
Abstract
Brucella abortus is the etiological agent of brucellosis, a disease causing human public health problems as well as major economic losses in domestic animal industries. In this study, the enolase gene of B. abortus A19 was cloned, sequenced and expressed in Escherichia coli BL21. Bacterial-expressed enolase protein (His-eno) was purified and its ability to catalyze the conversion of 2-phosphoglycerate (2-PGE) to phosphoenolpyruvate (PEP) (hereon referred to as enolase activity) was analyzed. Michaelis constant (K m ) and maximum reaction velocity (V max ) of the reaction was determined to be 2.0 × 10−3 M and 178 μM l−1 min−1, respectively. Factors influencing the enolase activity of His-eno, such as pH, the presence of metal ions and temperature were investigated in vitro. The results showed that His-eno exhibited maximal enolase activity in pH 8.5 reaction buffer containing 10 mM MgSO4 at 37°C. In addition to studying the enzyme activity, binding assays were performed to provide insights into the function of His-eno on pathogenesis and immunity. His-eno exhibits fibronectin-binding ability in immunoblotting assay, suggesting that enolase may play a role in B. abortus colonization, persistence, and invasion of host tissue. Furthermore, Western blot demonstrated His-eno’s binding ability to 34 bovine B. abortus positive sera, suggesting that future studies may find enolase a useful as a diagnostic marker or a vaccine candidate for brucellosis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Brucella abortus, a facultative intracellular Gram-negative bacterium, is the most important Brucella species involved in bovine brucellosis, which is characterized by abortion and infertility of infected animals [1–3]. Brucellosis is also a zoonotic disease and can infect humans through contact with contaminated animal products [4]. Brucella does not have classical virulence factors such as exotoxins, cytolysins, capsule, fimbria, flagellum, plasmids, lysogenic phages, antigenic variation, endotoxic lipopolysaccharide (LPS), and inducers of host cell apoptosis [5]. A key step in the pathogenesis of B. abortus is its ability to invade phagocytic and non-phagocytic host cells. Virulent Brucella organisms can infect both nonphagocytic and phagocytic cells. The mechanism of invasion of nonphagocytic cells is not clearly established. However, a 41 kDa surface protein (SP41) and the O antigen of the LPS molecule have been reported to be involved in Brucella’s adherence and invasion of host cells [6, 7]. A considerable number of fibronectin-binding proteins from various bacterial species have been reported, most of these proteins were shown to be involved in colonization, invasion and adhesion to epithelial cells, which is a critical early step in infection [8]. Enolase (2-phospho-d-glycerate hydrolase) is an enzyme of the glycolytic pathway, which catalyzes a reversible conversion of 2-phosphoglycerate (2-PGE) to phosphoenolpyruvate (PEP). Recent studies suggested that enolases from eukaryotic and prokaryotic cells possess fibronectin-binding activity and play such roles in pathogenesis [9, 10], however, there has been no report regarding B. abortus enolase yet.
We were particularly interested in studying the enolase protein in B. abortus is because: (1) Enolase is reported as an important enzyme of the glycolytic pathway, therefore, we intent to understand this enzyme to find ways to control the infections by blocking the sugar metabolism pathway in B. abortus by targeting enolase. (2) Enolase has been found on the cell surface in a variety of microorganisms where it plays a critical role in bacterial colonization, persistence and invasion of host tissue [11–13] or acts as an immunogenic protein [14–17], so it is extremely important to study enolase from pathogenesis and immunity points of view. Therefore in this study, we cloned, expressed and characterized B. abortus enolase in hope to understand its roles in B. abortus metabolism, pathogenesis and immunity.
Materials and methods
Culturing of bacteria
B. abortus A19 was obtained from Chinese Veterinary Culture Collection Center (CVCC) and cultured in tryptic soy agar (TSA, Difco, NJ) or tryptic soy broth (TSB, Difco, NJ) at 37°C with 5% CO2. Escherichia coli strains DH5α (Invitrogen, Carlsbad, CA) and BL21 (DE3, Stratagene, La Jolla, CA) were grown on Luria–Bertani (LB) agar plates or broth at 37°C and transformants were selected on media supplemented with 50 μg/ml kanamycin. The expression vector pET-28a was purchased from Novagen (Madison, WI). Restriction enzymes were from MBI Fermentas (Hanover, MD). All chemicals used in this study were of analytical grade and purchased from Sigma (St. Louis, MO).
