
Abstract
Medium-chain-length polyhydroxyalkanoates (mcl-PHA) consisting of 3-hydroxyhexanoate (HHx), 3-hydroxyoctanoate (HO), 3-hydroxydecanoate, 3-hydroxydodecanoate, and high-content 3-hydroxytetradecanoate (HTD) was produced by knockout mutant Pseudomonas putida KT2442 termed P. putida KTOY06. When grown on 6 to14 g/L single-carbon-source tetradecanoic acid, P. putida KTOY06, which β-oxidation pathway was weakened by deleting genes of 3-ketoacyl-coenzyme A (CoA) thiolase (fadA) and 3-hydroxyacyl-CoA dehydrogenase (fadB), for the first time, produced several mcl-PHA including 31 to 49 mol% HTD as a major monomer. HHx contents in these mcl-PHAs remained approximately constant at less than 3 mol%. In addition, large amounts of oligo-HTD were detected in cells, indicating the limited ability of P. putida KTOY06 in polymerizing long-chain-length 3-hydroxyalkanoates. The mcl-PHA containing high HTD monomer contents was found to have both higher crystallinity and improved tensile strength compared with that of typical mcl-PHA.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyhydroxyalkanoates (PHA) are accumulated by a wide variety of microorganisms as carbon and energy storage materials (Taguchi and Doi 2004; Chen et al. 2001a; Hoffmann and Rehm 2004). There are now more than 100 different types of known basic building blocks for PHA polymers reported (Steinbüchel and Valentin 1995; Simon et al. 1983). PHA have received increased attention because of their potential applications in areas of tissue engineering, environmentally friendly packaging materials, and as a chiral hydroxyalkanoate (HA) pool (Chen and Wu 2005; Gursel and Hasirci 1995; Atkins and Peacock 1996; Zinn et al. 2001; de Roo et al. 2002; Hartmann et al. 2004). Among PHA, medium-chain-length PHA (mcl-PHA) consisting of monomers of 3-hydroxyhexanoate (HHx), 3-hydroxyoctanoate (HO), 3-hydroxydecanoate (HD), 3-hydroxydodecanoate (HDD), or even higher-chain-length monomers show amorphous and elastic properties compared with short-chain-length PHA (scl-PHA), which has a high degree of crystallinity and rigidness (Ouyang et al. 2007; Nakamura et al. 1991; Holmes 1985; Noda et al. 2004; Ashby et al. 2000). Properties of PHA copolymers depend strongly on the structure, monomer content, and distribution of monomer units, as well as the average molecular weight and molecular weight distribution (Noda et al. 2005). Various mcl-PHA with different functional side chains including carbon–carbon double and triple bonds (Lageveen et al 1988; Kim et al. 1998), acetoxy and ketone (Jung et al. 2000), aromatic groups (Fritzsche et al. 1990; Curley et al. 1996; Kim et al. 1999), or scl-PHA and mcl-PHA copolymer (scl-mcl-PHA; Chen et al. 2001b) have been produced with a hope to improve mcl-PHA properties and to exploit other applications.
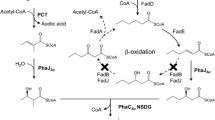
In this paper, the knockout mutant Pseudomonas putida KT2442 termed P. putida KTOY06, which β-oxidation pathway was weakened by knocking out fadA and fadB genes encoding 3-ketoacyl-coenzyme A (CoA) thiolase and 3-hydroxyacyl-CoA dehydrogenase, respectively, was cultivated in an attempt to produce mcl-PHA with structures closely resembling the fatty acids used as precursors for mcl-PHA formation. For the first time, as a specific example, tetradecanoic acid was used in this study for production of PHA containing 3HTD monomer. The physical properties of mcl-PHA with different 3HTD contents were characterized using gas chromatography (GC), gel permeation chromatography, differential scanning calorimetry (DSC), thermogravimetric analysis and stress–strain measurements.
