
Abstract
The transformation of trinitrotoluene (TNT) by several mutant strains of Clostridium acetobutylicum has been examined to analyze the maximal rate of initial transformation, determine the effects of metabolic mutations of the host on transformation rate, and to assess the cell metabolic changes brought about during TNT transformation. Little difference in the maximal rate of TNT degradation in early acid phase cultures was found between the parental ATCC 824 strain and strains altered in the acid forming pathways (phosphotransacetylase, or butyrate kinase) or in a high-solvent-producing strain (mutant B). This result is in agreement with the previous findings of a similar degradation rate in a degenerate strain (M5) that had lost the ability to produce solvent. A series of antisense constructs were made that reduced the expression of hydA, encoding the Fe-hydrogenase, or hydE and hydF, genes encoding hydrogenase maturating proteins. While the antisense hydA strain had only ∼30 % of the activity of wild type, the antisense hydE strain exhibited a TNT degradation rate around 70 % that of the parent. Overexpression of hydA modestly increased the TNT degradation rate in acid phase cells, suggesting the amount of reductant flowing into hydrogenase rather than the hydrogenase level itself was a limiting factor in many situations. The redox potential, hydrogen evolution, and organic acid metabolites produced during rapid TNT transformation in early log phase cultures were measured. The redox potential of the acid-producing culture decreased from −370 to −200 mV immediately after addition of TNT and the hydrogen evolution rate decreased, lowering the hydrogen to carbon dioxide ratio from 1.4 to around 1.1 for 15 min. During the time of TNT transformation, the treated acidogenic cells produced less acetate and more butyrate. The results show that during TNT transformation, the cells shift metabolism away from hydrogen formation to reduction of TNT and the resulting effects on cell redox cofactors generate a higher proportion of butyrate.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Trinitrotoluene (TNT) is a common nitroaromatic compound contaminating the surface and subsurface soil of many military sites, as a result of the accumulation of residues from explosives (Spain 1995). Data describing TNT presence in military sites have been compiled in several reviews (Ahmad and Hughes 2000; Spain 1995). The toxicity and mutagenicity of TNT has been widely known for decades (Won et al. 1976), and information on the toxicity and mutagenicity of synthetic and biodegradative intermediates also has been considered in efforts to remove toxicity and study the transformation of these compounds (Siciliano et al. 2000; Padda et al. 2000). Due to the high cost of incineration treatments for such soils, bioremediation, especially by strains of anaerobic bacteria such as Clostridium, has become a promising and cost-effective decontamination measure (Tan et al. 1992; Shin and Crawford 1995; Daun et al. 1998). The ability to monitor the soil communities and the ongoing metabolism is important in estimating the course of progress in decontamination of hazardous compounds (Frische and Hoper 2003; Eyers et al. 2006; Wu et al. 2008).
The prevalence of Clostridium in soil makes the study of these organisms relevant for bioremediation studies, and various Clostridium species have been reported to degrade TNT through alternative routes (Ahmad and Hughes 2000; Spain 1995; Zhang and Bennett 2005). This widely isolated species and related anaerobes are important contributors to the degradation of a variety of compounds in the oxygen-depleted environment of contaminated subsurface soil. Clostridium acetobutylicum is known to enzymatically reduce TNT primarily through the activity of a Fe-only hydrogenase, which transfers electrons to TNT through an iron–sulfur center (Watrous et al. 2003; Kutty and Bennett 2006; Ahmad and Hughes 2000; Hughes et al. 1998b; Khan et al. 1997). A more thorough understanding of the regulation of hydrogenase activity and the redox inputs to hydrogenase during TNT reduction may yield better techniques for accentuating the degradation of TNT.
In this study, we compared TNT reduction in C. acetobutylicum strains containing different genetic backgrounds. Since hydrogenase was found to be important in TNT degradation, we investigated if strains that produced an altered pattern of acids and solvents would have different abilities to degrade TNT at various stages of growth. The measurement of culture parameters and metabolic products during TNT degradation led to the finding that at maximal rates the TNT reduction was very competitive with cell processes and accounted for a significant fraction of the available reductant produced by glucose metabolism. The cell physiology interactions accompanying TNT degradation are discussed in relation to the distribution of the reduced metabolites produced by Clostridium cultures.
Materials and methods
Chemicals and reagents
2,4,6-trinitrotoluene, 2,6-dinitrotoluene, 2-amino-4,6-dinitrotoluene, and 4-amino-2,6-dinitrotoluene were purchased from ChemService (Westechester, PA); p-nitrotoluene and 2,4-dinitrotoluene were purchased from Sigma-Aldrich (St. Louis, MO); 2-hydroxylamino-4,6-dinitrotoluene, 4-hydroxylamino-2,6-dinitrotoluene, and 2,4-diamino-6-nitrotoluene were from SRI International (Menlo Park, CA).
All medium components for clostridium culture were obtained from Difco (Detroit, MI) or Sigma-Aldrich. All Restriction enzymes were obtained from New England Biolabs, Inc. (Beverly, MA). pCR2.1-TOPO vector from Invitrogen Corporation (Carlsbad, CA) was used for PCR product cloning. Automated DNA sequencing was performed by LoneStar automated DNA sequencing (LoneStar Laboratories Inc., Houston, TX).
Bacterial strains, plasmids, and growth conditions
All bacteria strains and plasmids are listed in Table 1.
Escherichia coli cultures were grown aerobically at 37 °C in Luria-Bertani medium, C. acetobutylicum was grown anaerobically at 37 °C in buffered clostridium growth medium (CGM) in a Forma Scientific anaerobic chamber (Thermo Forma, Marietta, OH) as described previously (Cai and Bennett 2011; Zhao et al. 2005). For E. coli recombinant strains, the medium was supplemented with ampicillin (100 μg/mL), chloramphenicol (35 μg/mL), kanamycin (50 μg/mL), or erythromycin (200 μg/mL) as appropriate. For C. acetobutylicum strains, erythromycin (40 μg/mL for solidified agar plate and 100 μg/mL for liquid medium) and thiamphenicol (25 μg/mL) were used when necessary. For long-term storage, E. coli strains were cultivated and stored as glycerol stocks at −80 °C. C. acetobutylicum strains were stored as lyophilized stocks at room temperature or glycerol stocks at −80 °C (Scotcher and Bennett 2005).