Preparation of bacterial genomic DNA of B. abortus A19
Brucella abortus A19 was cultured on TSA plates, harvested in saline (0.85% NaCl solution) and heat-killed at 80°C for 30 min in a water bath. Chromosomal DNA was isolated using Wizard Genomic DNA Purification Kit according to the manufacturer’s protocol (Promega, Madison, WI).
Expression and purification of B. abortus A19 enolase
The enolase gene was amplified by polymerase chain reaction (PCR) using primers Eno-F (5′-CGCGGATCC ATGACTGCAATCATCGACAT-3′) and Eno-R (5′-CGCGTCGAC TTAGAGCAACTTCAGTGCAC-3′). The primers were designed according to the enolase gene sequence of B. abortus 2308 (NC_007618) with BamHI and SalI site (underlined) inserted. PCR product was BamHI/SalI-digested and cloned into BamHI/SalI sites of pET 28a (+) (Novagen). The resulting plasmids, pET28a-Eno, was used to transform E. coli BL21. Expression of His-tagged enolase (His-eno) was induced by 1 mM IPTG, analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie blue staining. The recombinant protein His-eno was purified using HisTrap chelating high-performance columns (Amersham Pharmacia Biotech, Piscataway, NJ) and was quantitative determined using a BCA protein assay kit (Pierce, Rockford, IL).
Enolase activity and kinetics assay of His-eno
His-eno activity was determined by measuring the conversion of 2-PGE (Sigma) to PEP (Sigma) at 25°C as described [9] with modifications. A standard assay consists of adding 1 mM 2-PGE to 1.0 ml reaction buffer (100 mM HEPES buffer, pH 8.5, 7.7 mM KCl, 10 mM MgSO4, pre-warmed to 25°C), 15 μg of His-eno was then added to initiate the reaction. The reaction was monitored spectrophotometrically by measuring absorbance at 240 nm for the production of PEP at 1 min intervals for 10 min. The kinetics of the enolase activity was expressed as the production of PEP. Rabbit muscle enolase (Sigma) was used in place of His-eno as a positive control for the assay. The enzyme activity was also measured with 2.5, 5.0, 10.0 and 20.0 μg of His-eno to test whether enzyme quantity is related to the activity.
To study enolase enzyme kinetics, varying concentrations of 2-PGE substrate (0.5, 1.0, 1.5, or 2.0 mM) were used for the assay. Michaelis–Menten kinetics of His-eno showed that His-eno was able to fully convert 2-PGE to PEP for all four substrate concentration levels. V max and K m for His-eno were determined from double-reciprocal Lineweaver–Burk plots.
Effects of pH, metal ions and temperature on enolase activity of His-eno
Modifications to the standard spectrophotometry enzyme kinetics assay were made to study the effect of pH, metal ions, and temperature on His-eno activity. As for the pH experiment, reaction buffers with pH values ranging from 5.0 to 10.0 in 0.5 increments were used for the assay. To examine the effect of metal ions, 10 mM of MgSO4 in the original reaction buffer was replaced with 10 mM of ZnSO4, MnCl2, AlCl3, NiSO4, CuSO4, HgCl2, CrCl3 or nothing. To test the thermostability of His-eno, the enzyme was pre-incubated for 1 h at 25, 37, 50, 62 or 75°C prior to the assay. Enolase activity in each of the above tests were compared to that of a standard reaction at 25°C involving 1.0 mM 2-PGE substrate, 15 μg of His-eno, and the standard reaction buffer at pH 8.5 containing 10 mM MgSO4. All the reactions were performed in triplicate.
