
Abstract
A PCR-based method for the quantitative detection of Lentinus edodes and Trametes versicolor, two ligninolytic fungi applied for wastewater treatment and bioremediation, was developed. Genomic DNA was used to optimize a PCR method targeting the conserved copper-binding sequence of laccase genes. The method allowed the quantitative detection and differentiation of these fungi in single and defined-mixed cultures after fractionation of the PCR products by electrophoresis in agarose gels. Amplified products of about 150 bp for L. edodes, and about 200 bp for T. versicolor were purified and cloned. The PCR method showed a linear detection response in the 1.0 μg–1 ng range. The same method was tested with genomic DNA from a third fungus (Phanerochaete chrysosporium), yielding a fragment of about 400 bp. Southern-blot and DNA sequence analysis indicated that a specific PCR product was amplified from each genome, and that these corresponded to sequences of laccase genes. This PCR protocol permits the detection and differentiation of three ligninolytic fungi by amplifying DNA fragments of different sizes using a single pair of primers, without further enzymatic restriction of the PCR products. This method has potential use in the monitoring, evaluation, and improvement of fungal cultures used in wastewater treatment processes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The use of ligninolytic fungi for treatment of effluents with recalcitrant organic matter and xenobiotics has received considerable attention in the last 15 years. For instance, the pulp and paper industry, which discharges effluents rich in lignin derivatives and other poly-phenol contaminants, has led the search for new microbial consortia capable of resisting and transforming these compounds in monitored systems (Estrada-Vázquez et al. 1998; Ortega-Clemente et al. 2004; Poggi-Varaldo 1994, 1995). The morphological similarities of ligninolytic fungi, and the lack of methods for assessment and analysis of fungal biomass in defined-mixed cultures in bioreactors, necessitates development of alternative methods of biomass monitoring.
Laccase is one of the major enzymes involved in delignification (Leonowicz et al. 1999; Mayer and Staples 2002). This enzyme oxidizes phenolic compounds in an oxygen-dependent process requiring neither manganese ions nor H2O2 (Regalado et al. 1999; Robles et al. 2000). Laboratory scale bioreactors efficient in lignin- and color-removal developed by our research group, use two white-rot fungi known to produce laccase: Lentinus edodes and Trametes versicolor (Ortega Clemente et al. 2004; Ramirez-Canseco et al. 2000). However, complete characterization of the reactors, and proper understanding of biochemical performance, requires appropriate monitoring of the defined-mixed culture used.
PCR facilitates research to assess the genetic diversity of fungal populations using ribosomal DNA (Brown et al. 1993; Larena et al. 1999; Lecomte et al. 2000; Jasalavich et al. 2000). However, these methods sometimes require further steps of analysis of PCR products, such as denaturing gradient gel electrophoresis (DGGE) or restriction fragment length polymorphism (RFLP) analysis. Although detection by rDNA sequences is most commonly used, there are reports of the identification of fungi through genes such as ligninase H8 and glyceraldehyde-3-phosphate dehydrogenase (Johnston and Aust 1994; Kreuzinger et al. 1996). In this paper we report a PCR-based protocol targetting the laccase gene, detecting white-rot fungi in pure and defined-mixed cultures without the need for further analysis of the PCR products.
Materials and methods
Strains and culture conditions
Escherichia coli DH5α (Woodcock et al. 1989) was used for DNA cloning and manipulation. DH5α was grown at 37°C in solid or liquid Luria-Bertoni (LB) medium (pH 7.0); 50 μg/ml ampicillin was used when required. L. edodes CDBB-H-985, T. versicolor CDBB-H-1051, and Phanerochaete chrysosporium CDBB-H-298 were obtained from the CINVESTAV-IPN Collection. Fungi were grown on potato dextrose agar or 2% malt extract (ME) agar plates and 1-cm diameter dried circles with mycelia were excised. For T. versicolor and L. edodes single cultures, 100 ml 3% ME (pH 4.7) was inoculated with five mycelia circles and incubated at 100 rpm, for 7–10 days at 28°C. For P. chrysosporium single culture, 100 ml yeast extract-peptone-glucose broth (pH 4.5–4.8) was inoculated with five mycelia circles and shaken at 150 rpm, for 4 days at 39°C. Mycelia were homogenized and 15 ml was used to inoculate 100 ml selective mycologic medium (MM) for T. versicolor, and 100 ml Kirk medium (KM) pH 4.5 for L. edodes and P. chrysosporium. Flasks were incubated at 100 rpm for 12 days at 28°C, except for P. chrysosporium (39°C). Defined-mixed cultures of T. versicolor and L. edodes were grown in 3% ME, MM and KM, and incubated at 100 rpm for 13 days at 28°C. On day 1, 0.205 g dry weight mycelium of each fungus was added to 100 ml medium. Samples were taken on the 1st, 4th, 7th, 10th and 13th day of incubation.