Materials and methods
Bacterial strain and growth conditions
Knockout mutant Pseudomonas putida KT2442 termed Pseudomonas putida KTOY06 (P. putida KTOY06) in which β-oxidation pathway was weakened by knocking out genes of fadA and fadB (Ouyang et al. 2007) as donated by Dr. Ouyang SP of Dept of Biology, Tsinghua University, China. Genes fadA and fadB encoding 3-ketoacyl-CoA thiolase and 3-hydroxyacyl-CoA dehydrogenase in P. putida KT2442, respectively, catalyzed the last two steps in the β-oxidation pathway in P. putida KT2442. There were at least two sets of genes encoding putative proteins that were supposed to have catalytic abilities of 3-ketoacyl-CoA thiolase and 3-hydroxyacyl-CoA dehydrogenase in the genome of P. putida KT2442 available in the National Center for Biotechnology Information, including fadB2x- and fadB (Locus tag PP_2214 and PP_2136)-encoded 3-hydroxyacyl-CoA dehydrogenase and fadAx- and fadA (Locus tag PP_2215 and PP_2137)-encoded 3-ketoacyl-CoA thiolase. The method to generate the defined gene knockout mutant was previously reported (Schäfer et al. 1994). Two deoxyribonucleic acid (DNA) fragments, fadB′ and fadA′, corresponding to the partial 5′ sequence of fadB and the partial 3′ sequence of fadA, was inserted into suicide plasmid pK18mobsacB (Schäfer et al. 1994) for gene knockout, resulting in new plasmid termed pSPK11. Plasmid pSPK11 was transformed into Escherichia coli S17-1 by electroporation. Transconjugation of P. putida strains and E. coli S17-1 harboring recombinant plasmids were carried out as described (Fiedler et al. 2002).
P. putida KTOY06 with weakening β-oxidation was cultivated in Luria–Bertani (LB) medium using single-carbon-source tetradecanoic acid for production of mcl-PHA containing high 3HTD. Two-step shake-flask cultivation was conducted for mcl-PHA production: The seed culture was grown in the LB medium containing (g/L) 10 tryptone (Oxoid, UK), 5 yeast extract (Oxoid, UK), and 10 sodium chloride (pH 6.5) overnight at 30°C and 200 rpm, and then 5% (v/ v) of the seed culture was transferred into a 1,000-mL shake flask containing 350 mL LB medium (pH 6.5); the cells were cultivated at 30°C, 200 rpm for 12 h. Subsequently, tetradecanoic acid was added to promote mcl-PHA formation.
PHA extraction and purification
Bacterial cells were harvested by centrifugation (8,000 × g, 15 min). The cells were washed with hot ethanol and distilled water and then lyophilized. Intracellular PHA polymers were isolated from lyophilized cells by hot chloroform extraction at 100°C for 4 h, followed by filtration through a Whatman #1 paper filter to remove the cellular debris. PHA dissolved in chloroform was then precipitated with tenfold volume quantity of ice-cold ethanol. The collected PHA was dried in vacuo at 40°C for 24 h to completely eliminate the solvent.
Gas chromatography analysis
The intracellular PHA contents and PHA composition were determined by GC (Agilent 6890, USA). Both lyophilized cell and the extracted mcl-PHA were subjected to methanolysis in the presence of 3% H2SO4 (v/v); the resulting methyl esters of mcl-PHA were analyzed by GC. Benzoic acid was used as an internal standard (Braunegg et al. 1978).
Non-mcl-PHA HTD extraction
Bacterial cells that were cultured with 10 g/L tetradecanoic acid were harvested and washed as described above and resuspended with 100% ethanol. The cells were disrupted by sonication subsequently, after removal of cell debris by centrifugation (12,000 × g, 20 min), and the supernatant was collected and dried in an oven. The dried matter was dissolved in ethanol, filtered, and dried again. Finally, non-mcl-PHA HTD was obtained in a powder form.
NMR analysis of non-mcl-PHA HTD
The 1H nuclear magnetic resonance (NMR) spectra were recorded at room temperature in d-chloroform (CDCl3; 20 mg/mL) on a Bruker AV 400 MRI spectrometer to determine the chemical structure of non-mcl-PHA HTD. Tetramethylsilane was used as the internal standard.
Gel permeation chromatography analysis
Average molecular weights were estimated by GPC using a Waters 1525 pump with a combination of four styragel columns series (Styragel HR, 5 μm). A 2414 differential refractive index detector and UV detector were employed. Chloroform was used as eluent at a flow rate of 1.0 mL/min. The sample concentration was 2 mg/mL, and the injection volume was 50 μL. The calibration curve was generated with polystyrene standards (Sim et al. 1997).
