
Abstract
Many protocols to extract DNA directly from soil samples have been developed in recent years. We employed two extraction methods which differed in the method of lysis and compared these methods with respect to yield, purity and degree of shearing. The main focus was on the specific isolation of DNA from different microorganisms, especially DNA from actinomycetes, as these cells are very difficult to lyse, in contrast to non-actinomycetes. Thus, we used both methods to isolate DNA from Pseudomonas, Arthrobacter and Rhodococcus and from soil spiked with the respective microorganisms. Both methods rendered high DNA yields with a low degree of shearing, but differed in the type of cells that were lysed. By one protocol (utilizing enzymatic lysis) only DNA from the Gram-negative Pseudomonas strain could be obtained whereas, by the other protocol (utilizing mechanical lysis), all microorganisms that were used could be lysed and DNA extracted from them. Using a combination of both protocols, DNA from those organisms could be obtained selectively. Furthermore, one of the protocols was modified, resulting in higher DNA yield and purity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microorganisms offer a huge potential for new biocatalysts for industrial and commercial applications. The classic approach to gain access to new biocatalysts is to cultivate microorganisms from soil samples in different media and screen for the desired enzyme activity. However, only a very small proportion of the total microbial community of a soil sample can be isolated by cell cultivation in standard media (Torsvik et al. 1990; Jacobsen and Rasmussen 1992); and current estimates indicate that approximately 99% of microorganisms occurring in nature cannot be cultivated by standard techniques (Amann et al. 1995). An alternative method is to isolate DNA directly from the microorganisms present in the soil without prior culturing. This DNA can be used for the construction of DNA libraries and to directly clone functional genes from environmental samples. Many protocols for isolating DNA from environmental samples have been developed, which vary with respect to the degree of shearing, purity and quantity of the isolated DNA. The DNA may be extracted from soil samples directly (Selenska and Klingmüller 1991; Tebbe and Vahjen 1993; Wikstrom et al. 1996), or the microorganisms of the soil sample can be extracted first and lysed subsequently (Steffan et al. 1988). Direct extraction of DNA introduces less bias than methods which are based on cell extraction prior to lysis (von Wintzingerode et al. 1997). To extract DNA, the microorganisms in the soil can be lysed mechanically by bead-beating (Moré et al. 1994; Berthelet et al. 1996; Purdy et al. 1996), sonication (Degrange and Bardin 1995), freeze-thawing (Lee et al. 1996), grinding in liquid nitrogen (Johnston and Aust 1994; Volossiouk et al. 1995) or enzymatically, using enzymes such as proteinase K (Wikstrom et al. 1996; Zhou et al. 1996), lysozyme (Porteous et al. 1994) or pronase. A combination of these treatments may also be applied (Tsai and Olson 1991; Picard et al. 1992; Tebbe and Vahjen 1993). In addition, SDS or other detergents are frequently added.
The construction of environmental DNA libraries presents special problems. Escherichia coli does not recognize approximately 80% of actinomycete promoters (Strohl 1992) and the G+C content of the DNA to be cloned and expressed may differ significantly. Hence, a great number of genes present in the sample are unlikely to be adequately expressed in E. coli. However, if a library is to be screened for enzymatic activities, the number of functionally expressed genes should be as high as possible, i.e. DNA from organisms that can barely be expressed should be excluded in the respective host. Thus, it is desirable to exclude actinomycete genes when E. coli is used as host. However, actinomycetes possess interesting enzymatic activities; and, although the genes for these are rarely expressed in E. coli, this DNA could be expressed in an alternative host, for example Streptomyces.
In this work, two methods for the extraction of DNA were compared with regard to DNA yield, DNA shearing and purity, using Gram-positive and Gram-negative microorganisms. Moreover, it was investigated whether DNA from all applied strains could be extracted in similar amounts or whether a bias of DNA originating from different species could be obtained by the choice of DNA-extraction method.
