
Abstract
Aldehyde inhibitors such as furfural and 5-hydroxymethylfurfural (HMF) are generated from biomass pretreatment. Scheffersomyces stipitis is able to reduce furfural and HMF to less toxic furanmethanol and furan-2,5-dimethanol; however, the enzymes involved in the reductive reaction still remain unknown. In this study, transcription responses of two known and five putative alcohol dehydrogenase genes from S. stipitis were analyzed under furfural and HMF stress conditions. All the seven alcohol dehydrogenase genes were also cloned and overexpressed for their activity analyses. Our results indicate that transcriptions of SsADH4 and SsADH6 were highly induced under furfural and HMF stress conditions, and the proteins encoded by them exhibited NADH- and/or NADPH-dependent activities for furfural and HMF reduction, respectively. For furfural reduction, NADH-dependent activity was also observed in SsAdh1p and NAD(P)H-dependent activities were also observed in SsAdh5p and SsAdh7p. For HMF reduction, NADPH-dependent activities were also observed in SsAdh5p and SsAdh7p. SsAdh4p displayed the highest NADPH-dependent specific activity and catalytic efficiency for reduction of both furfural and HMF among the seven alcohol dehydrogenases. Enzyme activities of all SsADH proteins were more stable under acidic condition. For most SsADH proteins, the optimum temperature for enzyme activities was 30 °C and more than 50 % enzyme activities remained at 60 °C. Reduction activities of formaldehyde, acetaldehyde, isovaleraldehyde, benzaldehyde, and phenylacetaldehyde were also observed in some SsADH proteins. Our results indicate that multiple alcohol dehydrogenases in S. stipitis are involved in the detoxification of aldehyde inhibitors derived from lignocellulosic biomass conversion.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Conversion of low-cost lignocellulosic biomass to ethanol has become an attractive option for alternative transportation fuels because lignocellulosic biomass is the most abundant renewable organic material on earth and its use will not release net carbon dioxide into the earth's atmosphere (Sánchez and Cardona 2008; Vertes et al. 2010). Saccharomyces cerevisiae is the most widely used microorganism for crop-based bioethanol production from hexose sugars; however, the wild-type S. cerevisiae strains cannot ferment pentose sugars that are abundantly present in lignocellulosic hydrolysates (Hahn-Hägerdal et al. 2007; Lin and Tanaka 2006). Although the genetically engineered S. cerevisiae strains with pentose metabolizing genes from other yeasts or bacteria can ferment xylose to ethanol, the yield and productivity of ethanol from xylose fermentation of the best engineered strain are still far below expectation (see review by Kim et al. 2013). Among the natural pentose-fermenting yeasts, Scheffersomyces (Pichia) stipitis has shown the most promise for industrial ethanol production from lignocellulosic biomass. S. stipitis can not only ferment xylose with better ethanol yield and productivity but also ferment a wider range of sugars (such as cellobiose) to ethanol than the genetically engineered xylose fermentation S. cerevisiae strains (du Preez and Prior 1985; Kim et al. 2013; Parekh and Wayman 1986; Slininger et al. 2006). Meanwhile, the complete genome sequence revealed that S. stipitis has developed various cellulases and hemicellulases which can break down lignocellulose into monomeric sugars, offering a very useful promise for simultaneous saccharification and fermentation of lignocellulosic materials for ethanol production (Antoni et al. 2007; Jeffries et al. 2007; Weber et al. 2010). Recently, ethanol production from lignocellulosic biomass was studied using S. stipitis (Cho et al. 2010; Lin et al. 2012) or S. stipitis cocultured with S. cerevisiae (Li et al. 2011; Rudolf et al. 2008; Yadav et al. 2011).
Prior to ethanol fermentation by microorganisms, complex lignocellulosic biomass needs to be hydrolyzed to simple sugars via pretreatment and hydrolysis processes. During the pretreatment and hydrolysis processes, lignocellulosic biomass releases not only sugars but also numerous fermentation inhibitors (such as aldehydes, lignin derivatives, and acetic acid) that hinder microbial cell growth and subsequent fermentation (Jönsson et al. 2013; Klinke et al. 2004; Liu and Blaschek 2010). Thereafter, this poses a significant challenge for bioethanol production from lignocellulosic biomass. Among the numerous fermentation inhibitors, furfural and 5-hydroxymethylfurfural (HMF), derived from pentoses and hexoses, respectively, are the representative aldehyde inhibitors (Antal et al. 1991; Heer and Sauer 2008; Jönsson et al. 2013; Larsson et al. 1999; Lewkowski 2001; Taherzadeh et al. 1997). Remediation of inhibitory compounds by various physical and chemical methods has been studied, but it was too expensive for practice use (see review by Liu and Blaschek 2010). Overcoming the inhibitory effect of furfural and HMF on yeast is a key factor for cost-efficient bioethanol production from lignocellulosic biomass.
Previous studies have found that many microorganisms possess the ability to detoxify furfural and HMF. Stress response and in situ detoxification of furfural and HMF to the less toxic compounds furanmethanol (FM) and furan-2,5-dimethanol (FDM) were extensively studied in S. cerevisiae (see review by Liu 2011). The detoxification reaction was NADH- and/or NADPH-dependent and catalyzed by multiple enzymes, especially alcohol dehydrogenases (ADH) such as ScAdh6p (Sc for S. cerevisiae), ScAdh7p, and mutated ScAdh1p (Almeida et al. 2008; Heer et al. 2009; Laadan et al. 2008; Liu and Moon 2009; Liu et al. 2008; Petersson et al. 2006). Similar to S. cerevisiae, S. stipitis is able to convert furfural and HMF to FM and FDM, respectively, in the lag phase. Once furfural and HMF were reduced to a certain low concentration, cell growth recovered and ethanol production resumed (Bellido et al. 2011; Liu et al. 2004, 2005; Slininger et al. 2009). However, the enzymes involved in reduction of furfural and HMF in S. stipitis remain unknown. Now that alcohol dehydrogenases are most probably involved in the furfural and HMF detoxification, we selected all the seven SsADH (Ss for S. stipitis) genes as research targets (Table 1) based on previous reports (Cho and Jeffries 1998; Passoth et al. 1998) and recently annotated S. stipitis genome sequences (Jeffries et al. 2007). The objectives of this study were to (1) investigate the transcription response of the SsADH genes to furfural and HMF, (2) test their enzyme activities and properties for reduction of furfural and HMF in details, and (3) test their enzyme activities in brief for reduction of other six aldehydes—some of them are inhibitors generated during pretreatment of lignocellulosic biomass.