Preparation of solvent-cast films
The mcl-PHA films were prepared by a conventional solvent-casting method (Zheng et al. 2005): The films formed were dried in vacuo at 40°C for 48 h to completely remove the solvents. Samples of PHA solvent-cast films were stored at room temperature for approximately 1 week previous to thermal analysis and stress–strain measurements.
Thermal analysis
DSC was performed with a TA-Q100 DSC (USA) analyzer equipped with a mechanical cooler system. It was calibrated with an indium standard. Each sample weighing 1.5–2.0 mg was encapsulated in an aluminum pan and heated from −80 to 100°C for the first scan. After maintaining at 100°C for 1 min, the molten sample was quenched to −80°C and heated from −80 to 100°C again as the second scan. A heating rate of 10°C/min was used throughout the process. Melting temperature (T m) and apparent heat of fusion (ΔH m) were recorded from endothermal peak value and area of the first scan, respectively. T g was recorded from the inflection point of the second heating scan (Xu et al. 2006).
The thermal stability analysis of the polymers was performed using a Q-50 instrument (TA Instrument, USA) under a nitrogen atmosphere of 50 mL/min. The sample was heated from 40 to 500°C at a heating rate of 10°C/min. Samples of 3–5 mg were loaded each time (Xu et al. 2006).
Mechanistic properties
The films were cut into dumbbell-shaped specimens with a width of 4 mm and a thickness of about 100 μm. The stress–strain measurements of films were carried out using a CMT-4000 universal testing machine (Shenzhen SANS, China) at room temperature. Speed of the cross-head was 5 mm/min (Luo et al. 2006).
Results
Production of mcl-PHA in P. putida KTOY06
Two-step cultivation was conducted to produce mcl-PHA containing 3HTD monomer. P. putida KTOY06 was first grown in LB for forming biomass, and tetradecanoic acid was added to promote 3HTD production (Table 1). With increasing cultivation time from 48 to 72 h in the presence of 12 g/L tetradecanoic acid, cell growth increased from 3.1 to 3.9 g/L, respectively. A maximal 3HAs (but not PHA) content detected by GC was 84.5 wt.% in biomass grown for 60 h in tetradecanoic acid. The PHA contained 43 mol% HTD as a major intracellular monomer. When cells were grown for 60 h in the presence of 6, 10, and 14 g/L tetradecanoic acid, 46, 61, and 63 wt.% HA was obtained, respectively. The highest HTD content in the PHA was 49 mol% obtained from growth in 6 g/L tetradecanoic acid. It appears that the higher the tetradecanoic acid concentration, the more the cell growth. In addition, the higher the tetradecanoic acid concentration, the more the HA contents detected by GC. It is interesting to note that longer incubation time did not lead to more HTD monomer. On the other hand, higher tetradecanoic acid reduced HTD content in PHA (Table 1), as it is evident that HTD content decreased from 49 to 31 mol% when the tetradecanoic acid concentration increased from 6 to 14 g/L.
Beside HTD, other PHA monomers including HHx, HO, HD, and HDD were found in GC studies. HD, as the second major monomer after HTD, makes up 22–27 mol% of PHA. Contents of HO and HDD, which were approximately the same in PHA, remained almost equal at less than 20 mol% regardless of changing incubation time and tetradecanoic acid concentration. While, HHx content was almost constant in all samples at around 3 mol% under all growth conditions (Table 1). These phenomena indicated that the β-oxidation pathway was only partially inactivated in P. putida KTOY06, and this explains the formation of HDD, HD, HO, and HHx that were the products of multiple β-oxidation reactions.
