
Abstract
Leuconostoc carnosum 4010 is a protective culture for meat products. It kills the foodborne pathogen Listeria monocytogenes by producing two class IIa (pediocin-like) bacteriocins, leucocin A and leucocin C. The genes for leucocin A production have previously been characterised from Leuconostoc gelidum UAL 187, whereas no genetic studies about leucocin C has been published. Here, we characterised the genes for the production of leucocins A and C in L. carnosum 4010. In this strain, leucocin A and leucocin C operons were localised in different plasmids. Unlike in L. gelidum, leucocin A operon in L. carnosum 4010 only contained the structural and the immunity genes lcaAB without transporter genes lcaECD. On the contrary, leucocin C cluster included two intact operons. Novel genes lecCI encode the leucocin C precursor and the 97-aa immunity protein LecI, respectively. LecI shares 48 % homology with the immunity proteins of sakacin P and listeriocin. Another leucocin C operon lecXTS, encoding an ABC transporter and an accessory protein, was 97 % identical with the leucocin A transporter operon lcaECD of L. gelidum. For heterologous expression of leucocin C in Lactococcus lactis, the mature part of the lecC gene was fused with the signal sequence of usp45 in the secretion vector pLEB690. L. lactis secreted leucocin C efficiently, as shown by large halos on lawns of L. monocytogenes and Leuconostoc mesenteroides indicators. The function of LecI was then demonstrated by expressing the gene lecI in L. monocytogenes. LecI-producing Listeria was less sensitive to leucocin C than the vector strain, thus corroborating the immunity function of LecI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bacteriocins are ribosomally synthesised anti-microbial peptides produced by bacteria. Because of their activity against foodborne pathogens and food spoilage bacteria, bacteriocins and bacteriocin-producing cultures have been noticed as potential natural preservatives (Mills et al. 2011). Many lactic acid bacteria (LAB), for example, Lactococcus, Lactobacillus and Leuconostoc, have been reported to secrete bacteriocins. LAB bacteriocins are generally divided into two main groups, namely, class I (modified bacteriocins, mainly lantibiotics) and class II (unmodified bacteriocins) (Rea et al. 2011). Class II bacteriocins can further be divided into four subgroups, of which class IIa (pediocin-like) is comprised of bacteriocins sharing a highly conserved N-terminal sequence YGNGV/L and characterised by their high activity against the food pathogen Listeria monocytogenes (Nissen-Meyer et al. 2009).
Class II bacteriocins do not undergo post-translational modifications and are thereby structurally simple. Generally, only genes encoding the bacteriocin, immunity protein and transporters are required for the production of class II bacteriocins. Most bacteriocins are synthesised as a precursor carrying an N-terminal signal peptide, which is cleaved by the transporter during the exportation of bacteriocin. The most common type of Class II bacteriocin signal peptide is the double-glycine leader (Nes et al. 2007). Class IIa bacteriocins kill targeted cells by binding to mannose phosphotransferase permease, leading to a bacteriocin-permease pore, and thus disrupting the proton motive force of the cells and depleting the ATP pool. The self-protection system of class IIa bacteriocins requires an intracellular immunity protein, which binds to the bacteriocin-permease complex blocking the pore (Kjos et al. 2011). Synthesis of some class IIa bacteriocins is under the control of a three-component regulatory system, which include a sensor (histidine protein kinase), a response regulator and an induction factor (peptide pheromone) (Drider et al. 2006).
Many Leuconostoc strains produce one or more bacteriocins (Papathanasopoulos et al. 1997; Vaughan et al. 2001). Leuconostoc carnosum 4010 is a commercial bioprotective strain for vacuum packed meats (Budde et al. 2003). It produces two different class IIa bacteriocins, leucocin A and leucocin C. The genes for leucocin A have previously been characterised from L. carnosum Ta11a and Leuconostoc gelidum UAL 187 (Felix et al. 1994; van Belkum and Stiles 1995). The leucocin A gene cluster in L. gelidum UAL 187 is composed of the structural and the immunity genes lcaAB and the ABC transporter genes lcaECD. In contrast, the knowledge of leucocin C is fairly limited. Even though the amino acid sequence of the secreted 43-aa leucocin C peptide and its activity against Listeria have been published (Fimland et al. 2002b; Papathanasopoulos et al. 1997), there are no reports about genetics of leucocin C. A study about cross-immunity of class IIa bacteriocins showed that leucocin A immunity protein does not protect the cell against leucocin C, indicating that the two leucocins are independent bacteriocins and should have their own production-immunity systems (Fimland et al. 2002a). However, none of the genes typically needed for class IIa bacteriocin production, i.e., dedicated translocators for secretion, an immunity protein or a regulatory system, has been found for leucocin C production.
In this study, our aims were to localise the gene cluster for leucocin C production in L. carnosum 4010 genome, to characterise the genes and to demonstrate their explicit functions. This study also aimed to produce leucocin C in a heterologous host Lactococcus lactis for development of new strains with improved anti-listerial activity.
