
Abstract
Using a DNA-mediated transformation technique, a molecular breeding approach to isolate Pleurotus ostreatus strains with enhanced productivity of its versatile peroxidase MnP2 was conducted. A recombinant mnp2 construct under the control of P. ostreatus sdi1 expression signals was introduced into the wild-type P. ostreatus strain by cotransformation with a carboxin-resistant marker plasmid. A total of 32 transformants containing the recombinant mnp2 sequence were isolated in a screening with specific amplification by PCR. Productivity of MnP2 in the recombinants was evaluated by the decolorization ability of Poly R-478 on agar plates in the absence of Mn2+. Recombinant P. ostreatus strains with elevated manganese peroxidase (MnP) productivity were successfully isolated. One of the recombinants, TM2-10, was demonstrated to secrete recombinant MnP2 predominantly on a synthetic medium containing 15 mM ammonium oxalate, which was confirmed by reverse transcription PCR (RT-PCR) and isozyme profile analysis using anion-exchange chromatography. The benzo[a]pyrene-removing activity by fungal treatment was also analyzed using the isolated recombinant strains.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite its property as the most abundant aromatic polymer on the earth, plant cell wall lignin is resistant to biological attack by most microorganisms due to its heterogeneity and bulky structure (Sarkanen 1971). It is noteworthy, however, that white rot basidiomycetes degrade lignin and also recalcitrant environmental pollutants to carbon dioxide and water. Therefore, they have been a focus of research interest in terms of effective utilization of plant biomass resources and application to bioremediation of polluted environments containing various hazardous compounds (Kirk and Farrell 1987; Gold and Alic1993; Cullen 1997; Duran and Esposito 2000).
Several Pleurotus and Bjerkandera species are known to have distinct characteristic from other white rot basidiomycetes: they secrete peroxidases with an extraordinarily wide range of substrate specificity (Mester and Field 1998; Camarero et al. 1999a,b; Kamitsuji et al. 2005b). These peroxidases are called versatile, or “hybrid,” peroxidases because they have properties similar to those of the two popular fungal peroxidase families, namely, lignin peroxidases (LiPs) and manganese peroxidases (MnPs). The versatile peroxidases (VPs) have an Mn2+-binding site and efficiently oxidize Mn2+ to Mn3+, just like what typical MnPs do. Also, like LiPs, they directly oxidize phenolic and nonphenolic compounds including veratryl alcohol (Camarero et al. 1999a,b; Ha et al. 2001). Among the VPs, Pleurotus ostreatus MnP2 was demonstrated to be unique since it can directly oxidize high-molecular-weight compounds such as Poly R-478 and RNaseA (Kamitsuji et al. 2005b). We demonstrated that a tryptophan residue conserved in all the VPs and LiPs was essential for the oxidation of veratryl alcohol and Poly R-478, but not of Mn2+, by MnP2, based on a chemical modification with N-bromosuccimide (Kamitsuji et al. 2005b). It is of interest to elucidate the molecular mechanism by which MnP2 oxidizes bulky substrates directly. MnP3, another major extracellular peroxidase in P. ostreatus, has less reactivity with Poly R-478, and it does not decolorize the dye in the absence of Mn2+ (Kamitsuji et al. 2004). Anion-exchange chromatography and reverse transcription PCR (RT-PCR) analyses in liquid culture using synthetic media composed of defined chemicals demonstrated that the expression of MnP2 and MnP3 was differentially regulated (Kamitsuji et al. 2005a, unpublished result), suggesting distinct physiological functions of the two MnP isozymes with different catalytic functions.
In the present study, a molecular breeding approach to isolate P. ostreatus strains with elevated MnP2 productivity was carried out by introducing a recombinant mnp2 under the control of homologous expression signals.
Materials and methods
Strains and plasmids
P. ostreatus dikaryotic strain #261 (ATCC 66376) was used as the host strain throughout this study. It was grown on 3.9% potato dextrose agar (Nissui Co., Tokyo, Japan) for maintenance and preculture. For liquid culture, four pieces of mycelial mats (7 mm ∅) grown on 3.9% potato dextrose agar (Nissui Co.) plates for 6 days were cut off and inoculated onto 30 ml of the synthetic medium in a 300-ml Erlenmeyer flask. In all experiments, P. ostreatus strains were grown at 28°C in darkness.