Preparation of mycelia for DNA extraction
Mycelia of pure and defined-mixed cultures were rinsed with deionized water and filtered. Wet mycelium was dried for 30 min between filter papers and placed overnight into a hermetically sealed vessel containing CaCl2. Dry mycelium was frozen using liquid nitrogen, ground, transferred to 1.5 ml polypropylene tubes, and stored in a dried atmosphere at −70°C.
Genomic DNA extraction and purification
DNA was extracted using an adaptation of procedures reported by Ashktorab and Cohen (1992) and Challen et al. (1995): 1 ml extraction buffer (EB; 0.4 M KCl, 50 mM EDTA pH 8.0, 1% v/v Triton X-100) was added to 208 mg ground T. versicolor, 66 mg L. edodes or 79 mg P. chrysosporium. Mycelia (400 mg) from defined-mixed cultures were mixed with 200 mg P. chrysosporium and ground. EB (1 ml) was added to 200 mg of the mixture. Tubes were vortexed and incubated 30 min at 70°C, mixing every 5 min, and centrifuged for 15 min, at 20,817 g, 20°C; 0.8 ml of the supernatant was transferred to a fresh tube, and 8 μl RNase A (10 mg/ml) was added and incubated for 30 min at room temperature. DNA was extracted using phenol-chloroform and dissolved in TE buffer (pH 8). Samples were stored at −70°C.
PCR amplification of genomic DNA
The 50 μl PCR reaction mixture contained: 1× PCR buffer, 0.5 μM forward (5′-CAC TGG CAT GGG TTC TTC CAG-3′) and reverse (5′-ATG GCT GTG GTA CCA GAA AGT-3′) primers, 4.0 mM MgCl2, 0.05 μg genomic DNA template, and 1.25 U Taq DNA polymerase (Gibco-BRL, Rockville, Md.). The PCR program consisted of 2 min preheating at 95°C, 35 cycles of 30 s denaturing at 95°C, 30 s annealing at 55°C, 30 s extension at 72°C, and a final extension of 5 min at 72°C. Amplified products were analyzed in 2.5% agarose gels stained with ethidium bromide. Sensitivity, and the linear range of detection, were determined by amplification using genomic DNA of each fungus, starting with 1.0 μg and serial 1:10 dilutions down to 1.0 fg. PCR products were quantified using Kodak 1D Image Analysis Software v3.0.2 (Eastman Kodak, Rochester, N.Y.). Net intensity data from three independent assays for each DNA were adjusted to linearity using the shareware program Curve Expert v1.34 ((http://curveexpert.webhop.biz/ and Microsoft Excel (Microsoft, Seattle, Wash.).
DNA cloning, manipulation and purification
Enzymes for DNA modification were from Gibco-BRL. PCR products amplified from genomic DNA were fractionated in preparative agarose gels. Bands of interest were recovered and ligated into the SmaI site of pDL19 plasmid (Das 1990) using a cycle restriction-ligation protocol (Lund et al. 1996). The presence of an insert of about 200 bp for T. versicolor and 400 bp for P. chrysosporium candidates, was determined by KpnI and BamHI digestion. For L. edodes, BamHI and SstI were used to probe for a 150 bp insert.
DNA sequencing
pDL19 derivatives pDL19-TvC5, pDL19-LeC5 and pDL19-PcC4ch were sequenced using a BigDye Terminator Kit v2.0 (Applied Biosystems, Foster City, Calif.). Reactions were analyzed using a Perkin Elmer ABI-PRISM Model 310 (Perkin-Elmer/Applied Biosystems). The final sequence was analysed and assembled using Vector NTI v5.5 (InforMax, Frederick, Md.). Similarity searches were performed using the BLAST program (Altschul et al. 1997).