Analysis of fermentation products
Cell growth was monitored by A 600 with a Beckman DU-800 spectrophotometer. In a fermentor, cell growth was monitored by a cell density sensor. The concentrations of butanol, acetone, ethanol, butyrate, and acetate were determined using gas chromatography with a Hewlett-Packard 5890 Series II instrument (Hewlett-Packard Company, Palo Alto, CA) as described previously (Scotcher and Bennett 2005; Zhao et al. 2003, 2005). Glucose was measured by HPLC as described (Wang et al. 2011).
TNT assay in C. acetobutylicum culture
TNT concentration in the culture medium was followed by a spectrometric protocol modified from Jenkins et al. (Jenkins and Walsh 1992; Watrous et al. 2003). Freshly made sodium sulfite solution (0.2 g/mL) in 50 mM at pH 8.0 Tris–HCl buffer reacts with TNT solution to form a colored complex with a maximum absorption at A 414 that remains stable for hours under assay conditions. A 414 readings increase linearly up to a 500-μM TNT concentration.
Single C. acetobutylicum colonies from freshly grown plates were inoculated into buffered CGM medium with appropriate antibiotics and incubated overnight as seed cultures. An appropriate amount from seed culture was used for the subculture to yield a starting A 600 of 0.1. The culture was incubated at 37 °C and growth (A 600) measured periodically. TNT rate was assayed during early-exponential phase (A 600, 0.3), mid-exponential phase (A 600, 1.0), and stationary phase (24-h culture with A 600, varied) by diluting the culture at different times into fresh medium containing 400 μM TNT. At different time points, 250 μL samples were taken and immediately reacted with 750 μL sodium sulfite solution by vortex; the samples were further centrifuged at 14,000 rpm for 10 min, and the supernatant was used for the TNT spectrophotometric assay. The TNT degradation rate μ max for different strains were compared by calculating the rate of TNT decrease during the first 40 min.
In order to determine the effect of culture dilution on TNT degradation and confirm the linear range of the measurements, five C. acetobutylicum cultures were prepared to ultimately yield ×10, ×20, ×30, ×40, and ×50 dilutions, respectively. For each dilution sample, three subcultures were made. The appropriate amounts of C. acetobutylicum (A 600, 0.3) and CGM/TNT mixture, for a total volume of 10 mL, were transferred anaerobically into 15 mL culture tubes. The tubes were immediately vortexed, and a 0.25 mL sample of each tube was added to 0.75 mL assay buffer. The tubes were left to incubate at 37 °C, and 0.25 mL samples were again taken at 20 and 40 min. All operations were conducted in the anaerobic chamber, and all solutions were pre-equilibrated in the chamber for 24 h. Once the three time samples were collected and centrifuged at 13,000 rpm for 10 min, the A 414 absorbance of each supernatant was measured, and the dilution factors were considered to calculate the specific TNT degradation rates. Since the time of the initial rate measurement was short after the dilution compared with a typical lag phase recovery of growth rate, the measured rates are considered indicative of the metabolism of the cell at the time points used.
Plasmid constructions and transformation
Plasmids from E. coli were purified using QIAprep Miniprep protocols (Qiagen Inc., Valencia, CA). Plasmids from C. acetobutylicum were purified according to the protocol (Harris et al. 2002). Genomic DNA was purified from C. acetobutylicum using the Genomic DNA Purification Kit from Puregene (Gentra Systems, Minneapolis, MN). All commercial enzymes used in this study (Taq polymerase, restriction endonucleases, calf intestinal phosphatase, T4 DNA ligase, and Klenow fragment of DNA polymerase I) were used according to the manufacturers’ recommendations.
E. coli transformation followed the Sambrook protocol (Sambrook et al. 1989). As to C. acetobutylicum transformation, plasmid DNA was methylated by the Φ3TI methyltransferase to prevent restriction by the clostridial endonuclease Cac824I (Mermelstein and Papoutsakis 1993). This was achieved by transformation of the required plasmid into E. coli DH10β harboring vector pDHKM (Zhao et al. 2003), carrying an active copy of the Φ3TI methyltransferase gene. Electrotransformation of C. acetobutylicum was carried out according to a modification of the protocol (Mermelstein and Papoutsakis 1993); positive transformants were isolated on agar-solidified CGM supplemented with the appropriate antibiotic; and transformations were also confirmed by plasmid DNA purification.
Oligonucleotide sequences used are listed in Table 2. For antisense constructs, pSOS94 was used as the backbone plasmid. It was digested with BamH I and Ava I and ligated with the digested target antisense fragments (as-hydA, 175 bp; as-hydE, 244 bp; and as-hydF, 244 bp) to form pSOS-as-hydA, pSOS-as-hydE, and pSOS-as-hydF. pSOS 94 digested by BamH I and Ava I was treated and self-ligated to form the control plasmid pSOS-del. In order to overexpress the hydA gene, primers hydA-F and hydA-R were used to amplify the 1,989 bp fragment from C. acetobutylicum 824 genomic DNA. This new construct was further confirmed by sequencing before being ligated to pSOS94 digested by BamH I and Ava I to yield the plasmid pSOS-hydA.