Materials and methods
Strains, media, and chemicals
The yeast strains of S. stipitis CBS 6054 and S. cerevisiae INVSc1 (his3Δ1/his3Δ1 leu2/leu2 trp1-289/trp1-289 ura3-52/ura3-52) were purchased from the China Center of Industrial Culture Collection (Beijing, China) and Invitrogen (Carlsbad, CA, USA), respectively. Escherichia coli DH5α (Sangon Biotech, Shanghai, China) was used for cloning and recombinant DNA manipulation. Yeast strains were cultivated in either YPD medium (1 % yeast extract, 2 % peptone, and 2 % dextrose) or synthetic complete (SC) medium [6.7 g/L of yeast nitrogen base without amino acids but with ammonium sulfate (Difco, Detroit, MI, USA); 20 mg/L of adenine, arginine, cysteine, leucine, lysine, threonine, and tryptophan; and 10 mg/L of aspartic acid, histidine, isoleucine, methionine, phenylalanine, proline, serine, tyrosine, and valine (Sigma-Aldrich, St. Louis, MO, USA)] with or without uracil according to requirements. Competent cells of E. coli were grown on LB medium (1 % tryptone, 0.5 % yeast extract, 1 % NaCl, pH 7.0) amended with 100 mg/L ampicillin for plasmid selection. For the preparation of solid plate, 2 % agar was added into the medium. All reagents for medium preparation were purchased from Sigma-Aldrich (St. Louis, MO, USA), Difco (Detroit, MI, USA), or Sangon Biotech (Shanghai, China). All chemicals including the aldehydes of furfural, HMF, formaldehyde, acetaldehyde, isovaleraldehyde, benzaldehyde, phenylacetaldehyde, and cinnamaldehyde were provided by Best-Reagent Company (Chengdu, China).
Cell growth and metabolic response to furfural and HMF
For cell growth, S. stipitis CBS 6054 was incubated in YPD medium at 30 °C overnight with agitation of 200 rpm. Culture inocula of S. stipitis were prepared using freshly grown cells harvested at logarithmic growth phase. Inoculum cells of S. stipitis were incubated in fresh YPD medium containing 30 mM of furfural at a final concentration under microaerobic conditions with agitation of 200 rpm at 30 °C for 72 h. The concentration of furfural used in this study was determined according to a brief pretest using 20, 30, and 40 mM of furfural for treatment. To observe the different inhibition degree on cell growth and metabolic responses, the concentration of HMF was equivalent to furfural. Cell cultures lacking inhibitors were used as an untreated control. Cell growth of S. stipitis was monitored over time by measuring absorbance at OD600 using a UV-2802 spectrophotometer (Unico, NJ, USA). Metabolic conversion profiles including glucose, furfural, HMF, ethanol, FM, and FDM for each sample were analyzed using a Waters high-performance liquid chromatography equipped with an Aminex HPX-87 H column (Bio-Rad Laboratories, Hercules, CA) and a refractive index detector as previously described (Ma et al. 2012). The experiments were carried out in triplicate under each condition.
Gene expression response to furfural and HMF
Culture inocula of S. stipitis CBS 6054 were prepared as mentioned above. Inoculum cells were inoculated into fresh YPD medium and incubated under microearobic conditions as described above and until the OD600 reached about 1.0. At that time, furfural or HMF was added into the medium at a final concentration of 30 mM. The time point at the addition of an inhibitor was designated as 0 h. Cell samples were harvested at 0 and 2 h. Cells lacking inhibitor treatment served as an untreated control. Primers for qRT-PCR assay were designed (Supplementary material Table S1) using Primer 3 (Rozen and Skaletsky 2000). The total RNA was isolated from each of two biological and two technical replications using a protocol as previously described (Liu and Slininger 2007) and purified using an RNAclean kit (Tiangen Biotech Co., LTD, Beijing, China). RNA integrity was verified by gel electrophoresis and concentration was determined by a NanoDrop 2000C Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). Reverse transcription reactions were carried out using the procedures described previously (Liu et al. 2009a). For each qRT-PCR reaction, a RealMasterMix (SYBR Green) kit (Tiangen Biotech Co., Ltd, Beijing, China) was utilized and the PCR reaction was run on a Mastercycler® ep realplex system (Eppendorf, Hamburg, Germany). Data were analyzed using a defined profile as previously described (Liu et al. 2009b).
Gene cloning
Standard molecular biology techniques were performed to transform yeast as previously described (Sambrook and Russell 2001). All of the seven SsADH genes (refer to Table 1 for accession numbers) were individually amplified from S. stipitis using corresponding primers (Supplementary material Table S1). PCR products were verified by sequencing analysis (Sangon Biotech, Shanghai, China). The shuttle vector pYES2/NT B (Invitrogen, Carlsbad, CA, USA), carrying the URA3 selection marker and the galactose-inducible promoter of GAL1, was used to clone the coding region of the target genes at NotI and XhoI (or XbaI) sites individually. The individual pYES2/NT B construct with the ORF insert was transformed into E. coli DH5α, and the constructed plasmids (Supplementary material Fig. S1) were isolated and verified using diagnostic PCR method. The confirmed pYES2/NT B construct containing an individual ORF insert was transformed into INVSc1 using the lithium acetate method (Gietz et al. 1995). Transformants were selected on SC medium lacking uracil supplemented with 2 % glucose at 30 °C and then verified using diagnostic PCR method.