Escherichia coli JM109 (recA1 endA1 gyrA96 thi hsdR17 supE44 relA1 Δ(lac-proAB)/F′ [traD36 proAB +lacIq lacZΔM15]) was used as a host bacterium for standard recombinant DNA constructions and was grown on a Luria–Bertani medium. Plasmid pTM1, a pGEM-T-based (Promega) plasmid containing a carboxin-resistant marker gene (Honda et al. 2000), was used for the transformation of P. ostreatus.
A recombinant plasmid to express mnp2 was constructed as follows: a 1,906-bp mnp2 coding sequence was amplified from P. ostreatus genomic DNA, using primer molecules, MnP2-NcoI (5′-TCACTGGCCATGGCTTTCGCT-3′), which adapts a NcoI site at the start codon, and MnP2-KpnI (5′-TAGGTACCGGGGCATTTACGAAGG-3′), which adapts a KpnI site after the termination codon. Amplification conditions were 98°C for 30 s, 52°C for 1 min, and 72°C for 1 min. This cycle was repeated 30 times. Then the amplified fragment was inserted onto an expression plasmid pIpMc (Irie et al. 2001) with NcoI and KpnI to produce the plasmid pIpM2g (Fig. 1). It contained the coding sequence of mnp2 under the control of promoter and terminator sequences from the P. ostreatus sdi1 gene.

Physical map of the MnP2 expression plasmid pIpM2g. Shaded arrow represents the location and transcriptional direction of the genomic sequence encoding MnP2. Closed arrows indicate the promoter and terminator sequences from the P. ostreatus sdi1 gene. Thin line and dotted area indicate the pGEM-T vector and its Amp R marker sequence, respectively. The location of the primer molecules used for detection of the recombinant mnp2 sequence are indicated by thin arrows
Isolation of the P. ostreatus recombinants
The expression plasmid pIpM2g was introduced into the wild-type P. ostreatus by cotransformation with pTM1 by the PEG/CaCl2 method described previously (Irie et al. 2001), except for Novezyme234 that was substituted with a protoplasting enzyme purchased from Mushroom Experimental Station (Horst, The Netherlands). DNA-treated protoplasts were screened for carboxin resistance on a regenerating plate containing 2 μg/ml carboxin. Isolated colonies were transferred onto fresh 3.9% potato dextrose agar (Nissui Co.) plates containing the same concentration of carboxin.
Incorporation of the recombinant mnp2 was checked with specific amplification in a PCR experiment using primers M13 (5′-GTTTTCCCAGTCACGAC-3′) and MnP2R (5′-GGCGGTCCCTTGCAACATTGT-3′; Fig. 1), and genomic DNA was extracted from the isolates as a template. The amplification conditions were 98°C for 1 min, followed by 25 cycles at 98°C for 30 s, 57°C for 1 min, and 72°C for 1 min. After amplification, a 10-μl aliquot of each PCR product was analyzed by agarose gel electrophoresis. A 2,509-bp fragment was amplified from the strains containing the recombinant mnp2 sequence.
Screening of recombinants with elevated MnP2 activity
P. ostreatus MnP2 oxidizes and decolorizes Poly R-478 in the absence of Mn2+ (Kamitsuji et al. 2005b). Elevated Poly R-478 decolorizing activity should be observed when the production of MnP2 is enhanced in recombinants. In this context, to assess the production of the recombinant MnP2 by each transformant, we compared decolorizing activity on a specific GY medium: 1% glucose, 0.02% yeast extract, 1% Kirk's salts (without MnSO4; Tien and Kirk 1988), 2% agar, and 20 mM Na–phthalate buffer (pH 4.5), in addition to 0.1% Poly R-478. One piece of mycelial mat of each transformant (7 mm ∅) was inoculated at the center of an agar plate and was incubated for 10 days. The productivity of MnP2 in each transformant was evaluated by a change in the color of Poly R-478.