Materials and methods
Bacterial strains, plasmids and culture conditions
Strains and plasmids used in this study are listed in Table 1. L. carnosum 4010, Leuconostoc mesenteroides ATCC 8293 and L. lactis strains were grown in M17 (Oxoid Ltd. Basingstoke, UK) supplemented with 0.5 % (w/v) glucose (M17G) at 30 °C. M17G was supplemented with 1 μg nisin/ml for L. lactis transformant selection and 0.5 μg nisin/ml for plasmid maintenance and nisin induction. E. coli was grown in Luria-Bertani (10 g/l Bacto™ Tryptone, 5 g/l Bacto™ Yeast Extract, 10 g/l NaCl) agar or in broth with shaking at 37 °C. L. monocytogenes strains were routinely grown in brain heart infusion (BHI; Lab M, Lancashire, UK) agar or in broth with shaking at 30 °C. When appropriate, erythromycin (Erm) was used in concentrations of 250 μg/ml for E. coli and 5 μg/ml for L. monocytogenes. Lactobacillus rhamnosus 1/6 was grown on MRS agar (Lab M) at 37 °C.
Plasmid isolations
L. carnosum 4010 plasmid DNA was isolated according to Anderson and McKay's method for isolating large plasmids from Lactococcus with the following modifications (Anderson and McKay 1983). Cells of 50 ml culture were collected by centrifugation (10,000 rpm, 4 min, JA-14, Beckman Avanti™ J-251). Pelleted cells were resuspended in 3.8 ml sucrose-Tris-EDTA buffer and divided into two 15-ml Falcon tubes. Reagent volumes used for each tube were five times of that in Anderson and McKay's method for screening. For each tube, 485 μl of lysozyme (20 mg/ml) and 25 μl of mutanolysin (5,000 U/ml) were added before incubation at 37 °C for 1 h. After cell lysis by SDS, the tubes were vortexed for 1 min. Extraction by phenol (5 ml) was repeated. The clear aqueous layer was subsequently extracted with 5 ml of chloroform-isoamyl alcohol (24:1). DNA precipitation by isopropanol was carried out in a freezer for 1 h. Pelleted DNA was washed with 4 ml of 70 % ethanol and dissolved in 200 μl of sterile Milli-Q water. RNA in the obtained DNA sample was degraded by RNase (0.1 mg/ml).
Vectors and constructed plasmids from E. coli, L. lactis and L. monocytogenes were isolated with E.Z.N.A. plasmid mini kit (Omega Bio-Tek, Norcross, GA, USA). For plasmids from L. lactis or Listeria, lysozyme (20 mg/ml) was added to cell resuspension solution and incubated at 37 °C for 1 h before lysing the cells.
DNA techniques
Fragments for screening, cloning or sequencing were amplified by standard PCR with either Taq DyNAzyme™ II DNA polymerase or Phusion High-Fidelity DNA polymerase (Finnzymes, Espoo, Finland) in Eppendorf Mastercycler (Hamburg, Germany). Due to the lack of available nucleotide sequence for leucocin C, the PCR primers for amplifying leucocin C gene were designed according to its amino acid sequence. PCR primers used are listed in Table 2. PCR products and plasmid constructs were sequenced by outsourced DNA sequencing service (Institute of Biotechnology, University of Helsinki). In order to obtain complete sequence of large unknown DNA fragments, primer walking technique was used. DNA was separated in agarose gel containing ethidium bromide (0.5 μg/ml) by electrophoresis and visualised under UV light.
In Southern hybridisation, DNA was first transferred from gel to nylon membrane by vacuum blotting. Probes were labelled with digoxigenin (DIG) by standard PCR supplemented with DIG-labelled dUTP (Boehringer Mannheim, Germany). DNA on membrane was then hybridised and detected according to Boehringer Mannheim DIG labelling and detection kit.
Calf intestinal phosphatase, DNA ligase, polynucleotide kinase and restriction enzymes were used as recommended by the suppliers (Fermentas, Vilnius, Lithuania; New England Biolabs, Ipswich, MA, USA; Promega, Madison, WI, USA). Plasmids were transferred into E. coli, L. lactis and L. monocytogenes by electroporation with a Bio-Rad Gene Pulser device (Bio-Rad Laboratories, Richmond, CA, USA) essentially as described previously (Holo and Nes 1989; Park and Stewart 1990; Zabarovsky and Winberg 1990).
Clonings and gene expression constructs
To produce leucocin C and its immunity protein LecI in L. lactis, the mature part of the leucocin C gene lecC, with and without the immunity gene lecI, were amplified by PCR using Phusion High-Fidelity DNA polymerase (Finnzymes) with primer pairs LecC forw-LecI rev and LecC forw-LecC new rev, respectively. The amplicons were cloned as blunt-end fragments into the NaeI site in the secretion vector pLEB690 (Li et al. 2011), resulting in plasmids pLEB728 (lecC) and pLEB729 (lecCI). Blunt-end cloning into the NaeI site in pLEB690 results in an SSusp45 gene fusion, which enables secretion of a native mature protein with no extra amino acids in its N-terminus. As a control, the leucocin C immunity gene was amplified with the primers LecI forw-LecI rev and cloned as a BspHI-blunt fragment into the NcoI-SmaI cut expression vector pLEB688 (Li et al. 2011), resulting in the plasmid pLEB730. Ligation mixtures of the above-mentioned plasmid constructs were electroporated into L. lactis MG1363 (Gasson 1983) and the transformant colonies were selected with nisin. Plasmids were isolated from the transformants, and the correct constructions were verified by sequencing. The lecC and lecCI secretion/expression plasmids were then electroporated into L. lactis NZ9000 (Kuipers et al. 1998).
