbiol 23:1091–1095
Nogueira SV, Fonseca FL, Rodrigues ML, Mundodi V, Abi-Chacra EA, Winters MS, Alderete JF, de Almeida Soares CM (2010) Paracoccidioides brasiliensis enolase is a surface protein that binds plasminogen and mediates interaction of yeast forms with host cells. Infect Immun 78:4040–4050
Nogueira SV, Smith AA, Qin JH, Pal U (2012) A surface enolase participates in Borrelia burgdorferi-plasminogen interaction and contributes to pathogen survival within feeding ticks. Infect Immun 80:82–90
Nogueira SV, Backstedt BT, Smith AA, Qin J-H, Wunder EA, Ko A, Pal U (2013) Leptospira interrogans enolase is secreted extracellularly and interacts with plasminogen. PLoS ONE 8(10):e78150
Pal-Bhowmick I, Mehta M, Coppens I, Sharma S, Jarori GK (2007) Protective properties and surface localization of Plasmodium falciparum enolase. Infect Immun 75:5500–5508
Pancholi V (2001) Multifunctional α-enolase: its role in diseases. Cell Mol Life Sci 58:902–920
Pancholi V, Chhatwal GS (2003) Housekeeping enzymes as virulence factors for pathogens. Int J Med Microbiol 293:391–401
Park KS, Ono T, Rokuda M, Jang MH, Okada K, Iida T, Honda T (2004) Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect Immun 72:6659–6665
Patti JM, Allen BL, McGavin MJ, Hook M (1994) MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol 48:585–617
Qadri F, Alam MS, Nishibuchi M, Rahman T, Alam NH, Chisti J, Kondo S, Sugiyama J, Bhuiyan NA, Mathan MM (2003) Adaptive and inflammatory immune responses in patients infected with strains of Vibrio parahaemolyticus. J Infect Dis 187:1085–1096
Reed LJ, Muench H (1938) A simple method of estimating fifty percent endpoints. Am J Epidemiol 27:493–497
Schreiner SA, Sokoli A, Felder KM, Wittenbrink MM, Schwarzenbach S, Guhl B, Hoelzle K, Hoelzle LE (2012) The surface-localised α-enolase of Mycoplasma suis is an adhesion protein. Vet Microbiol 156:88–95
Sha J, Galindo C, Pancholi V, Popov V, Zhao Y, Houston C, Chopra A (2003) Differential expression of the enolase gene under in vivo versus in vitro growth conditions of Aeromonas hydrophila. Microb Pathog 34:195–204
Sha J, Erova TE, Alyea RA, Wang S, Olano JP, Pancholi V, Chopra AK (2009) Surface-expressed enolase contributes to the pathogenesis of clinical isolate SSU of Aeromonas hydrophila. J Bacteriol 191:3095–3107
Shimohata T, Takahashi A (2010) Diarrhea induced by infection of Vibrio parahaemolyticus. J Med Investig 57:179–182
Shinoda S (2011) Sixty years from the discovery of Vibrio parahaemolyticus and some recollections. Biocontrol Sci 16:129–137
Song Z, Li Y, Liu Y, Xin J, Zou X, Sun W (2012) α-Enolase, an adhesion-related factor of Mycoplasma bovis. PLoS ONE 7:e38836
Su YC, Liu C (2007) Vibrio parahaemolyticus: a concern of seafood safety. Food Microbiol 24:549–558
Toledo A, Coleman J, Kuhlow C, Crowley J, Benach J (2012) The enolase of Borrelia burgdorferi is a plasminogen receptor released in outer membrane vesicles. Infect Immun 80:359–368
Vongxay K, Wang S, Zhang X, Wu B, Hu H, Pan Z, Chen S, Fang W (2008) Pathogenetic characterization of Vibrio parahaemolyticus isolates from clinical and seafood sources. Int J Food Microbiol 126:71–75
Whiting G, Evans J, Patel S, Gillespie S (2002) Purification of native α-enolase from Streptococcus pneumoniae that binds plasminogen and is immunogenic. J Med Microbiol 51:837–843
Yang CK, Ewis HE, Zhang X, Lu CD, Hu HJ, Pan Y, Abdelal AT, Tai PC (2011) Nonclassical protein secretion by Bacillus subtilis in the stationary phase is not due to cell lysis. J Bacteriol 193:5607–5615
Yeung PM, Boor KJ (2004) Epidemiology, pathogenesis, and prevention of foodborne Vibrio parahaemolyticus infections. Foodborne Pathog Dis 1:74–88
Zhang A, Chen B, Mu X, Li R, Zheng P, Zhao Y, Chen H, Jin M (2009) Identification and characterization of a novel protective antigen, enolase of Streptococcus suis serotype 2. Vaccine 27:1348–1353
Acknowledgments
This work was funded by the National Key Technology Support Program (2012BAK08B07), Shanghai Municipal Science and Technology Commission technical standards (13DZ0502702), and Aquatic Three New Projects in Jiangsu Province (D2013-5-4).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 97 kb)
Rights and permissions
About this article
Cite this article
Jiang, W., Han, X., Wang, Q. et al. Vibrio parahaemolyticus enolase is an adhesion-related factor that binds plasminogen and functions as a protective antigen. Appl Microbiol Biotechnol 98, 4937–4948 (2014). https://doi.org/10.1007/s00253-013-5471-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5471-z