Real-time PCR quantification of gene expression in C. acetobutylicum
Cultures were grown to an A 600 of 0.3, at which point the cultures were treated with rifampicin, chloramphenicol, and phenylmethanesulfonyl fluoride at final concentrations of 30 mg/L, 50 mg/L, and 0.5 mM, respectively. Cultures were immediately placed on ice for 15 min, after which 7.5 mL were centrifuged at 10,000 rpm for 10 min at 4 °C. The supernatant was decanted and the cell pellets were re-suspended in 750 μL of a solution containing a 2:1 ratio of RNA Protect (Qiagen Inc. Valencia, CA) to PBS. After incubation for 5 min at room temperature, the cell suspensions were pelleted by centrifugation and stored at −80 °C. Cell pellets were thawed on ice, and the total RNA was extracted and purified using an RNeasy mini kit (Qiagen) with on column DNase I treatment according to the manufacturer’s instructions.
First-strand cDNA was synthesized from 0.15 μg total RNA using an Iscript cDNA synthesis kit (Bio-Rad, Hercules, CA) per the manufacturer’s instructions. Real-time PCR primers were designed using Primer3 software to amplify specific products that were between 100 and 170 bp in size. Each real-time PCR contained 12.5 μL iQ SYBR Green Supermix (Bio-Rad), 200 nM of each primer, 1 μL diluted cDNA template, and water to bring the final reaction volume to 25 μL. Real-time PCR was performed in a CFX96 PCR machine (Bio-Rad) using the standard program: 98 °C for 2 min (1 cycle) then 98 °C for 2 s and 55 °C for 5 s (40 cycles). Fluorescence data were collected and melting-curve analysis was performed following amplification to ensure that a single product was formed. The PCR efficiency for each primer set was determined by analyzing three replicates of 2-fold serial dilutions of cDNA. To determine relative expression levels, data were analyzed as described by Pfaffl (2001) by using CAC0905 and GapC as internal standards.
Measurement of redox potential
C. acetobutylicum ATCC 824 was grown anaerobically in a four-vessel bioreactor (DASGIP) on CGM with constant agitation at 37 °C. The redox and growth of the culture was recorded every 30 s, and the headspace was flushed with a constant stream of N2 with a flow rate of 0.1 L/min. The redox potential was measured using Broadly-James F-995 Redox FermProbe which measures the difference in potential between a platinum electrode in the fermentation media and an Ag/AgCl reference electrode. The electrodes are not calibrated because the zero points and slope of the half-cells do not change (Broadley-James FermProbe ORP Electrodes Manual). On-line growth reading was calibrated with a spectrophotometer. Once the cultures reached an A 600 of 0.25 then TNT was added in an acetonitrile solution through a septum port. As a control, the same amount of acetonitrile without TNT was added to other vessels in a similar manner.
Measurement of carbon dioxide and hydrogen gas production
The fermentation vessels received a constant flow of nitrogen gas which mixed with the headspace gases produced by the metabolizing organism. These gases were exhausted through a water-cooled off-gas condenser which was plumbed directly to an Agilent MicroGC 3000A that was used to measure nitrogen, carbon dioxide, and hydrogen gas levels. The gas analyzer was set to take samples every 5 min for approximately 1 h before the addition of TNT and continuing for about 4 h after the addition.
Results
The colorimetric TNT assay protocol
A simplified Jenkins TNT assay protocol was adapted in our experiments. Major nitro compounds and TNT metabolites do not interfere with our TNT assay, in which TNT showed an absorption peak at 414 nm while other partially reduced species did not. The other nitro compounds showed absorption peaks around 350 nm with a much lower intensity than the peak at 414 nm for TNT (data not shown). The measured TNT concentration in the culture declined with time during the assay, and the rate of decrease had diminished considerably after 1 h. It was found that sample dilution affected the calculated specific TNT degradation rate. Samples of clostridial cultures diluted in higher volumes of the CGM/TNT mixture exhibited increasing levels of specific TNT degradation rate up to a dilution factor of 40, and further dilution incurred no additional effect on the specific rate of transformation. Therefore, the maximal specific TNT degradation rate as measured by this method was used in the figures and tables.
TNT degradation rate remains relatively stable among different C. acetobutylicum strains
Strains of C. acetobutylicum were compared for their rate of TNT transformation. The strains analyzed were the wild-type ATCC 824; strain M5 (Clark et al. 1989; Stim-Herndon et al. 1996), a strain that has lost the pSOL plasmid and does not produce butanol; strain 824(pta), with a disrupted phosphotransacetylase gene, pta, which produces a lower level of acetate during the early phase of culture; strain 824(buk) with a disrupted butyrate kinase gene, buk, which produces butyrate at a lower level during the early phase of culture (Green et al. 1996); and 824(mutant B), a strain that has a disruption in the gene upstream of the solvent operon and has increased butanol formation (Nair et al. 1999). These strains could reveal differences in the TNT transformation rate correlated with the metabolite pattern of the cells, especially in the early log phase. Since the metabolic mutations affected the products of early metabolism, this change in metabolic pattern could in turn affect the relative proportion of hydrogen produced during the early log phase. For example, the difference in acetate vs. butyrate production can result in a different level of hydrogen formation and potentially alter the availability of reductant for TNT transformation. Also, since some of these strains, the buk and mutant H strains, have previously been shown to enter the solvent-producing phase earlier than the wild-type parent, it was anticipated they may have lower ability to transform TNT in the early or mid-log phase samples as they would be physiologically more like the early-solvent-producing state. Samples from cultures of each C. acetobutylicum strain (Table 1) were diluted in order to measure the initial rate of the TNT transformation of the specific culture. To measure the TNT degradation rate during different growth phases, samples were analyzed from cultures at A 600 of 0.3 (for early exponential phase), A 600 of 1.0 (for mid-exponential phase), and 24-h culture (for stationary phase culture, solventgenic phase). The initial rate of TNT transformation of each culture is shown in Fig. 1. The experiment shows the maximal rate of the reaction with different strains and cell preparations. It was found that all strains tested showed similar initial TNT degradation rate, ranging from 10,000 to 12,000 μM h−1 A −1600 during the early exponential phase. All strains tested showed the same trend of a decrease in the specific TNT degradation rate with the advancement of growth phase, with those entering or in the stationary-solvent-producing phase having the lowest specific rate of TNT transformation. The maximal specific TNT degradation rate may be limited by hydrogenase levels or by other factors such as the glucose uptake rate, growth rate, supply of reductant ferredoxin (Fd) reduced (Fdreduced), or other components. To investigate the effects of these other factors, analyses were done on the media before and during the time the TNT was being degraded.