MnP productivity of the selected strains was measured in liquid culture conditions using a synthetic medium containing 3% glucose, 1% Kirk's salts (without MnSO4; Tien and Kirk 1988), 270 mM MnSO4, and 20 mM Na–phthalate buffer (pH 4.5), in addition to 1 mM ammonium oxalate as a nitrogen source (Kamitsuji et al. 2005a). Three replicate cultures for each strain were kept stationary for 19 days. Then, MnP activity in the culture filtrate was measured as Mn2+-dependent guaiacol-oxidizing activity by the method described previously (Kofujita et al. 1991; Kamitsuji et al. 2004).
RT-PCR analyses of recombinant mnp2
Wild type and one of the recombinants, TM2-10, were inoculated on a synthetic medium containing 3% glucose, 1% Kirk's salts (without MnSO4; Tien and Kirk 1988), 270 mM MnSO4, and 20 mM Na–phthalate buffer (pH 4.5), in addition to 15 mM ammonium oxalate as a nitrogen source (Kamitsuji et al. 2005a). Three replicate cultures for each strain were kept stationary for 19 days. The MnP activity in the culture filtrate was measured on days 9, 14, and 19.
RT-PCR amplification of the recombinant and endogenous mnp2 transcripts was performed using total RNA from mycelia on day 12. Transcript abundance of the β-tubulin gene was also monitored as a control. Extraction of RNA and PCR conditions basically followed those described previously (Kamitsuji et al. 2005a). The primers to detect transcripts from each gene are described in Table 1. The reverse transcription was conducted with 70 ng of total RNA and 10 ng of oligo(dT)12–18 primer at 42°C for 30 min, followed by 95°C for 5 min to inactivate the reverse transcriptase. Then, 12.5 pmol of each specific primer was added for the PCR. The amplification program was as follows: 95°C for 5 min, followed by 30 cycles at 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min. After amplification, a 10-μl aliquot of each PCR product was analyzed by agarose gel electrophoresis.
Analysis of isozyme profiles of the extracellular MnP
The isozyme profile of MnP in the culture filtrate on day 14 using the synthetic medium containing 15 mM ammonium oxalate was analyzed. The culture filtrate was separated from mycelia by filtration with gauze. The culture filtrate from five flasks, approximately 125 ml, was concentrated to 300 μl by ultrafiltration using a Centriprep-30 microconcentrator (Millipore). Then, 100 μl of concentrate was subjected to anion-exchange chromatography on a Mono-Q (5/5) column (Amersham Biosciences). The isozymes were eluted with a linear gradient from 70 to 175 mM NaCl in 10 mM Na–succinate buffer (pH 5.0) over 120 min at a flow rate of 0.5 ml/min. The elution of the protein was monitored by measuring the absorbance at 407 nm specific to the heme protein. The elution profile of each sample was compared to that of standard MnP2 and MnP3 isozymes purified from culture filtrates of the wild-type strain (Kamitsuji et al. 2004).
Removal of benzo[a]pyrene by fungal treatment
The ability to remove benzo[a]pyrene from culture medium in representative strains was monitored as follows. Wild type and TM2-10, TM2-30, and TM2-31 were inoculated on a synthetic medium containing 1 mM ammonium oxalate (described above). Three replicate cultures for each strain were kept stationary for 16 days. Benzo[a]pyrene (25 ppm) was added to 8-day-old cultures, and the concentration of the remaining benzo[a]pyrene on day 16 was measured as follows. At first, 500 μl of the culture filtrate was removed for the measurement of MnP activity in the culture filtrate. Then, 30-ml tetrahydrofuran was added to each flask, and the remaining benzo[a]pyrene was extracted by shaking at 140 rpm for 24 h. After filtration in a glass filter, the filtrate was made up to 60 ml with fresh tetrahydrofuran and subjected to ET 150/4 Nucleosil 100-5 C18 PAH column (Macherey-Nagel GmbH & Co. KG) in buffer A (50% methanol/25% acetonitrile/25% water). The compounds were eluted with a linear gradient from 0 to 100% of buffer B (93% acetonitrile/7% tetrahydrofuran) over 15 min at a flow rate of 1 ml/min. The elution of the compound was monitored by measuring the absorbance at 260 nm specific to the aromatic compounds. In a control experiment using autoclaved mycelium, it was demonstrated that 96% of initial benzo[a]pyrene was recovered by the extraction procedure described above.