Preparation of rabbit anti-serum against His-eno
Rabbit anti-serum against His-eno was prepared by immunizing two New Zealand female rabbits (female, 2–3 kg) subcutaneously with 500 μg of His-eno emulsified in Montanide ISA 50 V (SEPPIC, France), three times at 4-week intervals. Blood samples were collected 2 weeks after the third injection. The titers of immunized sera were evaluated by enzyme-linked immunosorbent assay (ELISA) according to the standard methods, with a coating concentration of 100 ng purified His-eno per well on a 96 well ELISA plate.
Preparation and Western blot analysis of B. abortus A19 membrane proteins
To determine whether B. abortus A19 enolase was exported to the cell membrane, bacterial membrane proteins were isolated and analyzed by Western blot as previously described [18] with modifications. Briefly, mid-logarithmic phase B. abortus A19 in 200 ml TSB culture were collected by centrifuging at 8,000×g for 15 min at 4°C. The pellets were washed with phosphate buffered saline (PBS, pH 7.4), re-suspended in buffer A (15 mM Tris–HCl, pH 8.0, 0.45 M sucrose, 8 mM EDTA, and 0.4 mg/ml lysozyme), incubated at room temperature for 30 min, and centrifuged at 8,000×g for 15 min again. The pellet was re-suspended in buffer B (50 mM Tris–HCl, pH 7.6, 5 mM MgCl2, and 2 mM PMSF), chilled on ice and sonicated (10 s at 400 W, for a total of 30 times with 5 s breaks in between). Then the samples were centrifuged twice at 3,000×g for 15 min to pellet intact cells. The supernatant was ultra-centrifuged at 45,000 rpm for 90 min and the pellet was collected and re-suspended in 12 ml buffer C (50 mM Tris–HCl, pH 7.6 with 2 mM PMSF). The protein was quantitative determined using a BCA protein assay kit (Pierce, Rockford, IL).
For Western blot, protein sample (10 μg/lane) was subjected to SDS-PAGE, then transferred onto PVDF membranes (Amersham Pharmacia Biotech) using a semi-dry transfer cell (Bio-Rad) at 15 V for 15 min. The membrane was blocked with 5% skim milk in PBS-0.05% Tween 20 (PBST) at 4°C for overnight and washed three times in PBST and then incubated with rabbit anti-serum against His-eno (1: 3,000) at room temperature for 2 h. After washing, the membrane was incubated with horseradish peroxidase (HRP)-labeled anti-rabbit IgG (Sigma, 1:5,000) at room temperature for 1 h. The membrane was washed and then developed with 3, 3′-diaminobenzidine (DAB, Sigma) until optimum color development was observed.
Binding ability of His-eno to human fibronectin or bovine B. abortus positive sera by Western blot
For Western blot, 10 μg purified His-eno was loaded into each lane of a 12% acrylamide gel and subjected to SDS-PAGE. The proteins were then electrophoretically transferred onto a PVDF membrane and the membrane was blocked with 5% skim milk in PBST at 4°C for overnight and washed with PBST.
To determine the ability for His-eno to bind to human fibronectin (Sigma), the membrane was incubated with 10 μg/ml of human fibronectin in PBST at room temperature for 2 h. After washing, the membrane was incubated with monoclonal anti-human fibronectin antibody (Sigma, 1:5,000) in PBST at room temperature for 2 h, followed by incubation with HRP-conjugated anti-mouse IgG (Sigma, 1:10,000). The blotting band was detected using an ECL kit (Amersham Pharmacia Biotech) as described in the manufacturer’s protocol.
To determine the binding ability of His-eno to bovine B. abortus positive serum, the membrane was incubated with the bovine serum at room temperature for 2 h. The membrane was washed and incubated with peroxidase-labeled affinity purified antibody to bovine IgG (H + L) (1:3,000, KPL, Gaithersburg, MD) in PBST at room temperature for 1 h. The blotting bands were detected using an ECL kit as described above. A total of 34 bovine B. abortus positive sera, which were tested as B. abortus positive infection by a government approved brucellosis testing method, from Shanghai Center for Animal Disease Control and Prevention, China were tested. A serum sample from a healthy bovine was used as the negative control.