Comparison of TNT degradation rates among C. acetobutylicum strains with different genetic backgrounds (different solvent profiles) revealed no major differences. The rate of TNT transformation was assayed during the early exponential phase (A 600, 0.3), mid-exponential phase (A 600, 1.0), and stationary phase (24-h culture with A 600, varied) by diluting the culture at different times into fresh medium containing 400 μM TNT. Standard deviations are shown
In order to quantitate the glucose metabolism during the TNT transformation, the metabolites were analyzed from parental C. acetobutylicum ATCC 824 cultures transforming TNT at the high rates presented in Fig. 1. Since the amount of glucose utilization during the period of TNT transformation was low, we assessed the metabolism during this period by two independent measurements. We measured glucose directly and also measured the metabolic products of glucose metabolism (acetate and butyrate) independently. The TNT transformation rate measured in this series of five culture experiments gave a TNT transformation rate of 15.2 mM h−1 A −1600 .
Glucose uptake rates in three of the culture experiments were found by measurement of the acetate and butyrate concentrations before and after the TNT transformation period. The correlation to glucose utilization used the stoichiometric amount of acetate and butyrate produced by Clostridia cultures (A 600 = 0.3) within an hour of TNT addition. Since the stoichiometric ratio of metabolites produced to glucose consumed is 2:1 for acetate and 1:1 for butyrate, the molar increase observed in these two products during the first hour was used to determine the amount of glucose metabolism to acids during the period of TNT transformation. Ethanol and butanol were measured and discounted from the analysis as negligible. These measurements gave glucose uptake rates of 22, 26, and 34 mM h−1 A −1600 . In two of the experiments, the glucose uptake by Clostridia culture (A 600 = 0.3) was analyzed by HPLC directly. For each experiment, the glucose concentration values were measured both before and after the hour-long run with TNT. The rate of glucose consumption was directly determined from the concentration difference showing values of 17 and 14 mM h−1 A −1600 . The ratios of TNT to glucose metabolism in these TNT studies ranged from 0.45 to 1.0. So it appeared, a mole of TNT was reduced for about every 1 to 2 mol of metabolized glucose.
These relatively high ratios suggest that TNT can be reduced competitively against regular cellular metabolism and that when operating at the maximal rate TNT reduction is not a minor factor in the cells metabolic activities and can represent a significant distortion and sink for the cell’s reducing power. Thus, rather than being a minor side reaction of the cells metabolism, the TNT is an effective in vivo reactant with the redox carriers/enzymes such as Fdreduced-hydrogenase, and it can affect the cells metabolism during this time. The reaction with TNT not only can use a significant proportion of the cells reducing power, but in doing this it affects the overall pattern of metabolism and is in turn affected by competitors which influence the redox situation, e.g., the more effective diversion of redox to solvent producing redox pathways later in the culture. This perspective would place TNT among the group of dyes that can be reduced by Clostridium (Rao and Mutharasan 1987), and thus this work could have broader implications in the general control/understanding of redox regulation and response in these anaerobes. In the case of TNT, the reduction is irreversible and continues beyond an initial reduction step (Hughes et al. 1998a, b) while for many dyes the reaction is reversible and the reduced dye can transfer electrons to another carrier or redox reaction and affect the metabolite pattern. This feature is the well-known observation that violgen addition results in higher butanol levels in late growth cultures (Rao and Mutharasan 1987).
Redox and metabolite changes are observed during TNT transformation as shown by measurements of the products of the cell culture during the TNT reduction period
We found TNT addition alters the redox potential of the culture. Addition of TNT to cultures in exponential growth resulted in a dose dependent spike in the redox potential, which is shown in Fig. 2. This initial spike was followed by a persistent increase in the redox potential of the cultures receiving TNT when compared with the redox level of the control culture. The relative increase in redox potential lasted for the remainder of exponential growth indicating that TNT addition results in a change to the culture’s redox status for 3 h.

The effect of TNT addition on redox potential. The redox potential of C. acetobutylicum cultures increased in a dose-dependent matter after the addition of TNT. Acetonitrile or an acetonitrile/TNT solution was added to C. acetobutylicum cultures in the exponential phase of growth (A 600 of ∼0.25) to final concentrations of 0, 100, or 500 μM TNT. The cultures were grown in a bioreactor, and the redox potential of the culture was recorded every 30 s. Details are given in the “Materials and methods”
We observed addition of TNT resulted in a decrease of hydrogen production by cultures of C. acetobutylicum ATCC 824. TNT can act as an alternative electron acceptor for the hydrogenase enzyme, so the effect of TNT addition on H2 production in cultures during exponential growth was determined by measuring the H2/CO2 ratio. Figure 3 shows that there was a sharp reduction in the H2/CO2 ratio following the addition of TNT, which was due to a decrease in H2 production since CO2 production remained relatively constant. This provides further evidence that TNT can compete with the natural electron acceptors of the hydrogenase enzyme in actively growing cells and indicates that TNT reduction could alter the metabolic output of the cells.
We noted that when TNT was added, and the reaction proceeds over a period of time, a different metabolic pattern was observed. More butyrate and less acetate are observed. A culture grown under the conditions where the TNT degradation rate was analyzed (Fig. 1) was processed and subjected to gas chromatography to determine the metabolites present in the pre-TNT added culture and in the culture after 1 h when the starting TNT had been depleted. The culture continued to grow during this time and the metabolism of glucose continued during the 1-h period. Analysis of the metabolites formed during the period when TNT is being transformed shows a significant change in the proportions of acetate and butyrate. The metabolites formed during the period of TNT transformation were determined by subtracting the concentrations of metabolites at the time of TNT addition from those determined after 1 h of TNT transformation.