Results
Isolation of genetically modified P. ostreatus
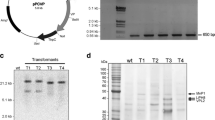
To obtain genetically modified P. ostreatus strains with elevated MnP2 productivity, the expression plasmid pIpM2g (Fig. 1) containing the coding sequence of mnp2 between the P. ostreatus sdi1 promoter and terminator sequences was constructed. sdi1 encodes iron–sulfur protein subunit of succinate dehydrogenase in the respiratory chain (Irie et al. 1998). In this context, its promoter is supposed to direct the transcription of the resulting recombinant gene with lower background level of endogenous ligninolytic enzymes compared to other homologous promoter sequences from lacs (Palmieri et al. 2000) or mnps (Cohen et al. 2001). Then, pIpM2g was introduced into the wild-type P. ostreatus strain by cotransformation with a carboxin-resistant marker plasmid pTM1 simultaneously. Protoplasts (108) prepared from vegetative mycelia were treated with a mixture of 5-μg pIpM2g and 1-μg pTM1 DNA by the PEG/CaCl2 method (see “Materials and methods”), and cells incorporating exogenous plasmids were screened on a carboxin-containing agar plate. In four repeated experiments, a total of 34 drug-resistant colonies were isolated as transformants and were subjected to PCR detection of the recombinant mnp2 sequence. PCR amplification was performed using the specific primers M13 and mnp2R (Fig. 1), and the reaction mixture was subjected to agarose gel electrophoresis (Fig. 2). Specific amplification of the recombinant mnp2 was observed for the transformant TM2-10 (lane 1), whereas no bands were observed for TM2-11 (lane 2) and the wild-type control (lane 3). These results suggested that TM2-10 is a cotransformant containing both the carboxin-resistant and recombinant mnp2 sequences, whereas TM2-11 contains only the pTM1 sequence. In this way, it was concluded that 32 transformants, out of 34 carboxin-resistant transformants, contained the recombinant mnp2 sequence.

PCR detection of the recombinant mnp2 sequence in the transformants. Incorporation of the recombinant mnp2 was checked with specific amplification in a PCR experiment followed by 0.7% agarose gel electrophoresis. The 2,509-bp fragment expected for the recombinant mnp2 sequence is indicated by an arrow. Samples are as follows: lane 1 TM2-10, lane 2 TM2-11, lane 3 wild-type host strain, lane 4 pTM1, lane 5 pIpM2g as a positive control, lane M EcoT14I-digested lambda DNA as a molecular standard. TM2-10 and TM2-11 are the representatives of carboxin-resistant transformants
Productivity of MnP2 in the recombinant strains
Unlike other typical MnPs, MnP2 decomposes and decolorizes the polymeric azo dye Poly R-478 directly in the absence of Mn2+ (Kamitsuji et al. 2005b). It was expected that the Mn2+-independent decolorizing activity for Poly R-478 would be enhanced in recombinant strains with elevated MnP2 productivity. We evaluated MnP2 productivity in all the 32 recombinants by monitoring Poly R-478 decolorizing activity on GY agar plates containing no Mn2+. Typical results of the representatives are shown in Fig. 3. In the wild-type strain, decolorization was observed only at a region close to the center of the plate where the inoculum had been placed. However, the decolorized area extended to the surrounding region in some recombinants (TM2-10, TM2-30, and TM2-31). Decolorizing activities as high as, and even less than, those of the wild-type strain were observed in TM2-16 and TM2-2, respectively. Among 32 recombinants, 13 were found to be elevated, 16 unchanged, and 3 with less Poly R-478 decolorizing activity, compared to the wild-type strain.