Synthesis of labeled probes
Biotin-labelled genomic and plasmid probes were synthesized by PCR in 50 μl reactions containing: 1× PCR buffer, 0.2 mM each of dATP, dGTP, dTTP and Biotin-14-dCTP-14 (KPL, Gaithersburg, Md.; cat. no. 60-01-00), 0.5 μM of each primer, 0.05 μg genomic DNA or 0.016 ng plasmid DNA, and 1.25 U Taq polymerase (Gibco BRL, 11615-028). Probe labeling was tested by slot-blot assay on Hybond N+ membranes (RPN 203B; Amersham, Piscataway, N.J.), using a KPL Southern hybridization kit (53-30-00). The β-lactamase probe was the 1,117 bp BglII fragment from pDL19. An AlkPhos Direct kit was used to cross-link the probe to alkaline phosphatase (Amersham, RPN 3680).
Southern-blot assay
All incubations and washes in the protocol were at 55°C. Slot-blot assays were performed using genomic and plasmid probes specific for each fungus: 5 μg plasmid or 40 μg genomic DNA were applied to a Hybond N+ membrane using a Hybri-slot filtration manifold (Gibco-BRL, 21052–014). DNA was UV cross-linked to the membrane for 10 min using a UVP M-20E transilluminator. Assays were performed with a KPL Southern hybridization kit (53-30-00) using biotinylated probes at a concentration of 50 ng/ml in blocking solution. The detection reaction was developed for 10 min using 3 ml LumiGLO (KPL) solution. Signal was recorded by 5–10 min exposures to film (Kodak, 601–1597). Southern blots using the β-lactamase probe were performed on same type of membranes using CDP-Star detection reagent and an AlkPhos Direct kit (Amersham, RPN3682 and RPN3680); 5 ng labeled probe was used per milliliter of hybridization buffer. Signal was detected by autoradiography after approximately 1 min.
GenBank accession numbers
The GenBank accession numbers of the sequences described are AY204502 for L. edodes; AY204503 for T. versicolor, and AY221632 for P. chrysosporium.
Results
DNA extraction, PCR amplification and restriction analysis
The PCR method developed here was based on a previously reported protocol designed to amplify laccase sequences in L. edodes and T. versicolor (D’Souza et al. 1996). The primer sequences used for this study corresponded to the first two consensus copper-binding regions in the N-terminal domains of laccases from white-rot fungi. This protocol was tested using genomic DNA, yielding two products of about 150 and 200 bp for L. edodes, and 200 bp for T. versicolor, as previously reported (Fig. 1; lanes 3, 5). We wanted to use these PCR products in a test to detect the presence of each fungus and quantify the variation of biomass in bioreactors. Since two of the PCR products were of similar size (Fig. 1), we searched for a restriction enzyme site in the reported sequence of the L. edodes 217 bp product (D’Souza et al. 1996) that could cut this fragment into two pieces, thus distinguishing it from the T. versicolor PCR product.

Electrophoresis of PCR products in 2% agarose gel. Lanes: M 25 bp DNA ladder; 1, 2 plasmid containing a unique PstI site; 3, 4 Lentinus edodes; 5, 6 Trametes versicolor. DNA in lanes 2, 4, and 6 was digested with PstI
In silico analysis indicated that a Pst I restriction site was present in the larger of the two L. edodes sequences, at position 112. This would cut this DNA molecule in two fragments of 107 bp and 110 bp. The Pst I site was not present in the T. versicolor 200 bp PCR product. Digestion of the PCR products with Pst I would thus produce three fragments for L. edodes: 150, 110 and 107 bp. Fragments of this size can be easily separated by electrophoresis and the amount of DNA in the bands measured by densitometry. However, the PCR product amplified from L. edodes DNA was not cut by Pst I (Fig. 1; lane 4), while a DNA control was digested under same conditions (Fig. 1, lane 2). Because Pst I digestion did not help to distinguish between L. edodes and T. versicolor PCR products, we decided to develop a suitable PCR protocol.
Optimization of the PCR protocol
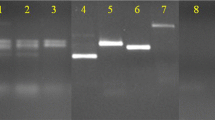
To optimize the PCR protocol, different MgCl2 concentrations (1–9 mM), annealing temperatures (49, 52, 54, 55, and 58°C), and number of PCR cycles (20, 30 and 40) were assayed. Since the PCR products we expected had a size of less than 500 bp, we changed the length of each PCR segment (denaturing, annealing, and extension) to 30 s. All these changes reduced the PCR program from 4.3 h to less than 2 h. We applied our optimal PCR protocol to single samples or mixtures of purified genomic DNA, obtaining, without any further enzymatic digestion, a major product of about 150 bp for L. edodes (Fig. 2, lane 1), 200 bp for T. versicolor (Fig. 2, lane 2), and 400 bp when genomic DNA of P. chrysosporium, a different ligninolytic fungus, was used (Fig. 2, lane 3). The same products were obtained when genomic DNA of L. edodes and T. versicolor were mixed and used as template (Fig. 2, lane 4), and also when genomic DNA of the three fungi were used as template (Fig. 2, lane 5).