As shown in the experiments of Fig. 2 showing the redox potential after TNT addition and in Fig. 3 where measurements of CO2 and hydrogen are included, we noted that when TNT was added the hydrogen production decreased. This would be expected if TNT is acting at the hydrogenase and taking some of the electrons that would normally go to hydrogen formation to instead reduce the TNT. We measured the acid metabolites formed during the time after TNT addition. A culture grown under the conditions where the TNT degradation rate was analyzed was processed and subjected to gas chromatography to determine the metabolites present in the pre-TNT added culture and in the culture 1 h after the TNT addition, a time when the starting TNT had been depleted. The culture continued to grow during this time, and the metabolism of glucose continued during the 1-h period and seemed unchanged in rate as judged by the total production of acids during this time. Analysis of the metabolites formed over the period during TNT transformation showed a significant change in the proportions of acetate and butyrate, with a large amount of butyrate being produced in comparison to very little acetate (Table 3).

The effect of TNT addition on hydrogen production. A TNT/acetonitrile solution was added to a culture of C. acetobutylicun grown in a bioreactor. The A 600 and redox were recorded every 30 s. The headspace of the culture was constantly flushed with N2 gas and the concentrations of H2 and CO2 present in the off-gas were measured using gas chromatography at 5-min intervals
Regulation of hydrogenase affects TNT degradation rate and solvent production
The maturation of active hydrogenase by other proteins has been investigated recently (King et al. 2006; McGlynn et al. 2007), and we sought to assess if reduction of expression of genes encoding these proteins by using antisense technology would generate a greater effect on reduction of hydrogenase activity than an antisense directed toward hydA itself. If so and if these low-concentration maturation proteins were limiting, we might thereby observe a much lower TNT transformation rate in strains with lowered expression of the maturation proteins. We chose hydE and hydF as genes to manipulate in this way and compared the results with the analogous reduction of hydA expression by a similar antisense construct. The previous work using hydA antisense constructs demonstrated the feasibility of the method and had shown a correlation of hydrogenase expression and activity with TNT degradation rate (Watrous et al. 2003). The construction of the homologous plasmid series is presented in Fig. 4a. Small fragments containing the upstream and start section of the coding regions of hydA, hydE, and hydF were targeted for antisense down-regulation. Meanwhile, the hydA gene was overexpressed based on the same backbone plasmid to increase hydrogenase activity. The scheme for making the constructs is shown in Fig. 4a. Figure 4b shows that down-regulation of hydA significantly reduced the specific TNT degradation rate while overexpression of hydA improved the specific TNT degradation rate to only a very small degree but not to the level of the extra dosage of the hydrogenase gene based on the higher copy number of the plasmid. Down-regulation of hydE reduced TNT degradation rate slightly while antisense against hydF did not show a decrease in TNT degradation activity. This result might indicate that hydF is present in excess under normal physiological conditions. Antisense down-regulation of hydA caused hydrogenase activity to decrease significantly. However, overexpression of hydA caused only a slight increase in hydrogenase activity under the assay conditions. All antisense constructs showed growth patterns similar to the control strain. A test for solvent production showed that all antisense strains produced slightly more butanol (as well as total solvent, data not shown) than the strain bearing the control plasmid. Reverse transcription polymerase chain reaction (RT-PCR) confirmed anti-sense down-regulation of target genes in all three strains (Fig. 4c). mRNA for hydA, hydE, and hydF in the antisense constructs was reduced by around 2- to 5-fold relative to the 824(pSOS-del) control. hydA and hydE underwent similar levels of reduction, but the down-regulation of hydF showed the largest expression decrease. RT-PCR also showed that hydA overexpression generated a large (∼50-fold) increase in hydA mRNA. Since those proteins, HydE and HydF, would be expected to be in lower concentration than HydA, it was anticipated that they would be more sensitive to antisense inhibition of expression and thus drive down the hydrogenase activity in the cells to perhaps even lower activity levels than were found in previous hydA antisense studies (Watrous et al. 2003). From the results, it appears that HydE and HydF are present in excess and even when expression of their genes is reduced as ascertained by RT-PCR, the effect does not substantially reduce the hydrogenase activity with the proportion of reduction of the hydrogenase being less than in the corresponding hydA antisense construct.

Antisense experiments show effects of hydrogenase on TNT degradation. a Plasmid construction required to regulate hydrogenase activity. The diagram shows schematically the constructions used in the formation of the plasmids studied in (b) and (c). Vector organizations and inserts for 824(pSOS-hydA), 824(pSOS-as-hydF), 824(pSOS-as-hydE), and 824(pSOS-as-hydA) are shown. The 824(pSOS-del) control plasmid, which lacks an insert, is also shown. b Measurement of the TNT degradation rate in cultures of the antisense and overexpression strains. The strains were grown and analyzed as described in Fig. 2. Regulation of hydrogenase and the level of cell reductant affects TNT degradation rates significantly. The simplified Jenkins TNT assay was used to find the maximal saturated rate of degradation for each strain. Standard deviations are shown. c The degree of antisense down-regulation of target genes is compared among three different 824 antisense strains. RT-PCR was used to amplify the target genes and compare their change in expression relative to the 824(pSOS-del) control. In a similar experiment, the expression of the hydA overexpression construct, 824(pSOS-hydA) yielded an increase of over 50-fold in the hydA RNA level compared with that of the wild-type cells. The relative expression levels were analyzed as described by Pfaffl (2001) by using CAC0905 and GapC as internal standards
Discussion
In the study presented in Fig. 1, the results show only a small reduction in the TNT transformation rate in the pta, buk, and mutant H strains during the medium log phase. It was anticipated that the different metabolite patterns might affect the availability of reductant for hydrogen formation and thus affect TNT transformation by the reductant going into the hydrogenase reaction. The small effect in the mid-log phase may have been due to more reductant going to butyrate in the pta mutant since this strain is known to form more butyrate early in the culture (Green et al. 1996) so there could be less available for hydrogenase mediated TNT reduction in this phase of the culture. In the buk and mutant H strain studies, a small reduction of the rate of TNT transformation was observed in the mid-log phase and this small effect could be due to an earlier transition to the more stationary, solvent phase physiological state where the cell has low ability to transform TNT (Green et al. 1996; Khan et al. 1997).