Protein expression, purification, and characterization
Clones were recovered in SC medium lacking uracil supplemented with 2 % glucose for initial yeast growth at 30 °C with agitation of 200 rpm. After overnight incubation, the fresh cells were transferred to an induction medium supplemented with 2 % galactose and 1 % raffinose and incubated at 30 °C with agitation of 200 rpm for 24 h. The induction cells were harvested and lysed using Y-PER® Plus reagent (Pierce, Rockford, IL, USA) following the manufacturer's instructions. Cell pellets were resuspended in the lysis solution and incubated at 25 °C with continuous shaking for 20 min. The solution was then centrifuged at 20 °C at 18,000×g to pellet the cells. The supernatant was collected and kept on ice. The lysis procedure was carried out twice and the supernatants were combined. The induced protein was purified from the supernatants using a Ni-NTA SefinoseTM Kit (Sangon Biotech, Shanghai, China). Protein concentrations for all samples were evaluated using a Modified Bradford Protein Assay Kit (Sangon Biotech, Shanghai, China), and a standard curve was created using bovine serum albumin (Sangon Biotech, Shanghai, China). Molecular weight of the purified proteins was estimated using 12 % SDS-PAGE gel electrophoresis and stained by Coomassie Brilliant Blue G-250 dye (Sangon Biotech, Shanghai, China). The remaining purified proteins were stored at 4 °C and all samples were used for enzyme activity assays within 2 days after protein purification.
Enzyme activity assay
All enzyme activities were assayed using a UV-2802 spectrophotometer (Unico, NJ, USA). Enzyme activity was determined by measuring a decrease in absorbance at 340 nm using the cofactor NADH or NADPH (Liu et al. 2008). The protein samples were kept on ice until use. All assays were carried out in a total volume of 500 μL at 30 °C for regular test. The reaction mixtures consisted of a final concentration of 10 mM furfural or HMF substrate and 100 or 200 μM (depending on enzyme activity) of cofactor in 100 mM potassium phosphate buffer (pH 7.2). The NADH concentration was increased to 3,000 μM and the substrate concentration was increased to 50 mM when acetaldehyde was used for Adh1p enzyme activity. All reagents were maintained in water bath at 30 °C prior to use. Kinetic experiments were performed using substrates in a concentration range of 0.5 to 10 mM. Kinetic parameters were estimated using the Lineweaver–Burk transformation of the Michaelis–Menten equation. The optimum pH for the specific enzyme activity was determined following incubation at pH levels ranging from 4.5 to 9.0. To determine the optimum temperature, enzyme activity was measured at temperature from 20 to 60 °C. One enzyme unit was defined as 1 μmol of NAD(P)H oxidized per minute, and specific activity was defined as the units of enzyme activity per milligram of protein. All assays were performed in triplicate.
Sequence analysis
Using the ClustalW method in the MegAlign program 5.0 (DNAStar, Inc. Madison, WI, USA), amino acid sequences of the seven SsADH proteins (refer to Table 1) from S. stipitis were aligned with ScAdh6p (accession number NM_001182831), ScAdh7p (accession number NM_001178812), and mutated ScAdh1p (accession number NM_001183340) from S. cerevisiae which have been demonstrated to exhibit enzyme activities for furfural and/or HMF reduction (Almeida et al. 2008; Laadan et al. 2008; Liu et al. 2008; Petersson et al. 2006). Cofactor binding and catalytic sites and regions were annotated according to previous reports (Baker et al. 2009; Larroy et al. 2002a, b; McKie et al. 1993; Valencia et al. 2004). The phylogenic tree of the above-mentioned proteins was constructed using the neighbor-joining method in the MEGA 5.10 (Tamura et al. 2011).
Results
Cell growth, metabolic conversion, and gene expression in response to furfural and HMF
Cell growth of S. stipitis was rapidly inhibited by both furfural and HMF at final concentrations of 30 mM (Fig. 1a). Furfural was more toxic to cell growth than HMF. The inhibition was much stronger with furfural than HMF, displaying a lag phase of approximate 16 and 4 h, respectively, and then cell growth resumed. Final cell density under furfural condition was significantly lower than that under both HMF and the untreated control conditions, and the final cell densities under the latter two conditions were almost equivalent. Consistent with cell growth performance, glucose consumption and ethanol production were delayed under furfural and HMF stress conditions, and the inhibition degree was stronger by furfural than HMF, too (Fig. 1b). Furfural and HMF were reduced to FM and FDM, respectively, under microaerobic fermentation conditions in this study, and none of them were detected in the untreated control samples (Fig. 1c). Within the first 4 h of lag phase, conversion of furfural to FM was faster than that of HMF to FDM. After that, cell growth recovered in HMF-treated condition and then the conversion of HMF to FDM accelerated gradually.

Cell growth and metabolic response to furfural and HMF. a Comparison of cell growth of S. stipitis CBS 6054 by estimating cell density at OD600 in response to 30 mM furfural (filled triangle), 30 mM HMF (filled square), and without inhibitors as control (filled circle). b Glucose consumption (filled symbol) and ethanol production (open symbol) in response to 30 mM furfural (triangle), 30 mM HMF (square), and without inhibitors as control (circle). c Concentrations of furfural (filled diamond) and HMF (filled triangle) along with their respective conversion products FM (open diamond) and FDM (open triangle). Mean values are presented with vertical error bars representing the standard deviations (n = 3)
Transcription analyses indicated that, under untreated control condition, SsADH1 was the highest expressed gene among the seven SsADH genes; transcription level of SsADH5 was about 85 % lower than that of SsADH1 and the levels of the remaining genes were very low (Fig. 2a, b). Under inhibitor stress conditions, SsADH4 and SsADH6 were significantly induced at 2 h time point after furfural or HMF was added into the medium. Transcription levels of SsADH4 and SsADH6 displayed more than 100- and 24-fold increase in response to furfural and 150- and 50-fold increase in response to HMF, respectively. The induced transcription level of SsADH4 by both furfural and HMF was higher than that of SsADH6 at 2 h time point. Under furfural stress condition, transcription levels of the remaining SsADH genes showed no significant difference with that of the control at 0 and 2 h time points, except for SsADH1 displaying higher transcription level at 2 h time point compared to the control (Fig. 2a). Similarly, transcription levels of SsADH2, SsADH5, and SsADH7 showed no significant difference with that of the control under HMF stress condition, but transcription of SsADH1 and SsADH3 was up-regulated at 2 h time point (Fig. 2b). Transcription level of SsADH3 was relatively low, despite its transcription was up-regulated at 2 h time point under HMF stress condition.