Results
Expression and purification of B. abortus A19 enolase
DNA sequence analysis of the PCR amplified B. abortus A19 enolase gene showed that the 1,278 bp gene has 100% sequence identity with that of B. abortus 2308 (NC_007618). Recombinant fusion protein His-eno was successfully expressed in E. coli BL21 as shown by a 49 kDa band in the SDS-PAGE analysis (Fig. 1, lanes 2 and 3). After purification with HisTrap chelating high-performance columns (Amersham), a single band of His-eno was identified (Fig. 1, lane 1).

Expression and purification of His-eno were identified with SDS-PAGE followed by Coomassie blue staining. M pre-stained protein marker (SM0671, Fermentas). Lane 1 a 49 kDa band of purified His-eno with HisTrap chelating high-performance columns (Amersham Pharmacia Biotech). Lane 2 and 3 total cellular proteins of E. coli BL21 transformed with expression plasmids pET28a-Eno. A 49 kDa expressed His-eno band in each lane was shown. Lane 4 Total cellular proteins of E. coli BL21 transformed with pET28a. No band of 49 kDa His-eno showed
Enolase activity and enzyme kinetic assay of His-eno
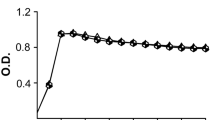
His-eno’s activity in catalyzing the conversion of 2-PGE to PEP was higher than that of rabbit muscle enolase as determined by the spectrophotometry enzyme kinetics assay (Fig. 2). His-eno activity is positively correlated with enzyme concentration of 2.5, 5, 10 or 20 μg. Michaelis–Menten plot was produced using the His-eno kinetics data with varying substrate concentrations. The plot was fitted to the equation V = V max S/(S + K m ) (Fig. 3), Michaelis Constant (K m ) and Maximum Reaction Velocity (V max ) were determined to be 2.0 × 10−3 M and 178 μM l−1 min−1, respectively, indicating high catalytic capability of His-eno for the dehydration of 2-PGE to PEP under our experimental condition.

Enolase activity was determined by measuring the conversion of 2-PGE to PEP. His-eno reached maximal enolase activity at 3 min, which was earlier than that of rabbit muscle enolase (positive control) at 10 min. The kinetics of the enolase activity was expressed as the production of PEP

Enzymatic characterization of purified His-eno. The conversion-rate (V) of 2-PGE (S) to PEP was measured at 240 nm using 15 μg of His-eno and various concentrations of 2-PGE (0.25–2 mM). Measurements were performed at 1 min intervals for 10 min. Data from three independent assays were plotted by the method of Michaelis–Menten and fitted to the equation V = V max ·S/(S + K m ). K m and V max for His-eno were determined to be 2 × 10−3 M and 178 μM l−1 min−1, respectively, by means of a Lineweaver–Burk plot (double-reciprocal plot). Data shown here were the mean values ± standard deviations
Effects of pH, metal ions and temperature on enolase activity of His-eno
His-eno exhibited maximal activity at pH 8.5 (Fig. 4). Comparing to having 10 mM Mg2+ in the reaction buffer, the relative activity of His-eno increased 10.6% when Mg2+ was replaced by Zn2+ of the same concentration (Table 1). Replacement with other ions such as Ni2+, Al3+ and Mn2+ or no salt in the buffer reduced enzyme activity by 90.5, 31.2, 9.6 and 57.0%, respectively, whereas replacement of Mg2+ by Cu2+, Hg2+ or Cr2+ completely inhibited the reaction (Table 1). The thermostability assay of His-eno revealed maximal enolase activity at 37°C. His-eno retained over 90% of the initial activity when pre-incubated at 50 or 62°C for 1 h. However, pre-incubation at 75°C caused His-eno to lose 100% activity (Fig. 5).