In Fig. 4b and in the RT-PCR studies 4c, we show the effects of modification of hydA expression and the effects of reduced expression of the genes encoding the hydrogenase maturation proteins HydE and HydF on TNT transformation. Since the level of hydrogenase activity (HydA), had been earlier correlated with TNT transformation rate (Watrous et al. 2003), we wished to assess the effect of antisense inhibition of expression of the hydrogenase maturation proteins (King et al. 2006; McGlynn et al. 2007) that have been recently been shown to be required for functional hydrogenase in vivo. It was anticipated the levels of these proteins might be more sensitive to antisense regulation than the highly expressed hydA gene which had been successfully modulated by the antisense technique (Watrous et al. 2003). The results suggested however that these proteins are able to function to mature and activate hydrogenase even when their expression level has been reduced significantly by antisense methods.
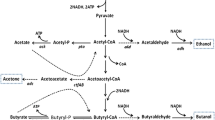
If one considers the total reductant available and that some of the redox goes to reduce the TNT, initially it might seem that the loss of this reducing power would lessen the amount of butyrate formed since formation of butyrate from pyruvate requires more reduction than formation of acetate. However, if we examine the process of reduction to form hydrogen and butyrate, we see a difference in the cofactor used. In the formation of hydrogen, the cofactor is Fdreduced generated by the pyruvate/Fd oxidoreductase in the conversion of pyruvate to acetyl-CoA, providing one Fdreduced per pyruvate consumed and for each CO2 released, and this is supplemented to some degree by conversion of some of the NADH formed in glycolysis via a NADH-Fd reductase. In the early-growth stage we are studying, there is no formation of the reduced solvents, ethanol and butanol and the enzymes forming these compounds are not induced under this growth condition. The normal ratio of acetate to butyrate 1:1 then gives a value of 1.3–1.4 for the H2/CO2 ratio and a use of more Fd than would be generated by the pyruvate/Fd reductase indicating use of some NADH to form Fdreduced for the extra hydrogen formation (Table 3). In this circumstance, the shift to more butyrate formation during the period of TNT reduction by the Fd-Fe hydrogenase in our anaerobic chamber experiments, can be considered as a result of a difference in the cofactor balance. With TNT, it appeared that blockage of the usual hydrogenase acceptance of reductant from Fdreduced was lessened, and thus the NADH-Fd reductase reaction did not proceed as well as normal. This situation then resulted in more NADH being available in the cell and since the NADH must be recycled, it is consumed by conversion of acetyl-CoA to butyrate. Since the cell cannot reduce acetyl-CoA directly due to a lack of aldehyde-alcohol dehydrogenase (e.g., AdhE), there are few options to recycle the NADH. It appeared the NADH was recycled to NAD+ through conversion of the acetyl-CoA to acetoacetyl-CoA, beta-hydroxylbutyryl-CoA, crotonyl-CoA, and butyryl-CoA. This route was already operating to a good extent in the cell since almost equivalent molar amounts of acetate and butyrate were formed during the normal acid phase culture and the normal proportion of acids yielded a hydrogen to carbon dioxide ratio of approximately 1.3–1.4 in the pre TNT culture (as shown in Fig. 3) and which is consistent with previous literature where uncontrolled pH batch early phase cultures were studied (Husemann and Papoutsakis 1990; Zhao et al. 2003). The pathway for acetyl-CoA to butyryl-CoA allowed consumption of two NADH per molecule of butyrate formed and relieved the cofactor imbalance between Fd and NADH since NADH is the reductant used in this pathway. The redistribution observed also points to the relative ability of TNT to both be reduced by hydrogenase and inhibit the reaction of hydrogenase by serving as an alternative to Fd. In this way, the addition of TNT effectively removes a portion of the normal role of hydrogenase while it acts as a competing substrate and leads to a build-up and redistribution of the substrate for the hydrogenase (i.e., Fdreduced) as shown in Fig. 5.

Diagram of redox cofactor utilization upon TNT addition. The NADH made during glycolysis is channeled to the formation of butyrate when the Fd-to-hydrogenase reaction is altered by the presence of TNT
The detailed redox calculations shown in Table 3 analyzes the redistribution of reductant from production of hydrogen to more production of butyrate during the time when TNT was present and being reduced. The level of TNT present (400 μM) would not consume much of the reductant available directly in this experiment, and the amount would be expected to be 1 mM or less based on the early reduction steps of TNT under these conditions as analyzed previously (Ahmad and Hughes 2000; Khan et al. 1997; Padda et al. 2000). Calculations of the observed reductant consumed in the formation of acids either before or during TNT transformation and the resulting reducing equivalents available for formation of hydrogen give values of 1.30 for the hydrogen/CO2 ratio before TNT addition and 1.05–1.12 during TNT transformation. These values are very close to the values measured experimentally and presented in Fig. 3, before TNT addition of 1.36 and during TNT transformation of 1.18. If the data are presented in a form where the fraction of total reductant formed from glucose metabolism, i.e., Fdreduced and NADH, that goes to either formation of hydrogen or butyrate is calculated, we can analyze the proportion of reductant that is consumed in butyrate formation in the culture during the 1-h period before TNT addition and it is 35 % of the total reductant. If the same calculation is done on the culture during the 1-h period of TNT transformation, a proportion of 44–47 % of the total reductant that goes into formation of butyrate is determined.