Non-mcl-PHA HTD were found together with mcl-PHA in P. putida KTOY06
With the normal intracellular PHA analysis procedure (Braunegg et al. 1978), it was observed that HTD contents ranged from a minimum of 39 wt.% to a maximum of 67 wt.% (Table 2) in samples obtained from Table 1. However, GC analysis revealed far less HTD contents when the PHA were extracted from lyophilized cells using hot chloroform in pure polymer forms, ranging from 24 to 42 wt.%, indicating the possible existence of free and/or oligo-HTD as intracellular substances. The MRI study showed that the free and/or oligo-HTD were oligo-HTD (data not shown). Subsequently, oligo-HTD average molecular weight was studied by GPC as described in “Materials and methods.” The weight-average molecular weights (M w), the number-average molecular weights (M n), and polydispersity (M w/M n) were 3,295, 2,670, and 1.23, respectively. Oligo-HTD that existed in cells indicated the limited ability of P. putida KTOY06 in polymerizing long-chain-length 3HA. So far, lengths of the carbon atom in mcl-PHA were found to be no more than 18, and the composition of long-chain mcl-HA, such as HD or HDD, has been less than 10 mol% in total PHA. Therefore, it can be assumed that the polymerization reaction does not favor longer-chain monomers, such as the HTD described here.
Characterization of mcl-PHA physical properties
Molecular weights of four mcl-PHA samples with 3HTD monomer contents of 31, 36, 43, and 49 mol%, respectively, were studied using GPC together with two HDD containing mcl-PHA (Table 3). The weight-average molecular weights (M w) were not much in difference, although the number-average molecular weights (M n) and polydispersity (M w/M n) of four HTD containing samples that were similar differ considerably from two the HDD containing mcl-PHA.
The glass transition temperature (T g), melting temperature (T m), and apparent heat of fusion (ΔH m) of all six mcl-PHA samples were not much different as revealed by DSC (Table 3). T g was approximately constant at around −40°C, similar to other mcl-PHA reported (Gross et al. 1989). When HTD contents increased from 31 to 49 mol%, T m increased from 58 to 67°C, compared with 53°C for 15% HDD containing mcl-PHA (Fig. 1). ΔH m also increased from 18 J/g for 15% HDD containing mcl-PHA to 30 J/g for 43% HTD containing PHA. It was completely different from the typical mcl-PHA, which are mostly amorphous (Noda et al. 2005; Gross et al. 1989; Preusting et al. 1990; Witholt and Kessler 1999). The increase in T m in accordance with an increase in the long-chain monomer contents signifies the increase in side chain crystallization, which also leads to dramatical change in the mcl-PHA mechanical properties (Table 3).

DSC thermographs of 3HTD and 3HDD containing mcl-PHA produced by P. putida KTOY06 and KT2442. a 3HTD 31 mol%; b 3HTD 36 mol%; c 3HTD 43 mol%; d 3HTD 49 mol%; e 3HDD 15 mol%; f 3HDD 39 mol%
The thermal stability of polymers is important for their melt processing. The temperature at 5% weight loss changed slightly as 3HTD content increased from 31 to 49 mol% (Data not shown); it was around 253°C. In comparison, polyhydroxybutyrate (PHB) and P(3HB–12 mol% 3HHx) degraded at 226 and 239°C, respectively (Fig. 2). The mcl-PHA thermal decomposition temperature was much higher than their melting temperature.

TGA thermograms for a PHB, b P(3HB–12 mol% 3HHx), and c mcl-PHA containing 31 mol% 3HTD
Tensile strength and Young’s modulus of the mcl-PHA ranged from 3 to 11 MPa and 4 to 75 MPa, respectively. There was not a clear relationship between mechanical property and polymer composition. This might be due to the inconsistent molecule weights that affect the polymer properties.
Discussion
P. putida KT2442 is a well-known mcl-PHA producer (Sun et al. 2006, 2007) with its whole genomic DNA sequenced (Weinel et al. 2002). The strain produces mcl-PHA from a wide range of carbon sources (Kellerhals et al. 2000). When a fatty acid is used as a sole carbon source, β-oxidation plays an important role both for cell growth and in providing intermediates for PHA synthesis (Fiedler et al. 2002).