To produce the leucocin C immunity protein LecI in L. monocytogenes, lecI gene was cloned with the promoter of pepR gene from L. rhamnosus strain 1/6 (Varmanen et al. 1998) into a cloning vector pTF1. Vector pTF1 harbours lactococcal pSH71 replicon (Gasson 1983), an Erm resistance gene, and multiple cloning sites from pBluescript. The promoter of pepR gene was chosen because we previously found it functional in various bacteria (unpublished results). The pepR promoter region was amplified by PCR with primer pair PpepR forw-PpepR rev and restricted with BcIl (cleavage site immediately after the RBS). The resulting 135-bp fragment was ligated with the BglII-cut lecI plasmid pLEB730 (BglII cleavage site 7 nt before lecI ATG). Then, from the PpepR-pLEB730 ligation mixture, the PpepR-lecI fragment was amplified by PCR with primers PpepR forw-LecI rev, restricted with EcoRI and ligated with EcoRI-SmaI-cut pTF1. The ligation mixture was electroporated into E. coli TG1. The resulting lecI expression plasmid pLEB732 was isolated and further electroporated into L. monocytogenes WSLC 1018. The plasmid was isolated from Listeria, and its correct construction was confirmed by sequencing. As a negative control, the vector pTF1 was electroporated into L. monocytogenes WSLC 1018.
Anti-bacterial activity
Bacteriocin activities were determined by conventional spot-on-lawn method. The indicator strains L. mesenteroides ATCC 8293, L. monocytogenes Scott A and L. monocytogenes WSLC 1018 were first grown overnight in M17G or BHI broth. Two hundred microlitres of the indicator cultures were added to 5 ml of M17G soft agar, poured onto a M17G agar plate and let dried. Ten-microlitre droplets of pasteurised supernatants (75 °C, 10 min) from L. lactis and L. carnosum overnight cultures were spotted onto the surface of the indicators, and the plates were incubated overnight at 28 °C.
SDS-PAGE and identification of the activity band
Proteins from 1-ml of the pasteurised cell-free overnight culture supernatants of the L. lactis strains LAC 409 (leucocin C) and LAC 360 (vector pLEB690) were concentrated with 10 μl of StrataClean resin (Agilent Technologies, Santa Clara, CA, USA). The resin beads were washed with sterile water, and the proteins were released in 10 μl of Bio-Rad Tricine sample buffer (Bio-Rad, Hercules, CA, USA). The concentrated supernatant samples containing the beads were analysed by tricine-SDS-PAGE essentially as described by Schägger (2006). Ten microlitres of samples were loaded into gels, except 2 μl of leucocin C sample that was loaded for inhibition test. Prestained Spectra Multicolor Low Range Protein Ladder (Thermo Scientific, Rockford, IL, USA) and 3.4 kDa pure nisin (Aplin and Barrett Limited, Bristol, UK) were used as protein size markers. Two identical gels were prepared of 16.5 % acrylamide resolving gel and 1 cm of 10 % acrylamide spacer gel. The electrophoresis was run at 4 °C with 30 V (about 20 mA) for the first 30 min and at 150 V for about 2 h (90 mA in the beginning, 55 mA in the end). After electrophoresis, the gels were fixed. One gel was stained with Silver Stain Plus kit (Bio-Rad) and the other with Coomassie Brilliant Blue R-250. The Coomassie-stained gel was used for identification of the bacteriocin band. After staining and destaining, the gel was washed for 2 h in distilled water and placed onto a BHI agar plate. Ten millilitres of BHI soft agar containing 200 μl of overnight L. monocytogenes WSLC 1018 culture was poured onto the plate. The plate was incubated overnight at 30 °C.
Leucocin C immunity assay
To determine the function of leucocin C immunity protein LecI, 3 μl of overnight Listeria carrying the lecI plasmid pLEB732 or the vector pTF1was added to 300 μl BHI + Erm5 broth containing different concentrations (0, 5, 10, 20, 50, and 100 μl/ml) of leucocin C-producing L. lactis culture supernatant in Bioscreen microtiter plates (100 wells). The plates were incubated in Bioscreen C (Labsystems, Helsinki, Finland) at 30 °C with constant shaking for 24 h. The optical density at 600 nm wavelength was measured every 2 h.
GenBank accession number
The sequence of the complete leucocin C gene cluster has been submitted to GenBank with accession number JQ061256.
Results
Localisation of leucocin A and C genes in L. carnosum 4010 genome
Since L. carnosum 4010 carries plasmids and class IIa bacteriocin genes are often plasmid-associated, the first aim was to study the loci of the leucocin A and leucocin C genes with Southern blotting by using leucocin A and C genes as probes. The probes were produced and labelled with PCR. Leucocin A gene lcaA was amplified with its immunity gene lcaB. The amplified fragments were confirmed to be the leucocin A and C genes by sequencing and translating to amino acids. The novel leucocin C gene was named lecC. Plasmid DNA of L. carnosum 4010 was isolated, run in agarose gel, transferred to nylon membrane and hybridised with DIG-labelled probes. L. carnosum 4010 was found to carry several plasmids (Fig. 1). The lecC probe hybridised with the largest of the plasmids and with the chromosomal band (Fig. 1a). The latter was most likely due to a partial degradation of large plasmid during plasmid isolation, resulting in plasmid fragments migrating with the chromosomal band in agarose gel. The lcaAB probe hybridised with two plasmid bands, presumably representing different forms of the second largest plasmid (Fig. 1b). Hence, the genes for the two leucocins were shown to be located in different plasmids, indicating that they are two independent bacteriocins in L. carnosum 4010.