Materials and methods
Bacterial strains
The bacterial strains used were Pseudomonas LU2023 (Entcheva et al. 2001), Arthrobacter LU9144 and Rhodococcus LU 9002 (Borneman et al. 1996). All strains were from BASF (Ludwigshafen, Germany) and were grown to the stationary phase in LB medium at 30 °C.
E. coli DH5α and S. antibioticus TÜ4 were used as controls for Gram-negative and Gram-positive organisms in the PCR reactions.
Soil DNA extraction and purification
For a modified version of Zhou’s method of DNA extraction (Zhou et al. 1996), soil samples (0.5 g) were mixed with 1.3 ml of extraction buffer (100 mM Tris-HCl, pH 8.0, 100 mM sodium EDTA, pH 8.0, 100 mM sodium phosphate, pH 8.0, 1.5 M NaCl, 1% hexadecylmethylammonium bromide) in 15-ml Falcon tubes. Three cycles of freezing in liquid nitrogen and thawing in a water bath at 65 °C were then applied to the suspensions. After the samples had cooled to 37 °C, 10 μl of proteinase K (20 mg/ml) was added and the samples were incubated at 37 °C for 30 min with horizontal shaking. Then, 150 μl of 20% SDS were added and the samples were incubated in a water bath at 65 °C for 2 h. After centrifugation at 3,200 g for 10 min at 4 °C, the supernatants were transferred to 15-ml Falcon-tubes. The soil pellets were washed by adding 1 ml of extraction buffer and 250 μl of 20% SDS, vortexed, incubated for 10 min at 65 °C and centrifuged as before. The aqueous phases were extracted with chloroform and precipitated with 0.7 vol. of isopropanol at −20 °C for 1 h. After centrifugation at 20,800 g for 25 min at 4 °C, the pellets were washed with ice-cold 70% ethanol, recentrifuged, dried and resuspended in 100 μl of TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0).
For PCR application, the DNA was either purified three times with the Wizard DNA clean-up system (Promega, Madison, Wis.) or purified once with the Wizard DNA clean-up system and subsequently by agarose gel electrophoresis. After gel electrophoresis, DNA was eluted with the Gel-extraction kit (Biozym, Hess. Oldendorf, Germany), according to the manufacturer’s instructions.
For a modified version of Moré’s method of DNA extraction (Moré et al. 1994), soil samples (0.5 g) were mixed with 500 μl of 100 mM sodium phosphate buffer (pH 8.0) in 2-ml Eppendorf tubes. Then, 2 g of glass beads (0.10–0.25 mm diameter) and 250 μl of a 10% SDS solution (100 mM NaCl, 500 mM Tris, pH 8.0, 10% SDS) were added. Each tube was shaken at 8,000 strokes/min for 10 min in a bead-mill (Retsch, Haan, Germany) before being centrifuged at 20,800 g for 3 min at room temperature. The supernatant was kept on ice, the soil pellet was washed with 250 μl of sodium phosphate buffer, mixed and treated for 2 min in an ultrasonic bath. After centrifugation, the supernatants were pooled, mixed 5:2 with 7.5 M ammonium acetate, precipitated for 5 min on ice and centrifuged at 20,800 g for 3 min.
The supernatant was precipitated with 2.5 vol. of ethanol, according to standard protocols (Sambrook et al. 1989). For PCR applications, the DNA obtained by ethanol precipitation was purified three times with the Wizard DNA clean-up System.
Silica-based purification of DNA
Using a modified version of the silica-based purification of DNA (Höss and Pääbo 1993), the supernatant of the ammonium acetate precipitation obtained by the method of Moré could be purified directly. For this, 2 vol. of extraction buffer (5 M guanidine thiocyanate, 50 mM Tris, pH 8.0, 25 mM NaCl, 20 mM EDTA, 1.3% Triton X-100) and 0.1 vol. of silica were added to the supernatant. After vortexing, the tubes were centrifuged at 1,000 g for 3 min and the supernatant was discarded. Then, 2 vol. of wash buffer (5 M guanidine thiocyanate, 0.1 M Tris, pH 8.0, 25 mM NaCl ) were added, the silica was resuspended by gentle vortexing and the tube was centrifuged at 1,000 g for 3 min. The supernatant was again discarded and the pellet was washed with 1.5 ml of 70% ethanol and dried. The DNA was eluted by the addition of 100 μl of TE buffer, incubation at 55 °C for 10 min and centrifugation at 20,800 g for 1 min. The DNA-containing supernatant was transferred to a fresh tube.