Effect of pH on enolase activity of His-eno. The maximal enolase reactivity of His-eno occurred at pH 8.5, with reaction temperature of 37°C and 10 mM MgSO4 in the reaction buffer. One unit is defined as the conversion of 1 μmol of substrate 2-PGE per minute at 37°C

Thermostability assay of His-eno. 15 μg of His-eno was pre-incubated at 25, 37, 50, 62 or 75°C, respectively for 1 h for the assay. The results showed that the enolase activity of His-eno was thermally stable at 25, 37, 50 and 62°C but was completely inhibited by a pre-incubation of the enzyme at 75°C. The experiment was performed in triplicate
Preparation of rabbit anti-serum against His-eno and Western blot analysis of membrane proteins
Rabbit anti-serum against His-eno was isolated and tested for ELISA titers as 1:10,000. The anti-serum was used for Western blot analysis of B. abortus A19 membrane proteins, using whole cell proteins and His-eno as the positive controls. Both total cellular proteins (lane 1) and membrane proteins (lane 2) of B. abortus A19 showed positive binding bands of 45 kDa (Fig. 6), suggesting that enolase is a membrane associated protein. Positive control of lane 3 with loading of His-eno showed a 49 kDa band. Lane 4 without any protein loading showed no binding band.

Rabbit anti-serum against His-eno binds to membrane proteins of B. abortus A19 by Western blot. Lane M pre-stained protein marker (SM0671, Fermentas). Lane 1 immunoblot of total cellular proteins of B. abortus A19. Lane 2 immunoblot of isolated membrane protein of B. abortus A19. Lane 3 immunoblot of purified of His-eno. Lane 4 no protein addition to the lane 4, showed no blot band to rabbit anti-serum against His-eno
Binding ability of His-eno to human fibronectin or bovine B. abortus positive sera
The results of Western blot demonstrated that His-eno has both binding ability to human fibronectin (Fig. 7) and bovine B. abortus positive sera (Fig. 8, complete set of data not shown).

Binding ability of His-eno to human fibronectin. Lane M pre-stained protein marker (SM0671, Fermentas). Lane 1 human fibronectin was immunoblotted with His-eno. Lane 2 no protein addition to the lane 2, showed no blot band to His-eno

Binding ability of His-eno to bovine B. abortus positive sera. Lane M pre-stained protein marker (SM0671, Fermentas). Lane 1–13 13 bovine B. abortus positive sera showed binding band to His-eno. Data for other 21 bovine sera not shown but results were the same for all 34 sera. Lane 14 normal bovine serum showed no binding band to His-eno
Discussion
In the present study, enolase of B. abortus A19 was cloned, expressed and characterized. The expressed recombinant enolase, His-eno, successfully catalyzes the conversion of 2-PGE to PEP with K m and V max values of 2.0 × 10−3 M and 178 μM l−1 min−1, respectively, at 25°C in pH 8.5 reaction buffer with 10 mM Mg2+.
His-eno maximal activity occurs at pH 8.5, a condition more suitable to alkalescent condition, which might be due to the structure and characteristics of enolase. Previous report indicated that the enolase of Lactobacillus crispatus were localized on the cell surface at pH 5 but released into the medium at alkaline pH, indicating that lactobacilli rapidly modify their surface properties in response to changes in pH [19]. It is possible that pH in the environment has similar effect on B. abortus enolase as well, which warrants future investigations for the biological significance. In addition, we demonstrated that Ni2+, Cu2+, Hg2+ and Cr3+ strongly inhibited the activity while Zn2+ and Mg2+ were the most potent activators. This result was consistent with a previous study showing that magnesium is a co-factor that stabilizes the enolase dimer formed by two subunits [20]. In this study, B. abortus enolase showed also a relatively broad thermostability from 25 to 62°C that is beneficial in prompting the process of infection at different temperatures.
Membrane proteins of B. abortus A19 were isolated and subjected to SDS-PAGE, and rabbit anti-serum against His-eno was used for Western blot. The membrane proteins reacted with the rabbit anti-serum of His-eno, suggesting there is enolase protein in the isolated membrane proteins. Combined with the result of His-eno binding to human fibronectin, we believe B. abortus enolase play roles in the colonization and/or invasion of the pathogen. Further investigations of B. abortus enolase function on brucella colonization and invasion will be performed.