P. putida KTOY06 was a strain of knockout mutant of P. putida KT2442, which β-oxidation was weakened by knocking out genes of fadA and fadB. In this study, mcl-PHA containing high HTD content was produced for the first time when tetradecanoic acid was utilized as a single carbon source. HTD content in mcl-PHA could be controlled to range from 31 to 49 mol%, depending on growth conditions (Table 1). Because tetradecanoic acid is the immediate precursor of HTD, its major share in the mcl-PHA indicated that tetradecanoic acid was largely turned into HTD because of the weakening of the β-oxidation. One would expect that the HDD will be the second major monomer unit if the PHA polymerase has the same substrate affinity to the HTD and HDD precursors. However, the results showed did not confirm this suggestion (Table 1). It was observed that HTD was the first major monomer fraction and HD the second instead of HDD, suggesting that HD is a substrate more favorable for PHA polymerase in P. putida KTOY06. The remaining β-oxidation activity was able to turn HTD into HDD, HD, HO, and HHx, leading to the formation of the mcl-PHA copolymer (Table 1). It can be estimated that the completely removal of the β-oxidation activity could possibly lead to the formation of homopolymer PHA consisting only of the fatty acids related structures, such as tetradecanoic acid leads to polyHTD, dodecanoic acid forms polyHDD, and so on.
Long-time cultivation (such as 72 h) could lead to the change of the carbon source to nitrogen source ratio. Cells tend to accumulate more PHA with oversupply of the carbon source. Reduction in carbon source concentration because of long-time cultivation favored cell growth but not PHA production. Therefore, it was observed a reduced total HA after 72 h or cultivation (Table 1).
Sample 2 cultivated in 12 g/L tetradecanoic acid concentration produced a maximum of 84% total HA, compared with samples 4, 5, and 6 that showed 46, 61, and 63% total HA production under 6, 10, and 14 g/L tetradecanoic acid, respectively; a total HA content drop from 84 to 63% was observed for 12 and 14 g/L tetradecanoic acid, respectively, demonstrating a two high concentration of tetradecanoic acid (14 g/L) may not be good for PHA production either. Therefore, the cultivation condition of sample 2 to grow KTOY06 in other experiments for high PHA content should be chosen.
Significant amount of oligo-HTD was found in P. putida KTOY06 beside the polymerized mcl-PHA polymers (Table 2). This may imply the limited ability of P. putida KTOY06 in polymerizing long-chain-length 3HA.
High 3HTD content incorporated into mcl-PHA forces the polymers to have side chain crystallization, leading to an HTD content-dependent T m increase: As the higher the HTD content in the mcl-PHA, the higher the T m (Table 3, Fig. 1). This is completely different from typical mcl-PHA, which are mostly amorphous and do not have a clear T m (Noda et al. 2005; Gross et al. 1989; Preusting et al. 1990; Witholt and Kessler 1999). Mechanical properties of mcl-PHA containing more than 30% HDD or HTD showed a remarkable change in mechanical properties; they become normal elastic materials that are not sticky. They were easily precipitated by cold ethanol precipitation from their chloroform solution. Their Young’s modulus are similar or better that even the copolymers PHB–HHx. However, the materials did not show a clear relationship among HTD contents, tensile strength, and stress at break (Table 3). Thermal stability of the mcl-PHA in terms of thermal decomposition temperature was much higher than their melting temperature. At the same time, mcl-PHA are more thermostable than PHB and P(3HB–12 mol%HHx) (Fig. 2). This provides convenience for melt blending with other thermo-resistant polymers.
Two important factors will have effects on the mechanical properties of mcl-PHA. They are the pendant group chain length and the monomer composition in the mcl-PHA. The latter factor namely, the monomer composition, will have more impact on the mechanical properties of mcl-PHA. In this study, mcl-PHA with HTD as the major monomer did show any distinct feature, such as their better tensile strength than the traditional mcl-PHA, which is always sticky and has very weak tensile strength. The long side chain of HTD that can result into side chain crystallization of PHA is the reason of improved mcl-PHA properties.