PCR amplification of 23S rDNA fragment
An insertion of 100 bases within domain III of the 23S rRNA provides a phylogenetic marker for Gram-positive bacteria with a high G+C content (actinomycetes), whereas other eubacteria do not contain this insertion. To verify the type of DNA isolated, domain III of the 23S rDNA was amplified by PCR, using a mixture of PCR buffer (10 mM Tris-HCl, pH 8.8,50 mM KCl, 0.08% Nonidet P40, 2 mM MgCl2; MBI Fermentas, St. Leon-Rot, Germany), 2.5 mM of each deoxynucleotide triphosphate, 50 pmol each of the primers 1900 V (5′-CCTAAGYYGAGGC-3′) and 1028 R (5′-CCTTCTCCCGAAGTTACGG-3′), 1 μl of template DNA and 1 unit of Taq polymerase. After denaturation at 94 °C for 4 min, 30 cycles were performed as follows: 0.5 min denaturation at 94 °C, 1.5 min annealing at 46 °C and 2 min extension at 72 °C. After a final extension at 72 °C for 5 min, the samples were cooled to 8 °C. Then, 10 μl of each mixture were separated by agarose gel electrophoresis (Sambrook et al. 1989) in a 2% agarose gel. Genomic DNAs from E. coli and Streptomyces purified by the Qiagen genomic-tip 100 kit (Qiagen, Hilden, Germany) were used as control templates.
Results
Comparison of lysis methods
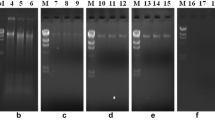
DNA was isolated from a mixture of different strains (Pseudomonas, Arthrobacter, Rhodococcus) and from autoclaved soil spiked with the same mixtures and was analyzed by gel electrophoresis. Two different lysis methods were compared, using pure cultures of Pseudomonas, Arthrobacter and Rhodococcus (Fig. 1a, b). The method according to Zhou was based on cell lysis by freeze-thaw cycles and treatment with proteinase K and hot SDS in the presence of high salt concentrations. In the method according to Moré, cells were lysed by SDS and mechanically disrupted by glass beads. These two methods were chosen as they both gave high DNA yields (data not shown) and significantly differed in their mode of action. Using the method according to Moré, DNA could be isolated from pure cultures of all strains tested (Fig. 1a, lanes 5–7). To study interactions between DNA and soil particles or the influence of soil particles on lysis efficiency, 0.5 g of autoclaved soil was added per 0.5 ml of pure culture (Fig. 1b). Following the addition of soil, the amount of DNA isolated from Pseudomonas was as high as from the pure culture (Fig. 1a, b, lane 5), but the lysis efficiency towards actinomycetes was strongly reduced when autoclaved soil was added (Fig. 1b, lanes 6 7).

Isolation of DNA. A comparison of different DNA isolation methods is shown for pure cultures (a) and pure cultures with autoclaved soil (b), with DNA separated in 1% agarose gels. Lane 1 1-kb ladder, lanes 2–4 Zhou method, lanes 5–7 Moré method, lanes 2, 5 Pseudomonas, lanes 3, 6 Arthrobacter, lanes 4, 7 Rhodococcus
The modified method according to Zhou was very efficient for Pseudomonas. High DNA yields were obtained and DNA shearing was low (Fig. 1a, b, lane 2). In contrast, the lysis efficiencies of Rhodococcus and Arthrobacter were very low, both with and without the addition of soil (Fig. 1a, b, lanes 3–4).