Electrophoresis in 2.5% agarose gel of PCR products using single or mixed genomic DNAs as templates. Lanes: M 50 bp DNA ladder; 1 L. edodes; 2 T. versicolor; 3 P. chrysosporium; 4 L. edodes + T. versicolor; 5 L. edodes + T. versicolor + P. chrysosporium; C PCR with no template
The sensitivity and linear range of detection of the PCR protocol on genomic DNA were determined as described in Materials and methods. When DNA of L. edodes was used, the 150 bp PCR product was detected down to 1 ng template (Fig. 3a), the 200 bp product was detected at 10 pg T. versicolor template (Fig. 3b), and the 400 bp product was detected at 0.1 ng P. chrysosporium template under the same conditions (Fig. 3c). For L. edodes, the linear range of detection was between 0.1 μg and 1.0 ng (R2 =0.98); for T. versicolor between 1 μg and 0.01 ng (R2 =0.97), and for P. chrysosporium between 0.1 μg and 0.1 ng (R2 =0.93). The lower detection threshold of 10 pg in the case of T. versicolor, versus 1 ng in L. edodes, suggested a higher primer binding affinity for T. versicolor genomic DNA. We performed a competition PCR with DNA of L. edodes and T. versicolor. This assay showed more PCR product from T. versicolor, even in the presence of a 10-fold excess of genomic DNA of L. edodes (data not shown).

Electrophoresis in 2.5% agarose gels of PCR products obtained using different concentrations of genomic DNA template. Lanes: M 50 bp DNA ladder, 1 1.0 μg, 2 0.1 μg, 3 0.01 μg, 4 1.0 ng, 5 0.1 ng, 6 0.01 ng, 7 1.0 pg, 8 0.1 pg, 9 0.01 pg. a L. edodes, b T. versicolor, c P. chrysosporium
Detection of L. edodes and T. versicolor by PCR in defined-mixed cultures
Two independent experiments of defined-mixed cultures of L. edodes and T. versicolor were carried out in three different liquid media. DNA was extracted from mycelia of cultures grown in KM, ME and MM. Samples were taken on days 1, 4, 7, 10 and 13. Purified DNA was quantified and subjected to PCR amplification. Analysis of the results showed that, although each medium were inoculated quantitatively with same amount of mycelium of each fungus, the 200 bp T. versicolor product was predominantly detected (Fig. 4), while the 150 bp L. edodes PCR product was less abundant. The 400 bp P. chrysosporium DNA band, used as a DNA extraction control, was seen as a faint band close to the 400 bp DNA marker (Fig. 4). The similar amount of PCR products obtained in each medium did not show any significant change in the relative proportion of the two fungi in culture during the 13 days of incubation.

Electrophoresis in 2.5% agarose gels of PCR products amplified from genomic DNA isolated from L. edodes and T. versicolor combined culture in different media. Lane numbers refer to days of incubation; M 100 bp DNA ladder, C PCR reaction without template. a Malt extract (ME), b Kirk medium (KM), c mycologic medium (MM)
Slot-blot analysis of DNA
To test the genomic origin of the amplified PCR products, a Southern-blot analysis was performed using Biotin-labeled probes. Genomic and plasmid DNA containing the PCR fragment of each fungus were used to synthesize the corresponding probes; 50 ng of genomic and plasmid probes versus L. edodes were specific for detection of 5 μg cloned DNA of this fungus (Fig. 5; lanes 1, 4). A similar result was obtained for the anti-T. versicolor probes (Fig. 5; lanes 3, 6); and the anti-P. chrysosporium probes (Fig. 5; lanes 2, 5). We detected no signal when 50 ng of each plasmid probe was used versus 40 μg genomic DNA.