Transcription levels of ADH genes in response to furfural and HMF. Comparison of transcription levels in transcript numbers of all SsADH genes in S. stipitis CBS 6054 in response to 30 mM furfural (a) and 30 mM HMF (b) 2 h after treatment (dark gray bars) compared with 0 h (blank bars) and 2 h (light gray bars) without inhibitor treatment as control. Mean values are presented with vertical error bars representing the standard deviations (n = 4)
Gene cloning and protein expression
The amplified fragment of the SsADH gene from S. stipitis CBS 6054 was inserted into designed restriction sites of the pYES2/NT B plasmid and the recombinant plasmids for overexpression of the seven SsADH genes under control of the galactose-inducible GAL1 promoter were constructed (Supplementary material Fig. S1). Agarose gel electrophoresis analysis of the newly constructed plasmids demonstrated that all the SsADH genes were successfully inserted into the pYES2/NT B plasmid (Supplementary material Fig. S2), and sequencing analysis confirmed that the inserted fragment was the corresponding SsADH gene. The newly constructed plasmids were transformed into S. cerevisiae strain INVSc1. The recombinant SsADH proteins were expressed in galactose induction medium and purified using the His-tag column chromatography for enzyme-specific analysis. Using the Protein Molecular Weight software (http://www.bioinformatics.org/sms/prot_mw.html), molecular weight of the SsADH proteins, His-tag amino acid sequence, and the corresponding fusion proteins were estimated (Table 2). SDS-PAGE analysis of the purified recombinant proteins indicated that the molecular weights of the expressed proteins ranged from 40 to 45 kDa, consistent with the estimated value of recombinant proteins (Supplementary material Fig. S3). Protein analysis indicated that all the SsADH genes were successfully expressed in S. cerevisiae.
Enzyme activity and kinetic parameters for furfural and HMF reduction
For furfural reduction, SsAdh4p, SsAdh5p, SsAdh6p, and SsAdh7p showed both NADH and NADPH-dependent activities, and SsAdh4p and SsAdh5p had relatively stronger reduction activities with cofactor NADPH than NADH (Table 3). SsAdh1p showed the highest NADH-dependent specific activity in furfural reduction, but no significant activity was observed with NADPH as cofactor (Table 3). For HMF reduction, NADPH-dependent activities were observed in SsAdh4p, SsAdh5p, SsAdh6p, and SsAdh7p, but no significant NADH-dependent activity was observed in all SsADH proteins. SsAdh4p displayed the highest NADPH-dependent specific activity for both furfural and HMF reduction.
Kinetic parameters of the SsADH proteins for furfural and HMF reduction were presented in Table 4. For furfural reduction, SsAdh1p and SsAdh7p showed relatively lower K m values (3.15 ± 0.21 and 1.94 ± 0.12, respectively) and higher catalytic efficiency (K cat/K m, 50.23 ± 3.42 and 51.09 ± 3.54, respectively) when NADH was used as the cofactor. However, SsAdh5p and SsAdh6p showed relatively higher V max (8.07 ± 0.54 and 9.29 ± 0.42, respectively) and catalytic activity (K cat, 323.00 ± 20.13 and 374.26 ± 15.51, respectively). When NADPH was used as a cofactor, SsAdh4p showed the lowest K m value (0.24 ± 0.02) and the highest values of V max (7.07 ± 0.55), K cat (285.22 ± 10.11), and K cat/K m (1,193.20 ± 98.16). Catalytic efficiency of SsAdh4p was higher than that of SsAdh5p and SsAdh6p by more than 2.4- and 35-fold, respectively. Catalytic efficiencies of SsAdh4p and SsAdh5p were significantly higher when NADPH was used as cofactor than that of all the five SsADH proteins when NADH was used as cofactor. For HMF reduction, SsAdh4p showed much lower K m value (0.26 ± 0.03) and much higher values of V max (9.66 ± 1.02), K cat (389.66 ± 22.15), and K cat/K m (1,516.17 ± 120.23) than that of SsAdh5p when NADPH was used as cofactor. Especially, catalytic efficiency of SsAdh4p was higher than that of SsAdh5p by more than 5-fold.
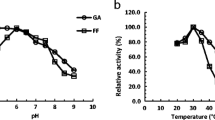
Effect of pH and temperature on enzyme activity for furfural and HMF reduction
Optimum enzyme activities for furfural and HMF were determined within the pH range of 4.5 to 9.0 (Fig. 3) and the temperature range of 20 to 60 °C (Fig. 4). The optimum enzyme activities were observed at pH 6.0 for most SsADH proteins with either NADH or NADPH as cofactor. Enzyme activities of all SsADH proteins were more stable in acidic condition than in alkaline condition. The optimal temperatures were 30 °C for most SsADH proteins with either NADH or NADPH as cofactor. More than 50 % enzyme activities remained at 60 °C for most SsADH proteins with either NADH or NADPH as cofactor, especially for NADPH-dependent furfural and HMF reduction activities in SsAdh5p.

Effect of pH on enzyme activities of SsADH proteins. Effect of pH on enzyme activities of SsAdh1p (a), SsAdh4p (b), SsAdh5p (c), and SsAdh6p (d) for reduction of furfural with NADH (filled diamond) or NADPH (filled squared) as cofactor as well as reduction of HMF with NADPH as cofactor (filled triangle) were measured in potassium phosphate buffer. Mean values are presented with vertical error bars of standard deviations (n = 3)

Effect of temperature on enzyme activities of SsADH proteins. Effect of temperature on enzyme activities of SsAdh1p (a), SsAdh4p (b), SsAdh5p (c), and SsAdh6p (d) for reduction of furfural with NADH (filled diamond) or NADPH (filled squared) as cofactor as well as reduction of HMF with NADPH as cofactor (filled triangle) were measured in potassium phosphate buffer. Mean values are presented with vertical error bars representing standard deviations (n = 3)
Enzyme activity for other aldehydes
In this study, enzyme activities for reduction of other six aldehydes were detected, including formaldehyde, acetaldehyde, isovaleraldehyde, benzaldehyde, phenylacetaldehyde, and cinnamaldehyde (Table 5), and some of them are inhibitors commonly detected in lignocellulosic hydrolysates (Liu 2011). SsAdh4p, SsAdh5p and SsAdh7p showed both NADH- and NADPH-dependent activities for reduction of formaldehyde and acetaldehyde. Formaldehyde reduction activity was also observed for SsAdh1p with NADH as cofactor and for SsAdh3p with NADPH as cofactor. NADH-dependent activity for acetaldehyde reduction was also observed for SsAdh1p and SsAdh2p. As for reduction of acetaldehyde, the highest activity was observed for SsAdh1p with NADH as cofactor, much higher than any other SsADH protein enzyme with either NADH or NADPH as cofactor in this study. For isovaleraldehyde reduction, NADH-dependent activity was observed for SsAdh3p, and NADPH-dependent activity was observed for both SsAdh4p and SsAdh5p. SsAdh3p and SsAdh7p showed NADH-dependent activities for benzaldehyde reduction. SsAdh5p showed both NADH- and NADPH-dependent activity for phenylacetaldehyde reduction; SsAdh3p possessed only NADH-dependent activity, and SsAdh4p and SsAdh7p exhibited only NADPH-dependent activity for the reduction of phenylacetaldehyde.