References
Ashby RD, Foglia TA, Solaiman DK, Liu C, Nunez A, Eggink G (2000) Viscoelastic properties of linseed oil-based medium chain length poly(hydroxyalkanoate) films: effects of epoxidation and curing. Int J Biol Macromol 28:355–361
Atkins TW, Peacock SJ (1996) The incorporation and release of bovine serum albumin from poly-hydroxybutyrate–hydroxyvalerate microcapsules. J Microencapsul 13:709–717
Braunegg G, Sonnleitner B, Lafferty RM (1978) A rapid gas chromatographic method for the determination of poly-b-hydroxybutyric acid in microbial biomass. Eur J Appl Microbiol Biotechnol 6:29–37
Chen GQ, Wu Q (2005) The application of polyhydroxyalkanoates as tissue engineering materials. Biomaterials 26:6565–6578
Chen GQ, Xu J, Wu Q, Zhang ZM, Ho KP (2001a) Synthesis of copolyesters consisting of medium-chain-length β-hydroxyalkanoates by Pseudomonas stutzeri 1317. React Funct Polym 48:107–112
Chen GQ, Zhang G, Park SJ, Lee SY (2001b) Industrial scale production of poly (3-hydroxybutyrate-co-3-hydroxyhexanoate). Appl Microbiol Biotechnol 57:50–55
Curley JM, Hazer B, Lenz RW, Fuller RC (1996) Production of poly(3-hydroxyalkanoates) containing aromatic substituents by Pseudomonas oleovorans. Macromolecules 29:1762–1766
de Roo G, Kellerhals MB, Ren Q, Witholt B, Kessler B (2002) Production of chiral R-3-hydroxyalkanoic acids and R-3-hydroxyalkanoic acid methylesters via hydrolytic degradation of polyhydroxyalkanoate synthesized by pseudomonads. Biotechnol Bioeng 77:717–722
Fiedler S, Steinbüchel A, Rehm, B (2002) The role of the fatty acid β-oxidation multienzyme complex from Pseudomonas oleovorans in polyhydroxyalkanoate biosynthesis: molecular characterization of the fadBA operon from P. oleovorans and of the enoyl-CoA hydratase genes phaJ from Pseudomonas oleovorans and Pseudomonas putida. Arch Microbiol 178:149–160
Fritzsche K, Lenz RW, Fuller RC (1990) An unusual bacterial polyester with a phenyl pendant group. Makromol Chem 191:1957–1965
Gross RA, DeMello C, Lenz RW (1989) Biosynthesis and characterization of poly (β-hydroxyalkanoates) produced by Pseudomonas oleovorans. Macromolecules 22:1106–1115
Gursel I, Hasirci V (1995) Properties and drug release behavior of poly(3-hydroxybutyric acid) and various poly(3-hydroxybutyrate-hydroxyvalerate) copolymer microcapsules. J Microencapsul 12:185–193
Hartmann R, Hany R, Geiger T, Egli T, Witholt B, Zinn M (2004) Tailored biosynthesis of olefinic medium-chain-length poly[(R)-3-hydroxyalkanoates] in Pseudomonas putida GPo1 with improved thermal properties. Macromolecules 37:6780–6785
Hoffmann N, Rehm BH (2004) Regulation of polyhydroxyalkanoate biosynthesis in Pseudomonas putida and Pseudomonas aeruginosa. FEMS Microbiol Lett 237:1–7
Holmes PA (1985) Application of PHB—a microbially produced biodegradable thermoplastic. Phys Technol 16:32–36
Jung K, Hany R, Rentsch D, Storni T, Egli T, Witholt B (2000) Characterization of new bacterial copolyesters containing 3-hydroxy-oxoalkanoates and acetoxy-3-hydroxyalkanoates. Macromolecules 33:8571–8575
Kellerhals MB, Kessler B, Witholt B (2000) Renewable long-chain fatty acids for production of biodegradable medium-chain-length polyhydroxyalkanoates (mcl-PHAs) at laboratory and pilot plant scales. Macromolecules 33:4690–4698
Kim DY, Kim YB, Rhee YH (1998) Bacterial poly(3-hydroxyalkanoates) bearing carbon–carbon triple bonds. Macromolecules 31:4760–4763
Kim YB, Kim DY, Rhee YH (1999) PHAs produced by Pseudomonas putida and Pseudomonas oleovorans grown with n-alkanoic acids containing aromatic groups. Macromolecules 32:6058–6064
Lageveen RG, Huisman GW, Preusting H, Ketelaar P, Eggink G, Witholt B (1988) Formation of polyesters by Pseudomonas oleovorans: effect of substrates on formation and poly-(R)-3-hydroxyalkanoates. Appl Environ Microbiol 54:2924–2932