Slot-blot analysis of PCR products. Lanes: W Empty slots, 1–3 Plasmid (5 μg) containing the PCR insert probed with 50 ng plasmid probe (5 min exposure), 4–6 plasmid (5 μg) containing the PCR insert versus 50 ng genomic probe (5 min exposure), 7–9 each genomic DNA (40 μg) versus 50 ng plasmid probe (20 h exposure), 10 26 ng anti-β lactamase DNA was used to probe 5 μg plasmid containing the PCR inserts (1 min exposure). Lanes: 1, 4, 7 anti-L. edodes probe; 2, 5, 8 anti-P. chrysosporium probe; 3, 6, 9 anti-T. versicolor probe. Rows: + Corresponding labelled probe (6 ng), Le L. edodes, Pc P. chrysosporium, Tv T. versicolor, − 5 μg pDL19 vector
DNA sequencing of cloned PCR fragments
Laccase PCR fragments were amplified and cloned. These fragments were subjected to DNA sequencing using a set of primers flanking the site of insertion. The 198 bp sequence of T. versicolor (GenBank accession number AY204503) contains two exons and one intron. It shares 94.4% identity with a laccase III gene sequence from T. versicolor (AY081188), and 98.5% identity with the 199 bp sequence reported previously (D’Souza et al. 1996). On the other hand, our L. edodes sequence (AY204502) showed 55.8% identity with a reported laccase gene of L. edodes (BD133566) and only 54.2% identity with the 217 bp sequence reported by D’Souza et al. (1996). Remarkably, sequencing of the 151 bp product of L. edodes (AY204502) revealed that this product contains three copies of the 21 bp copper-binding sequence I and two copies of the 18 bp copper binding sequence II (Cano-Ramirez 2001). The 378 bp sequence obtained for P. chrysosporium (AY221632) showed 51% identity with the sequence of the laccase III gene from T. versicolor (AY081188), and 48.1% identity with the sequence of L. edodes (BD133566). The same type of analysis showed that this sequence had 55.1% identity with T. versicolor and 48.9% identity with L. edodes sequences reported previously (D’Souza et al. 1996).
Discussion
PCR has proved useful in the amplification, cloning and differential expression analysis of laccase genes from ligninolytic fungi (D’Souza et al. 1996; Zhao and Kwan 1999). In this work we developed and optimized a PCR-based method applicable to the quantitative detection and differentiation of otherwise indistinguishable fungi used in the removal of pollutants from wastewater. Genomic DNA from L. edodes, T. versicolor and P. chrysosporium suitable for PCR analysis was extracted from dried mycelia following a simple mummification method using CaCl2. The amplification conditions used allowed differentiation among these fungi through different DNA size products in a convenient target amount range of 1.0 μg to 1 ng. Products of 151 bp for L. edodes, 198 bp for T. versicolor, and 378 bp for P. chrysosporium were obtained. Slot-blot analysis supported the notion that these PCR products were actually amplified from the cognate genome. DNA sequencing of cloned fragments indicated that the 198 bp PCR product contains two partial exons and one intron and was amplified from a T. versicolor laccase gene. In the case of P. chrysosporium, DNA sequencing and analysis of the 378 bp product suggests that it could belong to a laccase or a different gene harboring copper-binding regions, similar to the first two consensus regions in the N-terminal domains of laccases from white-rot fungi. Laccase expression has been reported for P. chrysosporium (Mougin et al. 2000) and other members of the same genus such as P. laevis (Bogan and Lamar 1996) and P. flavido-alba (De la Rubia et al. 2002). It is remarkable that DNA sequencing of the 151 bp product of L. edodes revealed that this product contains five copies of the consensus copper-binding domain of laccase (Cano-Ramirez 2001). One possibility to explain the origin of this product is that this molecule arose as an artifact during PCR amplification. However, in silico analysis to predict dimers and hairpin loops for each single primer, and duplexes between primers in the reaction, revealed no structure with a stable free energy that could explain the formation of the 151 bp product. Another possibility is that this molecule is amplified from a particular locus in the L. edodes genome. This hypothesis is supported in part by the specific recognition this molecule showed in the detection of cloned fragments in the slot-blot assay (Fig. 5, lanes 1–6). Unfortunately, under the conditions used in this work, we were unable to detect any signal in the slot-blot analysis of genomic DNA of these fungi (Fig. 5, lanes 7–9). To the best of our knowledge, there are few reports on molecular biology applications for detection of basidiomycetes in natural or artificial environments. Some studies use restriction enzyme analysis for detection of P. chrysosporium and other decay basidiomycetes (Jasalavich et al. 2000; Adair et al. 2002; Johnston and Aust 1994). There are also studies based on amplification of internal transcribed spacer (ITS) regions in nuclear ribosomal repeat units (Gardes and Bruns 1993; Nugent and Saville 2004). However, in the work of Gardes and Bruns (1993), the primers used cross-reacted with plant DNA sequences, whereas the pair of primers used by Nugent and Saville (2004) did not detect ligninolytic fungi. A different molecular approach to detect ligninolytic genes such as lignin and manganese peroxidase of white-rot fungi was unsuccessful (Stuardo et al. 2004). Therefore, the current study represents a first successful approach to easy and rapid detection and differentiation of white-rot fungi in mixed cultures.