Sequence analysis
All of the seven SsADH proteins were identified as zinc-dependent medium-chain dehydrogenase/reductase (MDR), which consists of approximately 350 residues of amino acids with one catalytic and one cofactor binding domains (Fig. 5). The typical sequence motif known as the zinc-containing ADH signature (GHEX2GX5(G,A)X2(I,V,A,C,S)) (segment 66–80 in SsAdh1p) for catalytic reaction was found in all the SsADH proteins, and the catalytic and structural zinc binding sites were marked with a triangle. The boxed region (GxxGxxG) (position 178 to 184 in SsAdh1p) is involved in cofactor binding, and the conserved glycine residues allow the cofactor to close to the main chain (Bottoms et al. 2002). The Asp residue (202 in SsAdh1p and SsAdh2p) marked with a star in Fig. 5 determines the specificity for NADH, which is bonded to oxygen atoms of the adenine ribose moiety of NADH (Fan et al. 1991). The Ser residue in the same alignment position determines the specificity for NADPH, which participates in the binding of the terminal phosphate group of NADPH (Valencia et al. 2004). Phylogenetic relationships derived from the deduced amino acid sequences of SsADH proteins from S. stipitis indicate that SsAdh4p, SsAdh6p, SsAdh3p, and SsAdh5p are more closely related, and that they are distinct to other SsADH proteins (Fig. 6). SsAdh7p is closely related to ScAdh6p and ScAdh7p from S. cerevisiae. SsAdh1p and SsAdh2p are more closely related to the mutated ScAdh1p from S. cerevisiae and are distinct to the other SsADH proteins in this study.

Alignment of amino acid sequences of ADH proteins. Residues in dark shade are identical or similar. The amino acids involved in the binding of the “catalytic zinc” are marked with solid inverted triangle, whereas the cysteine residues involved in the binding of the “structural zinc” are marked with open inverted triangle. Cofactor binding regions are boxed by bold black lines and a single binding site is marked with a star. Ss S. stipitis and Sc S. cerevisiae

Phylogenetic tree of ADH proteins. Ss S. stipitis and Sc S. cerevisiae
Discussion
Previous studies in S. cerevisiae demonstrated that alcohol dehydrogenases of ScAdh6p, ScAdh7p, and mutated ScAdh1p have NAD(P)H-dependent activities for furfural and/or HMF reduction (Almeida et al. 2008; Heer et al. 2009; Laadan et al. 2008; Liu and Moon 2009; Liu et al. 2008; Petersson et al. 2006). S. stipitis is also able to reduce furfural to FM and HMF to FDM, respectively. However, whether the alcohol dehydrogenases in S. stipitis have those kinds of reductive activities is unclear. To address the questions, we selected all the SsADH genes (either identified or putative) as targets and then carried out transcription analysis under furfural and HMF stress conditions. Furthermore, the SsADH genes were cloned and enzyme activities as well as kinetic parameters of the purified SsADH proteins were determined for furfural and HMF reduction in details. Finally, enzyme activities of the SsADH proteins for the reduction of other six aldehydes were studied in brief. To the best of our knowledge, this is the first report of SsADH genes involved in the detoxification of aldehydes derived from lignocellulosic hydrolysis in S. stipitis. Knowledge obtained in this study will aid to develop more inhibitor-tolerant strains of S. stipitis by genetic engineering.
Transcription analyses in this study demonstrated that SsADH4 and SsADH6 were highly induced by both furfural and HMF under microaerobic fermentation conditions, which suggests furfural and HMF may trigger a similar regulation mechanism for transcription response of SsADH4 and SsADH6 in S. stipitis. In comparison, ScADH6 and ScADH7 were significantly induced by furfural and HMF in S. cerevisiae; however, ScADH4 was not significantly induced under the same stress conditions (Liu et al. 2009a). Induced expression of ScADH6, ScADH7, as well as other furfural and HMF reductase genes was considered to be mainly co-regulated by oxidative stress-related transcription factors of Yap1p, Yap5p, and Yap6p under furfural and HMF stress conditions in S. cerevisiae (Ma and Liu 2010 and unpublished data), and the yeast activator protein (YAP) binding motifs (cis-acting elements) were found in the upstream sequences of the regulated genes (Ma and Liu 2010). Alriksson et al. (2010) demonstrated that the overexpression of the YAP1 gene increased resistance to coniferyl aldehyde, HMF, and spruce hydrolysate in S. cerevisiae. However, transcription factors and cis-acting elements involved in the regulation of SsADH4 and SsADH6 expression in S. stipitis remained unknown. CAP1 is a putative transcription factor gene involved in oxidative stress response in S. stipitis (Jeffries et al. 2007), which may have a similar function to YAP1 gene in S. cerevisiae (Jeffries and Van Vleet 2009). Whether the induced expression of SsADH4 and SsADH6 in S. stipitis is regulated by Cap1p deserves further investigation, and the molecular mechanism of transcription regulation including interaction of cis-acting element and trans-acting factor should be further studied in details.