To confirm that no DNA could be extracted from autoclaved soil, both DNA extraction methods were performed with autoclaved soil. No DNA could be extracted from autoclaved soil, using either extraction method, without the addition of pure cultures (data not shown).
To check the amount of DNA that could be extracted directly from soil samples, the DNA isolation methods according to Zhou and Moré were applied to soil samples.
The yield obtained by the method of Moré was comparable with the yield obtained by the method of Zhou, but the DNA was more fragmented when the method of Moré was applied.
Optimization of methods
DNA isolated by both methods was contaminated with humic compounds that interfered with PCR analysis and other reactions. Additionally, the last DNA precipitation step was relatively time-consuming. Therefore, it was investigated for both methods whether the DNA could be purified directly without prior precipitation. Accordingly, a direct silica-purification protocol (Höss and Pääbo 1993) was modified and performed instead of isopropanol/ethanol precipitation. After cell disruption and ammonium acetate precipitation of proteins using the Moré method, the supernatant could be treated directly with silica to obtain purer DNA with a yield comparable with that obtained by ethanol precipitation. The same purification procedure was performed with the aqueous phase from the Zhou protocol after chloroform extraction. In this case the DNA yield was very low in comparison with the isopropanol precipitation (data not shown).
Combination of methods
For the separation of Pseudomonas DNA from Rhodococcus and Arthrobacter DNA, the methods of Zhou and Moré were combined. To 0.5 g of autoclaved soil were added: (1) 1.8 ml of Pseudomonas, (2) 900 μl each of Arthrobacter and Rhodococcus and (3) 600 μl each of Pseudomonas, Arthrobacter and Rhodococcus, all cultures grown to the stationary phase. All samples were first lysed according to Zhou (Fig. 2, lanes 2–4). Subsequently, the remaining intact cells from the soil pellet were disrupted by the method of Moré (Fig. 2, lanes 5–7). Using freeze-thaw cycles, proteinase K and SDS treatment (i.e. the Zhou method) only Pseudomonas could easily be lysed (Fig. 2, lane 2). In Fig. 2, lane 4, the amount of extracted DNA is lower than in Fig. 2, lane 2, because the amount of Pseudomonas cells in that sample was 3-fold lower. Lysis of the cells by the method of Moré resulted in the disruption of both Pseudomonas cells and Rhodococcus and Arthrobacter cells (Fig. 2, lanes 5–7).

Isolation of DNA by a combination of the methods of Zhou and Moré, using 0.5-g autoclaved soil samples spiked with different cultures (Pseudomonas, Arthrobacter, Rhodococcus). First, DNA was extracted by the Zhou-method and, from the remaining cells, DNA was subsequently extracted by the Moré-method. DNA was separated in 1% agarose gels. Lane 1 1-kb ladder, lanes 2–4 Zhou method, lanes 5–7 Moré method, lanes 2, 5 autoclaved soil with 1.8 ml of Pseudomonas, lanes 3, 6 autoclaved soil with 900 μl each of Arthrobacter and Rhodococcus, lanes 4, 7 autoclaved soil with 600 μl each of Arthrobacter, Rhodococcus and Pseudomonas
Lysis of the different cell types was confirmed by PCR analysis of 23S rDNA, using specific primers. In the case of G+C-rich Gram-positive bacteria, a fragment of 350 bp was expected in contrast to a 250-bp fragment in the case of other eubacteria. In Fig. 3, lane 2, only one band with a size of 250 bp is visible when the template used for PCR was DNA isolated according to the method of Zhou from a mixture of all three strains in autoclaved soil. A band of the same size appeared when genomic DNA from E. coli served as template (Fig. 3, lane 5). When the remaining intact cells in the sample were subsequently lysed by the method of Moré and the extracted DNA amplified by PCR, using the same 23S rDNA primers, the main product had a size of 350 bp (Fig. 3, lane 3). This product was the same size as the PCR product that was amplified when Streptomyces DNA was used as template (Fig. 3, lane 4), confirming the presence of the 100-bp insertion. As this insertion is specific to Gram-positive bacteria with a high G+C content, the result proved the lysis of Arthrobacter and Rhodococcus cells.