References
Adair S, Kim SH, Breuil C (2002) A molecular approach for early monitoring of decay basidiomycetes in wood chips. FEMS Microbiol Lett 211:117–122
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Ashktorab H, Cohen RJ (1992) Facile isolation of genomic DNA from filamentous fungi. Biotechniques 13:198–200
Bogan BW, Lamar R (1996) Polycyclic aromatic hydrocarbon-degrading capabilities of Phanerochaete laevis HHB-1625 and its extracellular ligninolytic enzymes. Appl Environ Microbiol 62:1597–1603
Brown AE, Muthumeenakshi S, Sreenivasaprasad S, Mills PR, Swinburne TR (1993) A PCR primer-specific to Cylindrocarpon heteronema for detection of the pathogen in apple wood. FEMS Microbiol Lett 108:117–120
Cano-Ramirez C (2001) Quantitative detection of fungi Trametes versicolor and Lentinus edodes in defined mixed culture using a PCR method. B.Sc. Thesis. Universidad Autónoma Metropolitana-Xochimilco, Depto. del Hombre y su Ambiente. México DF, Mexico
Challen MP, Andrew JM, Daniel MC (1995) Facile extraction and purification of filamentous fungal DNA. Biotechniques 18:975–977
D’Souza TM, Boominathan K, Reddy CA (1996) Isolation of laccase gene specific sequences from white rot and brown rot fungi by PCR. Appl Environ Microbiol 62:3739–3744
Das A (1990) Overproduction of protein in Escherichia coli: vectors, hosts and strategies. In: Deutscher MP (ed) Methods in enzymology: guide to protein purification. Academic Press, San Diego, pp 93–112
De la Rubia T, Ruiz E, Perez J, Martinez J (2002) Properties of a laccase produced by Phanerochaete flavido-alba induced by vanillin. Arch Microbiol 179:70–73
Estrada-Vázquez C, Poggi-Varaldo HM, Hernández-Vera R, Fernández-Villagómez G, Magaña-Plaza I, Hernández-González A (1998) Biological post-treatment of an anaerobically-treated black liquor spills wastewater from Kraft pulp mills. In: Alleman JE, Blatchley ER, Hua I, Jacko RB, Javfert CT, Nies L (eds) 52nd Purdue industrial waste conference . Ann Arbor Sci, Chelsea, Mich., pp 493–503
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes application to the identification of mycorrhizae and rusts. Mol Ecol 2:113–118
Jasalavich CA, Ostrofsky A, Jellison J (2000) Detection and identification of decay fungi in spruce wood by restriction fragment length polymorphism analysis of amplified genes encoding rRNA. Appl Environ Microbiol 66:4725–4734
Johnston CG, Aust SD (1994) Detection of Phanerochaete chrysosporium in soil by PCR and restriction enzyme analysis. Appl Environ Microbiol 60:2350–2354
Kreuzinger N, Podeu R, Gruber F, Gobl F, Kubicek CP (1996) Identification of some ectomycorrhizal basidiomycetes by PCR amplification of their gpd (glyceraldehyde-3-phosphate dehydrogenase) genes. Appl Environ Microbiol 62:3432–3438
Larena I, Salazar O, Gonzalez V, Julian MC, Rubio V (1999) Design of a primer for ribosomal DNA internal transcribed spacer with enhanced specificity for ascomycetes. J Biotechnol 75:187–194
Lecomte P, Peros JP, Blancard D, Bastien N, Delye C (2000) PCR assays that identify the grapevine dieback fungus Eutypa lata. Appl Environ Microbiol 66:4475–4480
Leonowicz A, Matuszewska A, Luterek J, Ziegenhagen D, Wojtas-Wasilewska M, Cho NS, Hofrichter M, Rogalski J (1999) Biodegradation of lignin by white rot fungi. Fungal Genet Biol 27:175–185
Lund AH, Duch M, Pedersen FS (1996) Increased cloning efficiency by temperature-cycle ligation. Nucleic Acids Res 24:800–801
Mayer AM, Staples RC (2002) Laccase: new functions for an old enzyme. Phytochemistry 60:551–565