Metabolic analyses indicated that furfural and HMF were reduced to less toxic FM and FDM under microaerobic condition in vivo by S. stipitis. Enzyme activity tests indicated that reduction of furfural and HMF were catalyzed by multiple SsADH proteins in vitro. SsAdh4p exhibited the highest catalytic efficiency in vitro for both furfural and HMF reduction among all the SsADH proteins. Considering its highly induced transcription by both furfural and HMF, we have reason to believe that SsADH4 plays more important roles in adaptation response to furfural and HMF stress than the remaining SsADH genes in S. stipitis. Enzyme assays with other aldehydes in vitro indicated that reduction activity of alcohol dehydrogenase in S. stipitis is not specific to a certain aldehyde but to a broad range of aliphatic and aromatic aldehydes. Similarly, activities of aldehyde reductase for reduction of multiple aldehydes were observed in S. cerevisiae (Liu and Moon 2009). In this study, we also found that some alcohol dehydrogenases (for example SsAdh2p and SsAdh3p) may lack activity for furfural and/or HMF reduction, but can have activities for reduction of other aldehydes. These findings indicate that alcohol dehydrogenases in S. stipitis are involved in the detoxification of multiple aldehyde inhibitors derived from lignocellulosic biomass conversion. It should be pointed out that at least a dozen of aldehyde inhibitors are generated during the hydrolysis of lignocellulosic biomass (see review by Liu 2011) and some of them (such as coniferyl aldehyde, vanillin, glycolaldehyde, and methylglyoxal) were not tested in this study. Whether these kinds of aldehyde inhibitors derived from lignocellulosic hydrolysis can be reduced by alcohol dehydrogenases needs to be further studied.
NADH and/or NADPH can be used as cofactors for reduction of furfural and HMF by aldehyde reductases in S. cerevisiae (Almeida et al. 2008; Laadan et al. 2008; Liu et al. 2008; Petersson et al. 2006). In this study, we found that both NADH and NADPH were used as cofactors for reduction of furfural by multiple SsADH proteins in S. stipitis, and NADPH was used as cofactor for reduction of HMF by SsAdh4p and SsAdh5p in S. stipitis. Reduction of furfural and HMF will cause a deficit of cofactors, and accelerated regeneration of NAD(P)H can compensate this negative effect in S. cerevisiae (Gorsich et al. 2006; Liu et al. 2009a ). More SsADH proteins with good activities as well as more available cofactors (both NADH and NADPH) involved in the reduction of furfural could make S. stipitis detoxify furfural faster than HMF, which is consistent with metabolic conversion results observed in the same lag phase of 4 h after inhibitor addition as found in this study and previous reports (Liu et al. 2004, 2005).
Analysis of amino acid sequences of all SsADH proteins from S. stipitis and ScAdh6p, ScAdh7p, and the mutated ScAdh1p from S. cerevisiae found that the catalytic and structural zinc sites align together, which is consistent with other MDR proteins (Baker et al. 2009; Larroy et al. 2002a, b; McKie et al. 1993; Valencia et al. 2004). The cofactor binding region and single binding sites (Fig. 5) determine the preference for cofactors. SsAdh3p, SsAdh4p, SsAdh5p, and SsAdh6p have a more similar cofactor binding region (GxxGxxG), and all of them showed a preference for NADPH over NADH as evidenced by enzyme activity tests in this study. SsAdh1p contains a single amino acid difference with the mutated ScAdh1p in this cofactor binding region, and the sequence of amino acids of SsAdh2p in this region is identical to those in the mutated ScAdh1p. Moreover, the Asp that positions at the first star site in the amino acid sequences of SsAdh1p, SsAdh2p, and mutated ScAdh1p (Fig. 5) is considered to determine the specificity for NADH (Fan et al. 1991), which is consistent with the observed enzyme activity with NADH as cofactor in S. stipitis in this study and in a previous report in S. cerevisiae (Laadan et al. 2008). Phylogenetic analysis showed that the NADH-dependent SsADH proteins are more closely related, but are distinct to the NAD(P)H-dependent SsADH proteins (Fig. 6).
Reduction of furfural and HMF in fact is a kind of oxido-reductive reaction. Oxidoreductases represent a large family comprised of thousands of enzymes. This large family can be further grouped into three superfamilies, including long-chain dehydrogenase/reductase (approximately 385 residues but in few cases 900 residues), MDR (about 350 residues), and short-chain dehydrogenase/reductase (250–350 residues) superfamilies. Previous studies on S. cerevisiae showed that reduction of furfural and HMF is catalyzed not only by alcohol dehydrogenases, but also by multiple reductases, such as Ari1p, Ald4p, Ald6p, Gre2p, and Gre3p (Liu et al. 2008; Moon and Liu 2012; Park et al. 2011). According to these findings in the model yeast of S. cerevisiae, identification of alcohol dehydrogenases from S. stipitis for furfural and HMF reduction is just a beginning. More oxidoreductases involved in the detoxification of aldehyde inhibitors in S. stipitis should be identified in the future.