Luo RC, Chen JY, Zhang L, Chen JC, Chen GQ (2006) Polyhydroxyalkanoates copolyesters produced by Ralstonia eutropha PHB-4 harboring a low-substrate-specificity PHA synthase PhaC2Ps from Pseudomonas stutzeri 1317. Biochem Eng J 31:218–225
Nakamura S, Kunioka M, Doi Y (1991) Biosynthesis and characterization of bacterial poly(3hydroxybutyrate-co-3-hydroxy- propionate). Macromol Rep 28:15–24
Noda I, Satkowski MM, Dowrey AE, Marcott C (2004) Polymer alloys of Nodax copolymers and poly(lactic acid). Macromol Biosci 15:269–275
Noda I, Green PR, Satkowski MM, Schechtman LA (2005) Preparation and properties of a novel class of polyhydroxyalkanoate copolymers. Biomacromolecules 6:580–586
Ouyang SP, Liu Q, Fang L, Chen GQ (2007) Construction of pha-operon-defined knockout mutants of Pseudomonas putida KT2442 and their applications in poly(hydroxyalkanoate) production. Macromol Biosci 7:227–233
Preusting H, Nijenhuis A, Witholt B (1990) Physical characteristics of poly(3-hydroxyalkanoates) and poly (3-hydroxyalkenoates) produced by Pseudomonas oleovorans. Macromolecules 23:4220–4224
Schäfer A, Tauch A, Jäger, W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizble multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73
Sim SJ, Snell KD, Hogan SA, Stubbe J, Rha CK, Sinskey A (1997) PHA synthase activity controls the molecular weight and polydispersity of polyhydroxybutyrate in vivo. Nat Biotechnol 15:63–67
Simon R, Priefer U, Pühler A (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology 1:784–791
Steinbüchel A, Valentin HE (1995) Diversity of bacterial polyhydroxyalkanoic acids. FEMS Microbiol Lett 128:219–228
Sun ZY, Ramsay JA, Guay M, Ramsay, BA (2006) Automated feeding strategies for high-cell-density fed-batch cultivation of Pseudomonas putida KT2440. Appl Microbiol Biotechnol 71:423–431
Sun Z, Ramsay JA, Guay M, Ramsay BA (2007) Carbon-limited fed-batch production of medium-chain-length polyhydroxyalkanoates from nonanoic acid by Pseudomonas putida KT2440. Appl Microbiol Biotechnol 74:69–77
Taguchi S, Doi Y (2004) Evolution of polyhydroxyalkanoate (PHA) production system by “enzyme evolution”: successful case studies of directed evolution. Macromol Biosci 4:145–156
Weinel C, Nelson KE, Tummler B (2002) Global features of the Pseudomonas putida KT2440 genome sequence. Environ Microbiol 4:809–818
Witholt B, Kessler B (1999) Perspectives of medium chain length poly(hydroxyalkanoates), a versatile set of bacterial bioplastics. Curr Opin Biotechnol 10:279–285
Xu SL, Luo RC, Wu LP, Xu KT, Chen GQ (2006) Blending and characterizations of microbial poly(3-hydroxybutyrate) (PHB) with dendrimers. J Appl Polym Sci 102:3782–3790
Zheng Z, Bei FF, Deng Y, Tian HL, Chen GQ (2005) Effects of crystallization of polyhydroxyalkanoate blend on surface physicochemical properties and resulting biocompatibility for chondrocytes. Biomaterials 26:3537–3548
Zinn M, Witholt B, Egli T (2001) Occurrence, synthesis and medical application of bacterial polyhydroxyalkanoate. Adv Drug Deliv Rev 53:5–21
Acknowledgments
The strain P. putida KTOY06 was kindly donated by Dr. Ouyang SP. We are grateful to Li Ka Shing Foundation and State High Tech Project 863 Grant no. 2006AA02Z242 for financial support. We also like to thank Mr. Luo RC and Wu LP in the Multidisciplinary Research Center of Shantou University for their DSC and MRI studies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, W., Chen, GQ. Production and characterization of medium-chain-length polyhydroxyalkanoate with high 3-hydroxytetradecanoate monomer content by fadB and fadA knockout mutant of Pseudomonas putida KT2442. Appl Microbiol Biotechnol 76, 1153–1159 (2007). https://doi.org/10.1007/s00253-007-1092-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-1092-8