L. carnosum 4010 plasmids in agarose gels and Southern blots hybridised with DIG-labelled lecC (a) or lcaAB (b) probes. Leucocin A and C genes were located in different plasmids. Chr chromosomal band, Marker GeneRuler™ 1 kb DNA ladder (Fermentas)
Characterisation of leucocin C operons
Since leucocin C gene was shown to be located independently of leucocin A, the large leucocin C plasmid could also carry other leucocin C-dedicated genes. Thus, next aim was to characterise the flanking region of lecC gene by sequencing. Different approaches, including arbitrary primed PCR, inverse PCR and restriction Southern cloning strategy, were used to obtain lecC-flanking fragments for sequencing (data not shown).
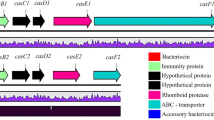
The best working strategies were restriction of the isolated plasmid DNA with AclI, HindIII or HpaI; self-ligation and inverse-PCR. First, HpaI-inverse PCR produced about 1-kb fragment containing an operon with two genes, the lecC structural gene and a putative immunity gene lecI downstream of lecC, and about 400 bp downstream of lecI including a putative transcription terminator. AclI-inverse PCR produced about 8-kb fragment containing the whole 5-kb lecC region: the lecCI operon and a complementary operon with three ABC-transporter genes lecXTS upstream of lecC (Fig. 2). The 8-kb AclI fragment also included a region about 3 kb downstream of lecS. No more lecC-related genes existed in this region. The closest gene to leucocin C cluster, about 540 bp downstream of lecS, was a 282-bp orf 74 % homologous to yaiI gene of L. lactis IL1403. To investigate the downstream region of lecI, HindIII-inverse PCR was used. In the 2.5-kb downstream area, only a plasmid mobilisation gene mob and some hypothetical genes were found. These genes matched to several Leuconostoc plasmids, e.g., L. mesenteroides J18 plasmid pKLE03 (accession number: NC_016821), which indicates that the sequencing of the entire lecC cluster was completed.

Organisations of the leucocin A and C gene clusters in L. carnosum 4010. Promoters are shown as black flags. Transcriptional terminators are shown as lollipop symbols. yaiI L. lactis gene with unknown function; S secretion gene lecS for ABC transporter; T translocator gene lecT for ABC transporter; X accessory gene lecX; C leucocin C precursor gene lecC; I immunity gene lecI; mob plasmid mobilisation gene; E' truncated accessory gene lcaE; A leucocin A precursor gene lcaA; B immunity gene lcaB. Restriction sites for AclI, HindIII and HpaI used in inverse PCR are shown. Solid line sequence in GenBank, dashed line unpublished/unknown sequences
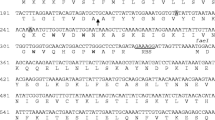
The five genes organised in two operons, lecXTS and lecCI, were identified to be class IIa-related genes by sequence homology. The mature part of lecC and the immunity gene lecI were novel genes without significant homology to any known genes. However, the protein sequence of LecI matched with other class IIa immunity proteins, with the best scores with listeriocin and sakacin P immunity proteins (48 % identity, 73 % similarity) (Fig. 3). Still, experimental evidence would be needed to confirm its functionality as an immunity protein. Interestingly, the organisations of the leucocin C operons, the 72-bp double-glycine type signal sequence of lecC and the sequence of the ABC transporter operon lecXTS were practically identical with those of leucocin A in L. gelidum (97 % identity). Therefore, the next aim was to determine whether the leucocin A operons in L. carnosum 4010 would be the same, i.e., whether the strain carries two sets of practically identical bacteriocin transporters.

Alignment of leucocin C immunity protein LecI with its closest relatives listeriocin (LisB), enterocin CRL 35 (EntA) and sakacin P (SakP) immunity proteins
Characterisation of leucocin A operons
Assumed leucocin A gene cluster was amplified by PCR with primers designed according to the leucocin A gene sequences from L. gelidum UAL 187. Surprisingly, the obtained 4.5 kb amplicon was somewhat smaller that the expected size (5 kb) of lcaA region in L. gelidum (result not shown). Sequencing of the PCR product showed that the amplicon contained identical leucocin A and its immunity genes lcaAB as in L. carnosum Ta11a (Fig. 2). However, the transporter operon lcaECD was missing due to a disruption by anIS30 family transposase in the accessory gene lcaE. The DNA sequence upstream of the transposase gene was not leucocin A-related but matched to a non-coding region of L. mesenteroides J18 plasmid pKLE03 (accession number: NC_016821). Therefore, the leucocin A gene cluster in L. carnosum 4010 only included the structural and immunity genes, but not transporters.