References
Almeida JR, Röder A, Modig T, Laadan B, Lidén G, Gorwa-Grauslund MF (2008) NADH- vs NADPH-coupled reduction of 5-hydroxymethyl furfural (HMF) and its implications on product distribution in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 78:939–945
Alriksson B, Horváth IS, Jönsson LJ (2010) Overexpression of Saccharomyces cerevisiae transcription factor and multidrug resistance genes conveys enhanced resistance to lignocellulose derived fermentation inhibitors. Process Biochem 45:264–271
Antal MJ, Leesomboon T, Mok WS, Richards GN (1991) Mechanism of formation of 2-furaldehyde from D-xylose. Carbohyd Res 217:71–85
Antoni D, Zverlov VV, Schwarz WH (2007) Biofuels from microbes. Appl Microbiol Biotechnol 77:23–35
Baker PJ, Britton KL, Fisher M, Esclapez J, Pire C, Bonete MJ, Ferrer J, Rice DW (2009) Active site dynamics in the zinc-dependent medium chain alcohol dehydrogenase superfamily. Proc Natl Acad Sci U S A 106:779–784
Bellido C, Bolado S, Coca M, Lucas S, González-Benito G, García-Cubero MT (2011) Effect of inhibitors formed during wheat straw pretreatment on ethanol fermentation by Pichia stipitis. Bioresour Technol 102:10868–10874
Bottoms CA, Smith PE, Tanner JJ (2002) A structurally conserved water molecule in Rossmann dinucleotide-binding domains. Protein Sci 11:2125–2137
Cho JY, Jeffries TW (1998) Pichia stipitis genes for alcohol dehydrogenase with fermentative and respiratory functions. Appl Environ Microbiol 64:1350–1358
Cho DH, Shin SJ, Bae Y, Park C, Kim YH (2010) Enhanced ethanol production from deacetylated yellow poplar acid hydrolysate by Pichia stipitis. Bioresour Technol 101:4947–4951
du Preez JC, Prior BA (1985) A quantitative screening of some xylose fermenting yeast isolates. Biotechnol Lett 7:241–248
Fan F, Lorenzen JA, Plapp BV (1991) An aspartate residue in yeast alcohol dehydrogenase I determines the specificity for coenzyme. Biochemistry 30:6397–6401
Gietz RD, Schiestl RH, Willems AR, Woods RA (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11:355–360
Gorsich SW, Dien BS, Nichols NN, Slininger PJ, Liu ZL, Skory CD (2006) Tolerance to furfural-induced stress is associated with pentose phosphate pathway genes ZWF1, GND1, RPE1, and TKL1 in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 71:339–349
Hahn-Hägerdal B, Karhumaa K, Fonseca C, Spencer-Martins I, Gorwa-Grauslund MF (2007) Towards industrial pentose-fermenting yeast strains. Appl Microbiol Biotechnol 74:937–953
Heer D, Sauer U (2008) Identification of furfural as a key toxin in lignocellulosic hydrolysates and evolution of a tolerant yeast strain. Microb Biotechnol 1:497–506
Heer D, Heine D, Sauer U (2009) Resistance of Saccharomyces cerevisiae to high concentrations of furfural is based on NADPH-dependent reduction by at least two oxireductases. Appl Environ Microbiol 75:7631–7638
Jeffries TW, Van Vleet JR (2009) Pichia stipitis genomics, transcriptomics, and gene clusters. FEMS Yeast Res 9:793–807
Jeffries TW, Grigoriev IV, Grimwood J, Laplaza JM, Aerts A, Salamov A, Schmutz J, Lindquist E, Dehal P, Shapiro H, Jin YS, Passoth V, Richardson PM (2007) Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat Biotechnol 25:319–326
Jönsson LJ, Alriksson B, Nilvebrant NO (2013) Bioconversion of lignocellulose: inhibitors and detoxification. Biotechnol Biofuels 6:16
Kim SR, Park YC, Jin YS, Seo JH (2013) Strain engineering of Saccharomyces cerevisiae for enhanced xylose metabolism. Biotechnol Adv. doi:10.1016/j.biotechadv.2013.03.004
Klinke HB, Thomsen AB, Ahring BK (2004) Inhibition of ethanol-producing yeast and bacteria by degradation products produced during pre-treatment of biomass. Appl Microbiol Biotechnol 66:10–16
Laadan B, Almeida JR, Rådström P, Hahn-Hägerdal B, Gorwa-Grauslund M (2008) Identification of an NADH-dependent 5-hydroxymethylfurfural-reducing alcohol dehydrogenase in Saccharomyces cerevisiae. Yeast 25:191–198
Larroy C, Fernández MR, González E, Parés X, Biosca JA (2002a) Characterization of the Saccharomyces cerevisiae YMR318C (ADH6) gene product as a broad specificity NADPH-dependent alcohol dehydrogenase: relevance in aldehyde reduction. Biochem J 361:163–172
Larroy C, Parés X, Biosca JA (2002b) Characterization of a Saccharomyces cerevisiae NADP(H)-dependent alcohol dehydrogenase (ADHVII), a member of the cinnamyl alcohol dehydrogenase family. Eur J Biochem 269:5738–5745
Larsson S, Palmqvist E, Hahn-Hägerdal B, Tengborg C, Stenberg K, Zacchi G, Nilvebrant N (1999) The generation of inhibitors during dilute acid hydrolysis of softwood. Enzyme Microb Technol 24:151–159
Lewkowski J (2001) Synthesis, chemistry and applications of 5-hydroxymethylfurfural and its derivatives. ARKIVOC 1:17–54
Li Y, Park JY, Shiroma R, Tokuyasu K (2011) Bioethanol production from rice straw by a sequential use of Saccharomyces cerevisiae and Pichia stipitis with heat inactivation of Saccharomyces cerevisiae cells prior to xylose fermentation. J Biosci Bioeng 111:682–686
Lin Y, Tanaka S (2006) Ethanol fermentation from biomass resources: current state and prospects. Appl Microbiol Biotechnol 69:627–642
Lin TH, Huang CF, Guo GL, Hwang WS, Huang SL (2012) Pilot-scale ethanol production from rice straw hydrolysates using xylose-fermenting Pichia stipitis. Bioresour Technol 116:314–319
Liu ZL (2011) Molecular mechanisms of yeast tolerance and in situ detoxification of lignocellulose hydrolysates. Appl Microbiol Biotechnol 90:809–825
Liu ZL, Blaschek HP (2010) Biomass conversion inhibitors and in situ detoxification. In: Vertès AA, Qureshi N, Blaschek HP, Yukawa H (eds) Biomass to biofuels: strategies for global industries. Wiley, Chichester, pp 233–259
Liu ZL, Moon J (2009) A novel NADPH-dependent aldehyde reductase gene from Saccharomyces cerevisiae NRRL Y-12632 involved in the detoxification of aldehyde inhibitors derived from lignocellulosic biomass conversion. Gene 446:1–10