The variation of hydrogenase activity observed fits with the finding of Watrous et al. (Watrous et al. 2003) and was shown again here, in that hydA antisense lowered the expression and activity of hydrogenase and TNT reduction activity. On the other hand, increasing the amount of hydrogenase through overexpression affected the rate of TNT transformation in cells to only a small degree, less than 2-fold, while the expression of the hydA gene as determined by RT-PCR was elevated by ∼50-fold. These results suggest the limitation on reducing capacity seems to be at least partially due to the availability of Fdreduced to transfer reducing equivalents to hydrogenase or TNT.
The results also fit with the observation shown in Fig. 1, in which the TNT-reducing activity is lower in cultures in the solventogenic phase, at a time when there is more diversion of electrons from Fd to NAD+. This situation is apparently due to increased activity of the Fd-NADH reductase, which is capable of lowering the amount of Fdreduced available for interaction with hydrogenase. Additionally, the lower amount of hydrogenase activity found in such cultures means that the capacity of the cells to transfer electrons from hydrogen or Fdreduced to TNT is reduced. The results of previous studies on the Fd of C. acetobutylicum and the level of the NADH-Fd reductase and the Fd-NAD reductase in cells grown under different conditions are consistent with this explanation (Girbal and Soucaille 1994; Vasconcelos et al. 1994).
These findings of the effect of competition of other factors and intracellular cofactor pathways on TNT reduction pave the way for more detailed investigation of the effects these parameters play in defining the rate of TNT degradation in the natural environment. This is particularly interesting in light of the analysis of natural mobile soluble electron carriers in native soil ecosystems where molecules such as quinones, phenazines, and humic acids exist or are made and released by a variety of soil organisms. The presence of these compounds that could act as competitors or inhibitors of Fdreduced reactions with hydrogenase and TNT. They could also influence the availability of Fdreduced and the shuttling of reductant to other pathways in the cell, e.g., those using NADH and limiting the availability of Fdreduced. This feature would mimic the situation in the later phase cultures where a lower rate of TNT transformation is observed. Thus, to define the “real-world” effectiveness of various microbial communities to degrade TNT, not only do we need to consider the major groups of direct degraders such as clostridia and their substrates (sugars and polymeric substrates such as cellulose) in the environment that enable growth but we also need to consider the community population and small redox molecules in the soil which may generate e-carrier competitors to the reaction with TNT and cell redox processes. Thus, the presence of these other natural electron carriers may play a role in limiting the performance of Clostridial TNT degradation in contaminated sites.
References
Ahmad F, Hughes JB (2000) Anaerobic transformation of TNT by Clostridium. In: Spain JC, Hughes JB, Knackmuss H-J (eds) Biodegradation of nitroaromatic compounds and explosives. Lewis Publishers/CRC Press, Boca Raton, pp 185–212
Cai X, Bennett GN (2011) Improving the Clostridium acetobutylicum butanol fermentation by engineering the strain for co-production of riboflavin. J Ind Microbiol Biotechnol 38(8):1013–1025
Clark SW, Bennett GN, Rudolph FB (1989) Isolation and characterization of mutants of Clostridium acetobutylicum ATCC 824 deficient in acetoacetyl-coenzyme A:acetate/butyrate:coenzyme A transferase (EC 2.8.3.9) and in other solvent pathway enzymes. App Environ Microbial 55:970–976
Daun G, Lenke H, Reuss M, Knackmuss HJ (1998) Biological treatment of TNT-contaminated soil. 1. Anaerobic cometabolic reduction and interaction of TNT and metabolites with soil components. Environ Sci Technol 32:1956–1963
Eyers L, Smoot JC, Smoot LM, Bugli C, Urakawa H, McMurry Z, Siripong S, El-Fantroussi S, Lambert P, Agathos SN, Stahl DA (2006) Discrimination of shifts in a soil microbial community associated with TNT-contamination using a functional ANOVA of 16S rRNA hybridized to oligonucleotide microarrays. Environ Sci Technol 40(19):5867–5873
Frische T, Hoper H (2003) Soil microbial parameters and luminescent bacteria assays as indicators for in situ bioremediation of TNT-contaminated soils. Chemos 50(3):415–427
Girbal L, Soucaille P (1994) Regulation of Clostridium acetobutylicum metabolism as revealed by mixed-substrate steady-state continuous cultures: role of NADH/NAD ratio and ATP pool. J Bacteriol 176(21):6433–6438
Green EM, Boynton ZL, Harris LM, Rudolph FB, Papoutsakis ET, Bennett GN (1996) Genetic manipulation of acid formation pathways by gene inactivation in Clostridium acetobutylicum ATCC 824. Microbiology 142(Pt 8):2079–2086
Harris LM, Welker NE, Papoutsakis ET (2002) Northern, morphological, and fermentation analysis of spo0A inactivation and overexpression in Clostridium acetobutylicum ATCC 824. J Bacteriol 184(13):3586–3597
Hughes JB, Wang CY, Bhadra R, Richardson A, Bennett GN, Rudolph F (1998a) Reduction of 2,4,6-trinitrotoluene by Clostridium acetobutylicum through hydroxylamino intermediates. Environ Toxicol Chem 17:343–348
Hughes JB, Wang CY, Yesland K, Richardson A, Bhadra R, Bennett GN, Rudolph F (1998b) Bamberger rearrangement during TNT metabolism by Clostridium acetobutylicum. Environ Sci Technol 32:494–500