Heterologous expression of leucocin C in L. lactis
In order to study the function of the newly found leucocin C immunity gene lecI, leucocin C peptide would be needed. A way to get the bacteriocin would be to produce it in a heterologous non-bacteriocinogenic host L. lactis. The mature part of the lecC gene was fused with the lactococcal signal sequence of usp45 in the plasmid pLEB690, under the control of the constitutive promoter P45 and the nisin-inducible promoter P nisZ . The lecC gene was cloned with and without the putative immunity gene lecI. To control that the immunity protein alone does not cause any inhibition, the lecI gene was cloned into pLEB688 with no SSusp45. The constructed plasmids were transferred into L. lactis MG1363 for constitutive expression and NZ9000 for nisin-inducible expression. The production of bacteriocin was determined by anti-bacterial assay on a plate. Leucocin C was effectively secreted by L. lactis strains, as shown by large halos on Listeria and Leuconostoc indicator plates (Fig. 4). Nisin-induced expression of leucocin C in NZ9000 was much better than constitutive expression from the promoter P45 and comparable with the wild type leucocin producer L. carnosum 4010. The halos of LecC and LecCI strains were of the same size, showing that co-expression of the putative immunity gene lecI had no significant effect on bacteriocin production. This efficient leucocin C-producing L. lactis could be used as the source of the bacteriocin for studying the function of the putative immunity gene lecI.

Anti-bacterial assay of L. lactis-producing leucocin C (LecC, LecCI) compared to the wild type leucocin A and C producer L. carnosum 4010. Indicator strains: a L. monocytogenes WSLC 1018, b L. monocytogenes Scott A, c L. mesenteroides ATCC 8293. Onto indicator lawn, 10 μl of cell-free overnight culture supernatants were spotted. All host strains were cultured with nisin (0.5 μg/ml) as selective agent for the plasmids and/or for nisin induction. Second row from top L. lactis MG1363 as the host. Third row from top nisin-inducible L. lactis NZ9000 as the host. V, plasmid vector pLEB690. LecI L. lactis MG1363 carrying lecI plasmid pLEB730
Identification of the bacteriocin band
To identify that the killing of the indicator strains by L. lactis LecC strains was due to a bacteriocin peptide, the proteins from culture supernatants were concentrated and analysed with SDS-PAGE. With silver staining, potential leucocin C band migrating between the 3.4 kDa nisin and the 10 kDa marker bands was observed (Fig. 5a). Even though the actual size of leucocin C is 4.6 kDa, the putative leucocin C band migrated slower than the 4.6 kDa marker band. However, as the 3.4 kDa nisin migrated slower than the 4.6 kDa band in the marker, the exact size determination according to the marker is not fully reliable. As expected, the putative leucocin C band could not be seen in Coomassie gel, since the loaded sample volume was only 2 μl instead of 10 μl in silver-stained gel. The lower sample volume was used because preliminary tests showed that 10 μl of leucocin C sample in gel causes too large inhibition for size identification (results not shown). The Coomassie gel was then placed into a Listeria soft agar plate for inhibition test. A large halo between the 4.6 and 10 kDa marker bands appeared in leucocin C lane (Fig. 5b), confirming the anti-bacterial activity of the visible leucocin C band in the corresponding silver-stained gel (Fig. 5a).

SDS-PAGE of supernatant concentrations from overnight cultures of leucocin C-producing L. lactis LAC409 (LecC) and the vector pLEB690 strain LAC360 (Vector). a Silver-stained gel. b Coomassie-stained gel on L. monocytogenes WSLC 1018 agar. Supernatant proteins adsorbed onto 10 μl StrataClean resin were released with 10 μl of SDS buffer and loaded into 16.5 % acrylamide gels. Ten microlitres of samples were loaded, except LecC for Coomassie/inhibition gel, 2 μl was loaded. A putative leucocin C band (black arrow) in a and corresponding inhibition zone in b appeared between 4.6 and 10 kDa. As bacteriocins may migrate somewhat differently in gel than marker bands, 1 μg of pure nisin (3.4 kDa) was used as a bacteriocin size marker. The actual size of leucocin C is about 4.6 kDa. Double bands of bacteriocins may indicate that different forms of bacteriocin, e.g., dimers or precursor peptides are present
LecI protects the cell against leucocin C
The last aim in this work was to confirm by experimental evidence, whether LecI was actually a functional immunity protein of leucocin C. The lecI gene was cloned with the constitutive lactobacillar promoter of pepR gene into the plasmid vector pTF1 in E. coli. The constructed plasmid and the empty vector were introduced into L. monocytogenes. The Listeria transformants were then grown with different concentrations of leucocin C-producing L. lactis culture supernatant. The growth curves are shown in Fig. 6. With no leucocin C in media, the vector and lecI strains grew equally. When leucocin C supernatant was added into the media, the growth of both strains retarded, but the retardation of the vector strain was significantly stronger than the LecI strain. With higher leucocin C concentrations (20, 50, and 100 μl supernatant per millilitre), the difference in growth of the two strains was less pronounced (data not shown), showing that the immunity capacity of LecI was limited. Yet, production of LecI protected Listeria cells against leucocin C, showing the immunity function of LecI.