Liu ZL, Slininger PJ (2007) Universal external RNA controls for microbial gene expression analysis using microarray and qRT-PCR. J Microbiol Methods 68:486–496
Liu ZL, Slininger PJ, Dien BS, Berhow MA, Kurtzman CP, Gorsich SW (2004) Adaptive response of yeasts to furfural and 5-hydroxymethylfurfural and new chemical evidence for HMF conversion to 2,5-bis-hydroxymethylfuran. J Ind Microbiol Biotechnol 31:345–352
Liu ZL, Slininger PJ, Gorsich SW (2005) Enhanced biotransformation of furfural and 5-hydroxy methylfurfural by newly developed ethanologenic yeast strains. Appl Biochem Biotechnol 121–124:451–460
Liu ZL, Moon J, Andersh BJ, Slininger PJ, Weber S (2008) Multiple gene-mediated NAD(P)H-dependent aldehyde reduction is a mechanism of in situ detoxification of furfural and 5-hydroxymethylfurfural by Saccharomyces cerevisiae. Appl Microbiol Biotechnol 81:743–753
Liu ZL, Ma M, Song M (2009a) Evolutionarily engineered ethanologenic yeast detoxifies lignocellulosic biomass conversion inhibitors by reprogrammed pathways. Mol Genet Genomics 282:233–244
Liu ZL, Palmquist DE, Ma M, Liu J, Alexander NJ (2009b) Application of a master equation for quantitative mRNA analysis using qRT-PCR. J Biotechnol 143:10–16
Ma M, Liu ZL (2010) Comparative transcriptome profiling analyses during the lag phase uncover YAP1, PDR1, PDR3, RPN4, and HSF1 as key regulatory genes in genomic adaptation to the lignocellulose derived inhibitor HMF for Saccharomyces cerevisiae. BMC Genomics 11:660
Ma M, Liu ZL, Moon J (2012) Genetic engineering of inhibitor-tolerant Saccharomyces cerevisiae for improved xylose utilization in ethanol production. Bioenerg Res 5:459–469
McKie JH, Jaouhari R, Douglas KT, Goffner D, Feuillet C, Grima-Pettenati J, Boudet AM, Baltas M, Gorrichon (1993) A molecular model for cinnamyl alcohol dehydrogenase, a plant aromatic alcohol dehydrogenase involved in lignification. Biochim Biophys Acta 1202:61–69
Moon J, Liu ZL (2012) Engineered NADH-dependent GRE2 from Saccharomyces cerevisiae by directed enzyme evolution enhances HMF reduction using additional cofactor NADPH. Enzyme Microb Technol 50:115–120
Parekh S, Wayman M (1986) Fermentation of cellobiose and wood sugars to ethanol by Candida shehatae and Pichia stipitis. Biotechnol Lett 8:597–600
Park SE, Koo HM, Park YK, Park SM, Park JC, Lee OK, Park YC, Seo JH (2011) Expression of aldehyde dehydrogenase 6 reduces inhibitory effect of furan derivatives on cell growth and ethanol production in Saccharomyces cerevisiae. Bioresour Technol 102:6033–6038
Passoth V, Schäfer B, Liebel B, Weierstall T, Klinner U (1998) Molecular cloning of alcohol dehydrogenase genes of the yeast Pichia stipitis and identification of the fermentative ADH. Yeast 14:1311–1325
Petersson A, Almeida JR, Modig T, Karhumma K, Hahn-Hägerdal B, Gorwa-Grauslund MF (2006) A 5-hydroxymethylfurfural reducing enzyme encoded by the Saccharomyces cerevisiae ADH6 gene conveys HMF tolerance. Yeast 23:455–464
Rozen S, Skaletsky H (2000) Bioinformatics methods and protocols. In: Krawetz S, Misener S (eds) Methods in molecular biology. Humana, Totowa, pp 365–386
Rudolf A, Baudel H, Zacchi G, Hahn-Hägerdal B, Lidén G (2008) Simultaneous saccharification and fermentation of steam-pretreated bagasse using Saccharomyces cerevisiae TMB3400 and Pichia stipitis CBS6054. Biotechnol Bioeng 99:783–790
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sánchez OJ, Cardona CA (2008) Trends in biotechnological production of fuel ethanol from different feedstocks. Bioresour Technol 99:5270–5295
Slininger PJ, Dien BS, Gorsich SW, Liu ZL (2006) Nitrogen source and mineral optimization enhance D-xylose conversion to ethanol by the yeast Pichia stipitis NRRL Y-7124. Appl Microbiol Biotechnol 72:1285–1296
Slininger PJ, Gorsich SW, Liu ZL (2009) Culture nutrition and physiology impact the inhibitor tolerance of the yeast Pichia stipitis NRRL Y-7124. Biotechnol Bioeng 102:778–790
Taherzadeh MJ, Eklund R, Gustafsson L, Niklasson C, Lidén G (1997) Characterization and fermentation of dilute-acid hydrolyzates from wood. Ind Eng Chem Res 36:4659–4665
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Valencia E, Larroy C, Ochoa WF, Parés X, Fita I, Biosca JA (2004) Apo and Holo structures of an NADPH-dependent cinnamyl alcohol dehydrogenase from Saccharomyces cerevisiae. J Mol Biol 341:1049–1062
Vertes A, Qureshi N, Yukawa H, Blaschek H (2010) Biomass to biofuels. Wiley, Chichester
Weber C, Farwick A, Benisch F, Brat D, Dietz H, Subtil T, Boles E (2010) Trends and challenges in the microbial production of lignocellulosic bioalcohol fuels. Appl Microbiol Biotechnol 87:1303–1315
Yadav KS, Naseeruddin S, Prashanthi GS, Sateesh L, Rao LV (2011) Bioethanol fermentation of concentrated rice straw hydrolysate using co-culture of Saccharomyces cerevisiae and Pichia stipitis. Bioresour Technol 102:6473–6478
Acknowledgments
This work was supported in part by the Scientific Research Fund of Sichuan Provincial Education Department (grant no. 13ZB0286) and the Talent Introduction Fund of Sichuan Agricultural University (grant no. 01426100).
Competing interests
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Menggen Ma and Xu Wang contributed equally to this paper.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 7,860 kb)
Rights and permissions
About this article
Cite this article
Ma, M., Wang, X., Zhang, X. et al. Alcohol dehydrogenases from Scheffersomyces stipitis involved in the detoxification of aldehyde inhibitors derived from lignocellulosic biomass conversion. Appl Microbiol Biotechnol 97, 8411–8425 (2013). https://doi.org/10.1007/s00253-013-5110-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5110-8