Poly R-478 decolorizing activity of the recombinants in the absence of Mn2+. To assess the production of recombinant MnP2 by the transformants, we compared their decolorizing activity on a Mn2+-deficient GY agar plate containing 0.1% Poly R-478. The mycelial mat of each transformant was inoculated at the center of the plate and incubated for 10 days. The productivity of MnP2 in each transformant was evaluated by a change in the color of Poly R-478
We measured the MnP activity in the culture filtrate of the recombinants on a synthetic medium containing 1 mM ammonium oxalate as a nitrogen source on day 19. As with the case of the GY medium, we previously demonstrated that endogenous MnP2 was predominantly produced by the wild-type P. ostreatus on this medium (Kamitsuji et al. 2005a). MnP activities of the wild type and TM2-2, TM2-10, TM2-16, TM2-30, and TM2-31 strains were 160, 80, 370, 110, 260, and 295 mU/ml, respectively. From these results, it was demonstrated that the Poly R-478 decolorizing ability on the GY agar plate was consistent with the MnP productivity of the analyzed strains in the liquid culture. It was also suggested that MnP productivity was enhanced in some of the recombinants by introducing the recombinant mnp2 gene. The transformants varied in their MnP productivity, as is often the case with recombinant gene expression in basidiomycetous fungi, where integrative transformation occurs, mostly, at multiple sites in the chromosome (Mayfield et al. 1994; Irie et al. 2001; Kajita et al. 2004).
Confirmation of recombinant MnP2 expression in TM2-10
To prove that the elevated MnP and Poly R-478 decolorizing activities were caused by expression of the recombinant MnP2, we conducted a time course assay of MnP activity on a synthetic liquid medium containing 15 mM ammonium oxalate (Fig. 4). We previously demonstrated that the wild-type P. ostreatus produces a small amount of MnP3 and no MnP2 in the above synthetic media containing high concentration of nitrogen source (Kamitsuji et al. 2005a). As shown in Fig. 4a, the wild-type strain secreted a small amount of MnP in this condition, which is consistent with the former results (Kamitsuji et al. 2005a). However, as the transformant TM2-10 produced a significant amount of MnP activity in the culture filtrate, it showed a 6.6-fold more MnP activity than the wild-type control on day 19. Analysis of the isozyme profile of the culture filtrate on day 14 was performed with anion-exchange chromatography (Fig. 4b). A small amount of MnP3 was detected for both the wild-type and recombinant strains; however, the expression of MnP2 was observed only for TM2-10. These results suggested that endogenous mnp2 was not expressed in this condition, and that MnP2 was produced by the expression of recombinant mnp2 in TM2-10.

Production of MnP2 on synthetic media containing 15 mM ammonium oxalate. a Time course of extracellular MnP activity. b Analysis of MnP isozymes in the culture filtrate on day 14 by anion-exchange chromatography. c RT-PCR analysis of transcript levels for recombinant mnp2 (rmnp2), endogenous mnp2, and β-tubulin (β-tub) on day 12
To confirm that the recombinant, but no endogenous, mnp2 was expressed in TM2-10, evaluation of transcripts from mnp2 genes was conducted using RT-PCR techniques (Fig. 4c). Specific primers corresponding to 5′ and 3′ untranslated region sequences (Table 1) were designed to detect transcripts from endogenous and recombinant mnp2 separately. Significant amounts of the recombinant mnp2 transcript were observed in TM2-10, but not in the wild-type strain. Moreover, transcript from endogenous mnp2 was not detected, whereas the levels of the β-tubulin transcript were similar in both strains. These findings clearly demonstrated that the elevated MnP productivity was caused by the introduced recombinant mnp2 in the transformant.
Removal of benzo[a]pyrene by fungal treatment
It was reported that polycyclic aromatic hydrocarbons (PAHs) were degraded by a crude MnP fraction from Nematoloma frowardii (Sack et al. 1997) and purified MnP from a litter-decomposing basidiomycetes Stropharia coronilla (Steffen et al. 2003). It was expected that the degradation activity of PAHs would be enhanced in the P. ostreatus recombinant strains. In this context, the removal of benzo[a]pyrene by fungal treatment with the wild-type and recombinant strains, TM2-10, TM2-30, and TM2-31, was carried out in a liquid culture using a synthetic medium containing 1 mM ammonium oxalate. Then, 25 ppm benzo[a]pyrene was added to 8-day-old cultures, and the concentration of the remaining benzo[a]pyrene after incubation for 8 days was measured by high-performance liquid chromatography. In each recombinant, more than twofold benzo[a]pyrene was removed compared to the wild-type strain (Fig. 5). It was also demonstrated that three recombinants produced elevated MnP activity in the culture filtrate compared with the wild-type strain. These results strongly suggested that the recombinant strains are superior at removing recalcitrant pollutants.