The major antigen that dominates the antibody response against B. abortus is the LPS, people used LPS as a marker to make diagnosis, however, there are cross-reactions of the Brucella O antigen with various bacteria, e.g. Yersinia enterocolitica O:9, Salmonella urbana group N, Vibrio cholerae, Francisella tularensis, E. coli O:157, and Stenotrophomonas maltophilia [21–23], therefore, serological diagnosis of brucellosis is still a challenge in human and animal disease. As Brucella is an intracellular parasite, the host immune response is not restricted to surface proteins, numerous outer and inner membranes, cytoplasmic, and periplasmic protein antigens of B. abortus have also been characterized, some are recognized by the immune system during infection and are useful in diagnostic tests [24–28]. To investigate whether there is enolase specific antibody in bovine B. abortus positive serum, we checked 34 such bovine sera by western blot analysis. All the sera tested showed binding bands to His-eno, suggesting that enolase is an immunogenic protein that may used as a diagnostic marker or potential vaccine candidate in the future.
In conclusion, bacterial expressed His-eno showed enolase activity of catalyzing the conversion of 2-PGE to PEP. His-eno has the ability to bind to human fibronectin and bovine B. abortus positive serum, which will benefit for future research concerning the role of enolase in B. abortus.
References
Boschiroli ML, Foulongne V, O’Callaghan D (2001) Brucellosis: a worldwide zoonosis. Curr Opin Microbiol 4:58–64
Pappas G, Akritidis N, Bosilkovski M, Tsianos E (2005) Brucellosis. N Engl J Med 352:2325–2336
Franco MP, Mulder M, Gilman RH, Smits HL (2007) Human brucellosis. Lancet Infect Dis 7:775–786
Corbel MJ (1997) Brucellosis: an overview. Emerg Infect Dis 3:213–221
Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (2006) The Genus Brucella. In The Prokaryotes: a handbook on the biology of bacteria. 3rd edn. New York, Springer, pp 315–456
Castaneda-Roldan EI, Ouahrani-Bettache S, Saldana Z, Avelino F, Rendon MA, Dornand J, Giron JA (2006) Characterization of SP41, a surface protein of Brucella associated with adherence and invasion of host epithelial cells. Cell Microbiol 8:1877–1887
Gorvel JP, Moreno E (2002) Brucella intracellular life: from invasion to intracellular replication. Vet Microbiol 90:281–297
de Greeff A, Buys H, Verhaar R, Dijkstra J, van Alphen L, Smith HE (2002) Contribution of fibronectin-binding protein to pathogenesis of Streptococcus suis serotype 2. Infect Immun 70:1319–1325
Esgleas M, Li Y, Hancock MA, Harel J, Dubreuil JD, Gottschalk M (2008) Isolation and characterization of alpha-enolase, a novel fibronectin-binding protein from Streptococcus suis. Microbiology 154:2668–2679
Sha J, Erova TE, Alyea RA, Wang S, Olano JP, Pancholi V, Chopra AK (2009) Surface-expressed enolase contributes to the pathogenesis of clinical isolate SSU of Aeromonas hydrophila. J Bacteriol 191:3095–3107
Pancholi V, Fischetti VA (1998) Alpha-enolase, a novel strong plasmin (ogen) binding protein on the surface of pathogenic streptococci. J Biol Chem 273:14503–14515
Kolberg J, Aase A, Bergmann S, Herstad TK, Rodal G, Frank R, Rohde M, Hammerschmidt S (2006) Streptococcus pneumoniae enolase is important for plasminogen binding despite low abundance of enolase protein on the bacterial cell surface. Microbiology 152:1307–1317
Briones G, Inon de Iannino N, Steinberg M, Ugalde RA (1997) Periplasmic cyclic 1,2-beta-glucan in Brucella spp. is not osmoregulated. Microbiology 143(Pt 4):1115–1124