Mougin C, Boyer FD, Caminade E, Rama R (2000) Cleavage of the diketonitrile derivative of the herbicide isoxaflutole by extracellular fungal oxidases. J Agric Food Chem 48:4529–4534
Nugent KG, Saville BJ (2004) Forensic analysis of hallucinogenic fungi: a DNA-based approach. Forensic Sci Int 140:147–157
Ortega-Clemente LA, Estrada-Vázquez C, Esparza-García F, Caffarel-Méndez S, Rinderknecht-Seijas N, Poggi-Varaldo HM (2004) Integrated biological treatment of recalcitrant effluents from pulp mills. Water Sci Technol 50:145–156
Poggi-Varaldo HM (1994) Pollution prevention and wastewater treatment in the pulp and paper industry. Water Wastewater Int 9:12–18
Poggi-Varaldo HM (1995) Anaerobic methanogenic consortia for pre-treatment and detoxification of pulp and paper mill wastewaters. In: Poggi-Varaldo HM, Rodríguez-Vázquez R (eds) Proceedings of the 1st International Meeting on Microbial Ecology. Editorial Facultad de Química UNAM, CONACyT Mexico DF, Mexico, pp 53–54
Ramirez-Canseco S, Poggi-Varaldo HM, Rodríguez-Vázquez R, Estrada-Vázquez C, Rinderknecht-Seijas N (2000) Fungal post-treatment of an anaerobically-treated black liquor-contaminated effluent from a Kraft pulp mill. In: Foresti E, Chemicharo CAL, Kato MT, Florencio L, Santos de Lima E (eds) Proceedings of the 6th Latin American workshop and seminar on anaerobic digestion, vol 2. Recife, Pernambuco. 5–9 September 2000. Editora Universitaria da UFPE, Recife, Pernambuco, pp 191–195
Regalado V, Perestelo F, Rodríguez A, Carnicero A, Sosa FJ, De la Fuente G, Falcón MA (1999) Activated oxygen species and two extracellular enzymes: laccase and aryl-alcohol oxidase, novel for the lignin-degrading fungus Fusarium proliferatum. Appl Microbiol Biotechnol 51:388–390
Robles A, Lucas R, Alvarez-de-Cienfuegos G, Gálvez A (2000) Phenol-oxidase (laccase) activity in strains of the hyphomycete Chalara paradoxa isolated from olive mill wastewater disposal ponds. Enzyme Microb Technol 26:484–490
Stuardo M, Vasquez M, Vicuna R, Gonzalez B (2004) Molecular approach for analysis of model fungal genes encoding ligninolytic peroxidases in wood-decaying soil systems. Lett Appl Microbiol 38:43–49
Woodcock DM, Crowther PJ, Doherty J, Jefferson S, DeCruz E, Noyer-Weidner M, Smith SS, Michael MZ, Graham MW (1989) Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res 17:3469–3478
Zhao J, Kwan HS (1999) Characterization, molecular cloning and differential expression analysis of laccase genes from the edible mushroom Lentinula edodes. Appl Environ Microbiol 65:4908–4913
Acknowledgements
The work described in this paper was partially supported by CINVESTAV-IPN, Accesolab México, BioRad México, and Kirkegaard & Perry Laboratories, USA. We thank Maria Guadalupe Aguilar-Gonzalez from the DNA Sequencing Unit (Departamento de Genética y Biología Molecular) and Carolina Miranda-Brito (Departamento de Fisiología, Biofísica y Neurociencias) for technical assistance, Daniel Castro-Roa for critical review of the manuscript, and Rosa Maria Cruces for clerical assistance. We wish to thank Dr. Ian Reid, formerly with PAPRICAN, Canada, for the kind donation of the strain of Trametes versicolor to one of the authors (H.M.P.-V.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
García-Mena, J., Cano-Ramirez, C., Garibay-Orijel, C. et al. A PCR method for the detection and differentiation of Lentinus edodes and Trametes versicolor in defined-mixed cultures used for wastewater treatment. Appl Microbiol Biotechnol 67, 524–531 (2005). https://doi.org/10.1007/s00253-004-1795-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-004-1795-z