Fungal treatment of benzo[a]pyrene and extracellular MnP activity. Ability to remove benzo[a]pyrene was monitored after fungal treatment with wild type and TM2-10, TM2-30, and TM2-31 strains for 8 days. Removal of benzo[a]pyrene and extracellular MnP activity are indicated by a shaded bar and diamond, respectively
Discussion
To date, several expression systems for a versatile peroxidase have been reported. Pleurotus eryngii VPL2 was expressed in an expression system using E. coli (Pérez-Boada et al.2002). Although the heterologously expressed VPL2 was reported to have kinetic properties similar to that of the fungal enzyme, it was not glycosylated, and an inefficient artificial folding process was required to achieve enzyme activity. VPL2 was also expressed in Aspergillus nidulans (Ruiz-Dueñas et al. 1999), but the yield was low. A homologous expression system has advantages over a heterologous one in terms of the production of an enzyme of interest in the native and active forms. It also allows the analysis of the physiological role of the gene product in the host organism by overexpression. Moreover, not only the expressed product, but also the recombinant strains themselves can be efficient biocatalysts in various industrial processes, including the decomposition of lignin and recalcitrant environmental pollutants.
We report here the molecular breeding of P. ostreatus transformants by introducing recombinant mnp2 constructs under the control of sdi1 expression signals. Strains with elevated MnP2 productivity were successfully isolated through a plate assay of Poly R-478 decolorizing activity in the absence of Mn2+. The expression of the recombinant mnp2 was confirmed by the predominant accumulation of the specific transcripts and also by the significant MnP2 productivity of the transformant TM2-10 in a high nitrogen synthetic medium. The expression system of the recombinant MnP2 will allow us to analyze the contribution of each amino acid residue to the activity of the enzyme using in vitro site-directed mutagenesis and elucidate the molecular structures responsible for its catalytic functions.
PAHs are a series of widespread environmental pollutants with condensed aromatic ring structures. PAHs, especially those with more than four rings, are known to be recalcitrant and resistant to microbial degradation (Cerniglia 1992). It was also reported that higher-molecular-weight PAHs were degraded by mycelial treatment with various white rot fungi (Bumpus 1989; Hammel et al. 1991; Wolter et al. 1997; Pickard et al. 1999). PAHs were degraded by a crude MnP fraction from N. frowardii (Sack et al. 1997). It was also reported that benzo[a]pyrene was oxidized by purified MnP from a litter-decomposing basidiomycetes S. coronilla (Steffen et al. 2003). Furthermore, in Phanerochaete chrysosporium, the addition of Mn2+ was beneficial, but not prerequisite, for the oxidation of PAHs (Zheng and Obbard 2002). On the other hand, the oxidation of PAHs was also reported using purified LiP from Ph. chrysosporium (Vazquez-Duhalt et al. 1994) and laccase from Trametes species with appropriate mediators (Collins et al. 1996; Johannes et al. 1996; Bohmer et al. 1998; Majcherczyk et al. 1998). Moreover, the contribution of P450 monooxygenase to PAH degradation was suggested by using specific inhibitors (Bezalel et al. 1996) and purified enzymes (Maspahy et al. 1999). It seems that white rot fungi have various pathways degrading PAHs, some of which seem to be part of the polymer decomposition system of the fungi. In this study, it was clearly demonstrated that the transformants removed benzo[a]pyrene more efficiently than the wild-type P. ostreatus (Fig. 5), suggesting that the overexpression of MnP2 is one of the effective ways to activate PAH degradation by the fungus. Although further works are required to determine the optimal condition for mycelial treatment of benzo[a]pyrene, it is expected that these recombinants would serve as an efficient biocatalyst to transform other recalcitrant compounds.
Overexpression of MnP2 may increase the possibilities of the enzyme to be utilized in various bioprocesses, e.g., as an immobilized enzyme in bioreactors. The productivity of the recombinant MnP2 in the present system was not high enough to meet the requirements for industrial applications. Development of a cultivation system for efficient production of MnP2 by the recombinant strains is now in progress.