Growth of LecI-producing Listeria in different leucocin C concentrations (0, 5, 10 μl of leucocin C-producing L. lactis supernatant per millilitre). LecI-producing cells tolerated leucocin C better than the vector strain. Vector Listeria-carrying pTF1. Error bars SD of the mean from three parallel cultures
Discussion
Lactic acid bacterium L. carnosum 4010 produces two class IIa bacteriocins, leucocin A and leucocin C. In this study, we have shown that leucocin C has its own genes for the production-immunity system, independently from the well characterised bacteriocin leucocin A. Until now, the leucocin C genes have remained unknown, even though the secreted leucocin C has been purified from L. mesenteroides strain 6, and the amino acid sequence of mature leucocin C has been determined (Fimland et al. 2002b). Most class IIa bacteriocins are encoded by plasmid-associated genes (Ennahar et al. 2000). Likewise, the leucocin C genes were shown to be plasmid-encoded in L. carnosum 4010. Leucocin A genes were found from another plasmid. However, the leucocin A region differed from the previously characterised leucocin A operon in L. gelidum UAL 187 (van Belkum and Stiles 1995). The signal peptide of leucocin A in the strain 4010 was identical with that of leucocin A in L. carnosum Ta11a, differing by 7 aa of LcaA leader in L. gelidum UAL 187 (Felix et al. 1994). In addition, in L. carnosum 4010, the transporter operon was almost entirely missing, and thus, the leucocin A region only contained the structural and the immunity genes, with 87 bp of the 5′ end of the accessory gene lcaE. As leucocin A is secreted by the strain (Budde et al. 2003), either the genes for leucocin A transporter are located somewhere else in the genome or leucocin A is secreted by some other transporter.
In most cases with class IIa bacteriocin genes, the bacteriocin structural genes are followed by an immunity gene in the same operon, and another operon(s) contains genes for secretion proteins (Ennahar et al. 2000). The leucocin C operons in L. carnosum 4010 followed this typical bacteriocin gene organisation. Two operons containing five genes were characterised. The organisations of lca operons in L. gelidum and lec operons in L. carnosum were the same. Moreover, the lecC signal sequence and the lecXTS operon-encoding accessory protein, translocator and secretion protein for leucocin C ABC transporter were virtually identical with the lca operon of leucocin A (Hastings et al. 1991; van Belkum and Stiles 1995). Since leucocin A operon in the strain 4010 does not contain dedicated transporter genes, leucocin A could possibly be secreted by the leucocin C transporter. However, further experiments are required to identify the actual leucocin A transporter.
According to their structures, leucocins A and C belong to different subgroups of class IIa bacteriocins (A in subgroup 2, C in subgroup 1) (Nissen-Meyer et al. 2009). Other class IIa bacteriocins of subgroup 1, e.g., sakacin P and listeriocin 743A, share high sequence similarity with leucocin C. Regardless of the sequence similarities, the organisations of these bacteriocin operons are different (Ennahar et al. 2000). Listeriocin 734A does not possess its own transporter genes and is secreted by the sec pathway (Kalmokoff et al. 2001). In the case of sakacin P, two genes sppK and sppR encode the regulatory proteins (Hühne et al. 1996). In L. carnosum 4010, regulatory genes were not present in the lecC-flanking regions.
In order to secrete class IIa bacteriocins in heterologous bacteria, naturally resistant L. lactis is a potential host candidate (Henderson et al. 1992; Kjos et al. 2009). Lactococci are resistant because class IIa bacteriocins do not bind to lactococcal mannose phosphotransferase permease, which serves as the target in sensitive cells (Kjos et al. 2009). The lactococcal sec-dependent secretion signal of usp45 has been used to replace the native signal sequences of enterocin P or pediocin genes (Borrero et al. 2011; Li et al. 2011). Here, in our study, lecC gene was fused with the SSusp45 in the secretion vector pLEB690, and leucocin C was secreted in L. lactis strains MG1363 and NZ9000. Large halos on indicator plates showed that leucocin C-producing L. lactis strains killed the indicators L. monocytogenes and L. mesenteroides (Fig. 4). Even though lactococci are naturally resistant to class IIa bacteriocins and hence their production would not require immunity factors, in some cases the co-expression of the cognate immunity gene has been reported to improve bacteriocin production. Co-expression of pediocin immunity gene has significantly increased the yields of heterologous pediocin in host L. lactis (Arqués et al. 2008). However, here in this study with leucocin C, there was no noticeable increase in LecC production with the co-expression of lecI (Fig. 4). As nisin promoter is known to be stronger than the P45 promoter (Li et al. 2011), leucocin C was also produced more with P nisZ in nisin-inducible strain NZ9000 than with the constitutive P45 promoter in the strain MG1363. Nisin-induced secretion of active leucocin C in L. lactis NZ9000 with leucocin C expression plasmids was comparable with the wild type leucocin producer L. carnosum 4010. To correspond the inhibition of the indicators with the bacteriocin-sized peptide, i.e., leucocin C, the proteins from the L. lactis cultures were concentrated, separated in SDS-PAGE and the gel was placed onto a Listeria plate. Putative leucocin C band was observed with silver staining, and the peptide was shown to be highly active in SDS gel after fixation, Coomassie staining and destaining (Fig. 5). The inhibition zone from leucocin C was somewhat smeared or doubled, suggesting that different forms, e.g., dimer or precursor peptide may also be present. Determining sizes of bacteriocins by SDS-PAGE is often inexact. Sizes of many bacteriocins, e.g., pediocin have been inaccurately estimated by SDS-PAGE (Bhunia et al. 1987; Yang et al. 1992). Therefore, the determined size between 3.4 and 10 kDa is sufficient to confirm the role of leucocin C peptide in the inhibition caused by heterologous L. lactis (Fig. 5). In conclusion, this study shows that L. lactis as a heterologous host is able to efficiently produce highly active leucocin C for potential use in food industry.