Husemann MH, Papoutsakis ET (1990) Effects of propionate and acetate additions on solvent production in batch cultures of Clostridium acetobutylicum. Appl Environ Microbial 56(5):1497–1500
Jenkins TF, Walsh ME (1992) Development of field screening methods for TNT, 2,4-DNT, and RDX in soil. Talanta 39(4):419–428
Khan TA, Bhadra R, Hughes J (1997) Anaerobic transformation of 2,4,6-TNT and related nitroaromatic compounds by Clostridium acetobutylicum. J Ind Microbiol Biotechnol 18:198–203
King PW, Posewitz MC, Ghirardi ML, Seibert M (2006) Functional studies of [FeFe] hydrogenase maturation in an Escherichia coli biosynthetic system. J Bacteriol 188(6):2163–2172
Kutty R, Bennett GN (2006) Studies on inhibition of transformation of 2,4,6-trinitrotoluene catalyzed by Fe-only hydrogenase from Clostridium acetobutylicum. J Ind Microbiol Biotechnol 33(5):368–376
McGlynn SE, Ruebush SS, Naumov A, Nagy LE, Dubini A, King PW, Broderick JB, Posewitz MC, Peters JW (2007) In vitro activation of [FeFe] hydrogenase: new insights into hydrogenase maturation. J Biol Inorg Chem 12(4):443–447
Mermelstein LD, Papoutsakis ET (1993) In vivo methylation in Escherichia coli by the Bacillus subtilis phage ϕ3T methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl Environ Microbial 59:1077–1081
Nair RV, Green EM, Watson DE, Bennett GN, Papoutsakis ET (1999) Regulation of the sol locus genes for butanol and acetone formation in Clostridium acetobutylicum ATCC 824 by a putative transcriptional repressor. J Bacteriol 181:319–330
Padda RS, Wang CY, Hughes JB, Bennett GN (2000) Mutagenicity of trinitrotoluene and its metabolites formed during anaerobic degradation by Clostridium acetobutylicum ATCC 824. Environ Toxicol Chem 19:2871–2875
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45
Rao G, Mutharasan R (1987) Altered electron flow in continuous cultures of Clostridium acetobutylicum induced by viologen dyes. Appl Environ Microbiol 53(6):1232–1235
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor
Scotcher MC, Bennett GN (2005) SpoIIE regulates sporulation but does not directly affect solventogenesis in Clostridium acetobutylicum ATCC824. J Bacteriol 187:1930–1936
Shin CY, Crawford DL (1995) Biodegradation of trinitrotoluene (TNT) by a strain of Clostridium bifermentans. In: Hinchee RW, Fredrickson J, Alleman BC (eds) Bioaugmentation for site remediation. Battelle Press, Columbus
Siciliano SD, Gong P, Sunahara GI, Greer CW (2000) Assessment of 2,4,6-trinitrotoluene toxicity in field soils by pollution-induced community tolerance, denaturing gel electrophoresis, and seed germination assay. Environ Toxicol Chem 19:2154–2160
Spain JC (1995) Biodegradation of nitroaromatic compounds. Annu Rev Microbiol 49:634–649
Stim-Herndon KP, Nair R, Papoutsakis ET, Bennett GN (1996) Analysis of degenerate variants of Clostridium acetobutylicum ATCC 824. Anaerobe 2:11–18
Tan EL, Ho CH, Griest WH, Tyndall RL (1992) Mutagenicity of trinitrotoluene and its metabolites formed during composting. J Toxicol Environ Health 36:165–172
Tummala SB, Welker NE, Papoutsakis ET (2003) Design of antisense RNA constructs for downregulation of the acetone formation pathway of Clostridium acetobutylicum. J Bacteriol 185(6):1923–1934
Vasconcelos I, Girbal L, Soucaille P (1994) Regulation of carbon and electron flow in Clostridium acetobutylicum grown in chemostat culture at neutral pH on mixtures of glucose and glycerol. J Bacteriol 176(5):1443–1450
Wang J, Zhu J, Bennett GN, San KY (2011) Succinate production from different carbon sources under anaerobic conditions by metabolic engineered Escherichia coli strains. Methods Eng 13(3):328–335
Watrous MM, Clark S, Kutty R, Huang S, Rudolph FB, Hughes JB, Bennett GN (2003) 2,4,6-trinitrotoluene reduction by an Fe-only hydrogenase in Clostridium acetobutylicum. Appl Environ Microbiol 69(3):1542–1547
Won WD, Salvo LHd, Ng J (1976) Toxicity and mutagenicity of 2,4,6-trinitrotoluene and its microbial metabolites. Appl Environ Microbiol 31:576–580
Wu Y, Luo Y, Zou D, Ni J, Liu W, Teng Y, Li Z (2008) Bioremediation of polycyclic aromatic hydrocarbons contaminated soil with Monilinia sp.: degradation and microbial community analysis. Biodegradation 19(2):247–257
Zhang C, Bennett GN (2005) Biodegradation of xenobiotics by anaerobic bacteria. Appl Microbial Biotechnol 67:600–618
Zhao Y, Hindorff LA, Chuang A, Monroe-Augustus M, Lyristis M, Harrison ML, Rudolph FB, Bennett GN (2003) Expression of a cloned cyclopropane fatty acid synthase gene reduces solvent formation in Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol 69(5):2831–2841
Zhao Y, Tomas CA, Rudolph FB, Papoutsakis ET, Bennett GN (2005) Intracellular butyryl phosphate and acetyl phosphate concentrations in Clostridium acetobutylicum and their implications for solvent formation. Appl Environ Microbiol 71(1):530–537
Acknowledgments
This project was supported by the Army Research Office, grant number W911NF0910119. Also, we would like to thank Mary Harrison and David French for assistance with this research and data management.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cai, X., Servinsky, M., Kiel, J. et al. Analysis of redox responses during TNT transformation by Clostridium acetobutylicum ATCC 824 and mutants exhibiting altered metabolism. Appl Microbiol Biotechnol 97, 4651–4663 (2013). https://doi.org/10.1007/s00253-012-4253-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4253-3