References
Bezalel L, Hadar Y, Fu PP, Freeman JP, Cerniglia CE (1996) Metabolism of phenanthrene by the white rot fungus Pleurotus ostreatus. Appl Environ Microbiol 62:2547–2553
Bohmer S, Messner K, Srebotnik E (1998) Oxidation of phenanthrene by fungal laccase in the presence of 1-hydroxybenzotriazole and unsaturated lipids. Biochem Biophys Res Commun 244:233–238
Bumpus JA (1989) Biodegradation of polycyclic hydrocarbons by Phanerochaete chrysosporium. Appl Environ Microbiol 55:154–158
Camarero L, Martínez MJ, Martínez AT (1999a) A search for ligninolytic peroxidases in the fungus Pleurotus eryngii involving alpha-keto-gamma-thiomethylbutyric acid and lignin model dimers. Appl Environ Microbiol 65:916–922
Camarero S, Sarkar S, Ruiz-Dueñas FJ, Martínez MJ, Martínez AT (1999b) Description of a versatile peroxidase involved in the natural degradation of lignin that has both manganese peroxidase and lignin peroxidase substrate interaction sites. J Biol Chem 274:10324–10330
Cerniglia CE (1992) Biodegradation of polycyclic aromatic hydrocarbons. Biodegradation 3:351–368
Cohen R, Yarden O, Hadar Y (2001) Transcript and activity levels of different Pleurotus ostreatus peroxidases are differentially affected by Mn2+. Environ Microbiol 3:312–322
Collins P, Kotterman MJJ, Field FA, Dobson ADW (1996) Oxidation of anthracene and benzo[a]pyrene by laccase from Trametes versicolor. Appl Environ Microbiol 162:4563–4567
Cullen D (1997) Recent advances on the molecular genetics of ligninolytic fungi. J Biotechnol 53:273–289
Duran N, Esposito E (2000) Potential applications of oxidative enzymes and phenoloxidase-like compounds in wastewater and soil treatment: a review. Appl Catal B Environ 28:83–99
Giardina P, Palmieri G, Fontanella B, Rivieccio V, Sannia G (2000) Manganese peroxidase isoenzymes produced by Pleurotus ostreatus grown on wood sawdust. Arch Biochem Biophys 376:171–179
Gold MH, Alic M (1993) Molecular biology of the lignin-degrading basidiomycete Phanerochaete chrysosporium. Microbiol Rev 57:605–622
Ha H-C, Honda Y, Watanabe T, Kuwahara M (2001) Production of manganese peroxidase peroxidase by pellet culture of the lignin-degrading basidiomycete, Pleurotus ostreatus. Appl Microbiol Biotechnol 55:704–711
Hammel KE, Green B, Gai WZ (1991) Ring fission of anthracene by a eukaryote. Proc Natl Acad Sci U S A 88:10605–10608
Honda Y, Matsuyama T, Irie T, Watanabe T, Kuwahara M (2000) Carboxin resistance transformation of the homobasidiomycete fungus Pleurotus ostreatus. Curr Genet 37:209–212
Irie T, Honda Y, Matsuyama T, Watanabe T, Kuwahara M (1998) Cloning and characterization of the gene encoding the iron–sulfur protein of succinate dehydrogenase from Pleurotus ostreatus. Biochim Biophys Acta 1396:27–31
Irie T, Honda Y, Watanabe T, Kuwahara M (2001) Homologous expression of recombinant manganese peroxidase genes in ligninolytic fungus Pleurotus ostreatus. Appl Microbiol Biotechnol 55:566–570
Johannes C, Majcherczyk A, Hüttermann A (1996) Degradation of anthracene by laccase of Trametes versicolor in the presence of different mediator compounds. Appl Microbiol Biotechnol 46:313–317
Kajita S, Sugawara S, Miyazaki Y, Nakamura M, Katayama Y, Shishido K, Iimura Y (2004) Overproduction of recombinant laccase using a homologous expression system in Coriolus versicolor. Appl Microbiol Biotechnol 66:194–199
Kamitsuji H, Honda Y, Watanabe T, Kuwahara M (2004) Production and induction of manganese peroxidase isozymes in a white-rot fungus Pleurotus ostreatus. Appl Microbiol Biotechnol 65:287–294
Kamitsuji H, Honda Y, Watanabe T, Kuwahara M (2005a) Mn2+ is dispensable for the production of active MnP2 by Pleurotus ostreatus. Biochem Biophys Res Commun 327:871–876