The function of a bacteriocin immunity protein can be demonstrated by expressing the putative immunity gene in a bacteriocin-sensitive host and to examine the increase of bacteriocin resistance. Previously, the functions of bacteriocin immunity genes of, e.g., enterocin A, leucocin A and sakacin P, have been investigated by expressing them in heterologous hosts (Fimland et al. 2002a). However, even though Listeria has been used as the model organism in proposing the mode of action of class IIa bacteriocins (Kjos et al. 2010), no immunity genes have been expressed and shown to function in Listeria. The gene lecI identified in this study was shown to confer immunity by expressing lecI in leucocin C-sensitive Listeria (Fig. 6). The sensitivity of LecI-producing Listeria towards leucocin C decreased, and this strain could grow in leucocin C-containing medium better than the vector strain. The immunity capacity was rather limited, as higher concentrations of leucocin C killed the LecI-Listeria as efficiently as the vector strain (data not shown). Limited immunity could be a consequence of a low expression level of lecI (not determined). Stronger expression could possibly have resulted in higher resistance, but the aim here was not to create Listeria strain with maximum bacteriocin resistance but to demonstrate the function of leucocin C immunity protein LecI.
Based on the common motifs in class IIa immunity proteins, they can be divided into three subgroups (Fimland et al. 2002a). The subgrouping of bacteriocins does not necessarily match with the subgrouping of the immunity proteins. For instance, pediocin and sakacin P bacteriocins are sorted into the same class IIa subgroup 1 (Nissen-Meyer et al. 2009), but their cognate immunity proteins belong to different subgroups: Ped-im in subgroup A and SakP-im in subgroup B. Immunity proteins in the subgroup B contain certain consensus motifs, e.g., the C-terminal SN/SIRYGY, which are also found in the amino acid sequence of LecI (Fig. 3) (Fimland et al. 2005). Therefore, based on the common consensus motifs and the highest identity, 48 %, with listeriocin and sakacin P immunity proteins, LecI is now suggested to be a member of subgroup B immunity proteins of class IIa bacteriocins. Leucocin A immunity protein is structurally different than LecI, and it belongs to subgroup A (Fimland et al. 2005). Therefore, it is not surprising that in the previous study leucocin A immunity protein did not protect the host against leucocin C (Fimland et al. 2002a). Leuconostoc strains producing the two leucocins need both immunity proteins.
References
Anderson DG, McKay LL (1983) Simple and rapid method for isolating large plasmid DNA from lactic streptococci. Appl Environ Microbiol 46:549–552
Arqués JL, Rodríguez JM, Gasson MJ, Horn N (2008) Immunity gene pedB enhances production of pediocin PA-1 in naturally resistant Lactococcus lactis strains. J Dairy Sci 91:2591–2594
Beasley SS, Takala TM, Reunanen J, Apajalahti J, Saris PEJ (2004) Characterization and electrotransformation of Lactobacillus crispatus isolated from chicken crop and intestine. Poult Sci 83:45–48
Bhunia AK, Johnson MC, Ray B (1987) Direct detection of an antimicrobial peptide of Pediococcus acidilactici in sodium dodecyl sulfate-polyacrylamide gel electrophoresis. J Ind Microbiol 2:318–322
Borrero J, Jiménez JJ, Gútiez L, Herranz C, Cintas LM, Hernández PE (2011) Use of the usp45 lactococcal secretion signal sequence to drive the secretion and functional expression of enterococcal bacteriocins in Lactococcus lactis. Appl Microbiol Biotechnol 89:131–143
Budde BB, Hornbaek T, Jacobsen T, Barkholt V, Koch AG (2003) Leuconostoc carnosum 4010 has the potential for use as a protective culture for vacuum-packed meats: culture isolation, bacteriocin identification, and meat application experiments. Int J Food Microbiol 83:171–184
Drider D, Fimland G, Héchard Y, McMullen LM, Prévost H (2006) The continuing story of class IIa bacteriocins. Microbiol Mol Biol Rev 70:564–582
Ennahar S, Sashihara T, Sonomoto K, Ishizaki A (2000) Class IIa bacteriocins: biosynthesis, structure and activity. FEMS Microbiol Rev 24:85–106
Felix JV, Papathanasopoulos MA, Smith AA, von Holy A, Hastings JW (1994) Characterization of leucocin B-Ta11a: a bacteriocin from Leuconostoc carnosum Ta11a isolated from meat. Curr Microbiol 29:207–212
Fimland G, Johnsen L, Dalhus B, Nissen-Meyer J (2005) Pediocin-like antimicrobial peptides (class IIa bacteriocins) and their immunity proteins: biosynthesis, structure, and mode of action. J Pept Sci 11:688–696
Fimland G, Eijsink VG, Nissen-Meyer J (2002a) Comparative studies of immunity proteins of pediocin-like bacteriocins. Microbiology 148:3661–3670