Feng Y, Pan X, Sun W, Wang C, Zhang H, Li X, Ma Y, Shao Z, Ge J, Zheng F, Gao F, Tang J (2009) Streptococcus suis enolase functions as a protective antigen displayed on the bacterial cell surface. J Infect Dis 200(10):1583–1592
Zhang A, Xie C, Chen H, Jin M (2008) Identification of immunogenic cell wall-associated proteins of Streptococcus suis serotype 2. Proteomics 8:3506–3515
Zhang A, Chen B, Mu X, Li R, Zheng P, Zhao Y, Chen H, Jin M (2009) Identification and characterization of a novel protective antigen, enolase of Streptococcus suis serotype 2. Vaccine 27:1348–1353
Pal-Bhowmick I, Mehta M, Coppens I, Sharma S, Jarori GK (2007) Protective properties and surface localization of Plasmodium falciparum enolase. Infect Immun 75:5500–5508
Eroles P, Sentandreu M, Elorza MV, Sentandreu R (1997) The highly immunogenic enolase and Hsp70p are adventitious Candida albicans cell wall proteins. Microbiology 143(Pt 2):313–320
Antikainen J, Kuparinen V, Lahteenmaki K, Korhonen TK (2007) pH-dependent association of enolase and glyceraldehyde-3-phosphate dehydrogenase of Lactobacillus crispatus with the cell wall and lipoteichoic acids. J Bacteriol 189:4539–4543
Terrier B, Degand N, Guilpain P, Servettaz A, Guillevin L, Mouthon L (2007) Alpha-enolase: a target of antibodies in infectious and autoimmune diseases. Autoimmun Rev 6:176–182
Caroff M, Bundle DR, Perry MB, Cherwonogrodzky JW, Duncan JR (1984) Antigenic S-type lipopolysaccharide of Brucella abortus 1119-3. Infect Immun 46:384–388
Caroff M, Bundle DR, Perry MB (1984) Structure of the O-chain of the phenol-phase soluble cellular lipopolysaccharide of Yersinia enterocolitica serotype O:9. Eur J Biochem 139:195–200
Corbel MJ, Stuart FA, Brewer RA (1984) Observations on serological cross-reactions between smooth Brucella species and organisms of other genera. Dev Biol Stand 56:341–348
Baldi PC, Wanke MM, Loza ME, Fossati CA (1994) Brucella abortus cytoplasmic proteins used as antigens in an ELISA potentially useful for the diagnosis of canine brucellosis. Vet Microbiol 41:127–134
Cassataro J, Pasquevich K, Bruno L, Wallach JC, Fossati CA, Baldi PC (2004) Antibody reactivity to Omp31 from Brucella melitensis in human and animal infections by smooth and rough Brucellae. Clin Diagn Lab Immunol 11:111–114
Goldbaum FA, Leoni J, Wallach JC, Fossati CA (1993) Characterization of an 18-kilodalton Brucella cytoplasmic protein which appears to be a serological marker of active infection of both human and bovine brucellosis. J Clin Microbiol 31:2141–2145
Lin J, Adams LG, Ficht TA (1996) Immunological response to the Brucella abortus GroEL homolog. Infect Immun 64:4396–4400
Gonzalez M, Andrew E, Folch H, Saez D, Cabrera A, Salgado P, Onate A (2009) Cloning, expression and immunogenicity of the translation initiation factor 3 homologue of Brucella abortus. Immunobiology 214:113–120
Acknowledgments
This work was supported by the fund of Chinese National Programs for Fundamental Research and Development (2010CB530202) and National Basic Fund for Research Institutes, which is supported by Chinese Academy of Agricultural Sciences (2010JB16). We also thank Ms. Siyu Ding from the University of Richmond for her helpful editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Han, X., Ding, C., Chen, H. et al. Enzymatic and biological characteristics of enolase in Brucella abortus A19. Mol Biol Rep 39, 2705–2711 (2012). https://doi.org/10.1007/s11033-011-1025-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-011-1025-6