Kamitsuji H, Watanabe T, Honda Y, Kuwahara M (2005b) Direct oxidation of polymeric substrates by multifunctional manganese peroxidase isoenzyme from Pleurotus ostreatus without redox mediators. Biochem J 386:387–393
Kirk TK, Farrell RL (1987) Enzymatic “combustion”: the microbial degradation of lignin. Annu Rev Microbiol 41:465–505
Kofujita H, Asada Y, Kuwahara M (1991) Arkil-aryl cleavage of phenolic β-O-4 lignin substructure model compound by Mn-peroxidase isolated from Pleurotus ostreatus. Mokuzai Gakkaishi 37:555–561
Majcherczyk A, Johannes C, Hüttermann A (1998) Oxidation of polycyclic aromatic hydrocarbons (PAH) by laccase of Trametes versicolor. Enzyme Microb Technol 22:335–341
Maspahy S, Lamb DC, Kelly SL (1999) Purification and characterization of a benzo[a]pyrene hydroxylase from Pleurotus pulmonarius. Biochem Biophys Res Commun 266:326–329
Mayfield MB, Kishi K, Alic M, Gold MH (1994) Homologous expression of recombinant manganese peroxidase in Phanerochaete chrysosporium. Appl Environ Microbiol 60:4303–4309
Mester T, Field JA (1998) Characterization of a novel manganese peroxidase-lignin peroxidase hybrid isozyme produced by Bjerkandera species strain BOS55 in the absence of manganese. J Biol Chem 273:15412–15417
Palmieri G, Giardina P, Bianco C, Fontanella B, Sannia G (2000) Pcupper induction of laccase isozymes in the ligninolytic fungus Pleurotus ostreatus. Appl Environ Microbiol 66:920–924
Pérez-Boada M, Doyle WA, Ruiz-Dueñas FJ, Martínez MJ, Martínez AT, Smith AT (2002) Expression of Pleurotus eryngii versatile peroxidase in Escherichia coli and optimization of in vitro folding. Enzyme Microb Technol 30:518–524
Pickard MA, Roman R, Tinoco R, Vazquez-Duhalt R (1999) Polycyclic aromatic hydrocarbon metabolism by white rot fungi and oxidation by Coriolopsis gallica UAMH 8260 laccase. Appl Environ Microbiol 65:3805–3809
Ruiz-Dueñas FJ, Martínez MJ, Martínez AT (1999) Heterologous expression of Pleurotus eryngii peroxidase confirms its ability to oxidize Mn2+ and different aromatic substrates. Appl Environ Microbiol 65:4705–4707
Sack U, Hofrichter M, Fritsche W (1997) Degradation of polycyclic aromatic hydrocarbons by manganese peroxidase of Nematoloma frowardii. FEMS Microbiol Lett 152:227–234
Sarkanen KV (1971) Lignin: occurrence, formation, structure and reactions. Wiley, New York
Steffen KT, Hatakka A, Hofrichter M (2003) Degradation of benzo[a]pyrene by the litter-decomposing basidiomycete Stropharia coronilla: role of manganese peroxidase. Appl Environ Microbiol 69:3957–3964
Tien M, Kirk TK (1988) Lignin peroxidase of Phanerochaete chrysosporium. Methods Enzymol 161:238–249
Vazquez-Duhalt R, Westlake DW, Fedorak PM (1994) Lignin peroxidase oxidation of aromatic compounds in systems containing organic solvents. Appl Environ Microbiol 60:459–466
Wolter M, Zadrazil F, Rrtens R, Bahadir M (1997) Degradation of eight highly condensed polycyclic aromatic hydrocarbons by Pleurotus sp. Florida in solid wheat straw substrate. Appl Microbiol Biotechnol 48:398–404
Zheng Z, Obbard JP (2002) Oxidation of polycyclic aromatic hydrocarbons (PAH) by the white rot fungus, Phanerochaete chrysosporium. Enzyme Microb Technol 31:3–9
Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Culture, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tsukihara, T., Honda, Y., Watanabe, T. et al. Molecular breeding of white rot fungus Pleurotus ostreatus by homologous expression of its versatile peroxidase MnP2. Appl Microbiol Biotechnol 71, 114–120 (2006). https://doi.org/10.1007/s00253-005-0136-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-005-0136-1