Fimland G, Sletten K, Nissen-Meyer J (2002b) The complete amino acid sequence of the pediocin-like antimicrobial peptide leucocin C. Biochem Biophys Res Commun 295:826–827
Gasson MJ (1983) Plasmid complements of Streptococcus lactis NCDO 712 and other lactic streptococci after protoplast-induced curing. J Bacteriol 154:1–9
Hastings JW, Sailer M, Johnson K, Roy KL, Vederas JC, Stiles ME (1991) Characterization of leucocin A-UAL 187 and cloning of the bacteriocin gene from Leuconostoc gelidum. J Bacteriol 173:7491–7500
Henderson JT, Chopko AL, van Wassenaar PD (1992) Purification and primary structure of pediocin PA-1 produced by Pediococcus acidilactici PAC-1.0. Arch Biochem Biophys 295:5–12
Holo H, Nes IF (1989) High-frequency transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl Environ Microbiol 55:3119–3123
Hühne K, Axelsson L, Holck A, Kröckel L (1996) Analysis of the sakacin P gene cluster from Lactobacillus sake Lb674 and its expression in sakacin-negative Lb. sake strains. Microbiology 142:1437–1448
Kalmokoff ML, Banerjee SK, Cyr T, Hefford MA, Gleeson T (2001) Identification of a new plasmid-encoded sec-dependent bacteriocin produced by Listeria innocua 743. Appl Environ Microbiol 67:4041–4047
Kjos M, Borrero J, Opsata M, Birri DJ, Holo H, Cintas LM, Snipen L, Hernández PE, Nes IF, Diep DB (2011) Target recognition, resistance, immunity and genome mining of class II bacteriocins from Gram-positive bacteria. Microbiology 157:3256–3267
Kjos M, Nes IF, Diep DB (2009) Class II one-peptide bacteriocins target a phylogenetically defined subgroup of mannose phosphotransferase systems on sensitive cells. Microbiology 155:2949–2961
Kjos M, Salehian Z, Nes IF, Diep DB (2010) An extracellular loop of the mannose phosphotransferase system component IIC is responsible for specific targeting by class IIa bacteriocins. J Bacteriol 192:5906–5913
Kuipers OP, de Ruyter PGGA, Kleerebezem M, de Vos WM (1998) Quorum sensing-controlled gene expression in lactic acid bacteria. J Biotechnol 64:15–21
Li R, Takala TM, Qiao M, Xu H, Saris PEJ (2011) Nisin-selectable food-grade secretion vector for Lactococcus lactis. Biotechnol Lett 33:797–803
Mills S, Stanton C, Hill C, Ross RP (2011) New developments and applications of bacteriocins and peptides in foods. Annu Rev Food Sci Technol 2:299–329
Nes IF, Yoon S, Diep DB (2007) Ribosomally synthesised antimicrobial peptides (bacteriocins) in lactic acid bacteria: a review. Food Sci Biotechnol 16:675–690
Nissen-Meyer J, Rogne P, Oppegård C, Haugen HS, Kristiansen PE (2009) Structure-function relationships of the non-lanthionine-containing peptide (class II) bacteriocins produced by Gram-positive bacteria. Curr Pharm Biotechnol 10:19–37
Papathanasopoulos MA, Krier F, Revol-Junelles AM, Lefebvre G, Le Caer JP, von Holy A, Hastings JW (1997) Multiple bacteriocin production by Leuconostoc mesenteroides TA33a and other Leuconostoc/Weissella strains. Curr Microbiol 35:331–335
Park SF, Stewart GS (1990) High-efficiency transformation of Listeria monocytogenes by electroporation of penicillin-treated cells. Gene 94:129–132
Rea MC, Ross RP, Cotter PD, Hill C (2011) Classification of bacteriocins from Gram-positive bacteria. In: Drider D, Rebuffat S (eds) Prokaryotic antimicrobial peptides: From genes to applications, 1st edn. Springer, New York, pp 29–53
Sambrook J, Russell DW (2001) Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, New York
Schägger H (2006) Tricine-SDS-PAGE. Nat Protoc 1:16–22
van Belkum MJ, Stiles ME (1995) Molecular characterization of genes involved in the production of the bacteriocin leucocin A from Leuconostoc gelidum. Appl Environ Microbiol 61:3573–3579
Varmanen P, Rantanen T, Palva A, Tynkkynen S (1998) Cloning and characterization of a prolinase gene (pepR) from Lactobacillus rhamnosus. Appl Environ Microbiol 64:1831–1836
Vaughan A, Eijsink VG, O'Sullivan TF, O'Hanlon K, van Sinderen D (2001) An analysis of bacteriocins produced by lactic acid bacteria isolated from malted barley. J Appl Microbiol 91:131–138
Yang R, Johnson MC, Ray B (1992) Novel method to extract large amounts of bacteriocins from lactic acid bacteria. Appl Environ Microbiol 58:3355–3359
Zabarovsky ER, Winberg G (1990) High efficiency electroporation of ligated DNA into bacteria. Nucleic Acids Res 18:5912
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wan, X., Li, R., Saris, P.E.J. et al. Genetic characterisation and heterologous expression of leucocin C, a class IIa bacteriocin from Leuconostoc carnosum 4010. Appl Microbiol Biotechnol 97, 3509–3518 (2013). https://doi.org/10.1007/s00253-012-4406-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4406-4