PCR amplification of domain III of 23S rRNA, using DNA isolated from a mixture (0.5 ml) of Pseudomonas, Arthrobacter and Rhodococcus from the stationary phase and 0.5 g of autoclaved soil. DNA was separated in a 2% agarose gel. Lane 1 100-bp ladder, lane 2 template DNA isolated by the method of Zhou, lane 3 template DNA isolated by the method of Moré, lane 4 template DNA from Streptomyces, isolated with the Qiagen Genomic DNA kit, lane 5 template DNA from Escherichia coli, isolated with the Qiagen Genomic DNA kit
To check whether the fractionated isolation of DNA from real samples is possible, the two DNA extraction methods were performed in combination. First the DNA isolation method according to Zhou was applied, then DNA was isolated from the remaining soil pellet by the method of Moré. The isolated DNA from both methods was used as template for PCR amplification of 23S rDNA. No PCR products from either template showed any 350-bp band (data not shown). As this result could be due to a bias of non-actinomycetes in the sample, 200 μl of Rhodococcus cells were added to the soil sample and DNA extraction methods were performed as before. PCR analysis of 23S rDNA as described above showed that it was not possible to obtain a fragment of 350 bp when using DNA isolated by the method of Zhou as template (Fig. 4, lane 2). However, when DNA isolated subsequently by the method according to Moré served as template, both a 250-bp fragment and a 350-bp fragment were visible on the agarose gel (Fig. 4, lane 3), indicating that DNA from Rhodococcus cells was extracted.

PCR amplification of domain III of 23S rRNA, using DNA isolated from a soil sample with the addition of 200 μl of Rhodococcus cells. DNA was separated in a 2% agarose gel. Lane 1 1-kb extension ladder, lane 2 template DNA isolated by the method of Zhou, lane 3 template DNA subsequently isolated by the method of Moré, lane 4 template DNA from Streptomyces, lane 5 template DNA from E. coli
Discussion
In this study, two different methods for the isolation of DNA from pure cultures, soil samples and soil samples spiked with pure cultures were compared, with respect to purity, yield, efficiency and selectivity of DNA extraction from different species.
Comparison of different DNA extraction methods
The comparison of the two different lysis methods concerning yield showed that the method according to Moré can be efficiently used to lyse both Pseudomonas cells and Rhodococcus and Arthrobacter cells. The method of Zhou obtains the best results for Pseudomonas, as the yield is very high and the DNA is less fragmented than after isolation according to Moré. However, the DNA isolated by this method is more contaminated with humic compounds than the DNA obtained by the Moré method for cell lysis. This contamination is visible in the brownish pellet. Furthermore, DNA from Rhodococcus and Arthrobacter can hardly be isolated by the method according to Zhou, whereas the method according to Moré is effective towards these strains. However, when soil is added to the pure cultures the lysis-efficiency towards Rhodococcus and Arthrobacter is strongly reduced, even with the Moré method. Presumably, the soil particles reduce the effectiveness of the glass beads and therefore DNA can only be efficiently extracted from cells that are easier to disrupt than Rhodococcus and Arthrobacter cells. The method of Zhou introduces a bias towards Gram-negative bacteria, whereas this bias is lower with the method according to Moré. Recent publications (La Montagne et al. 2002) comparing the methods of Zhou and Porteous (Porteous et al. 1994) show a lower DNA yield is obtained by the method of Porteous (ultrasonic, lysozyme) than with the method of Zhou, but the lower yield does not introduce a bias towards lower community diversity. However, the two methods that were compared here differ strongly in the kind of extraction procedure (freeze-thaw cycles, enzymatic vs bead-beating), whereas the methods compared by La Montagne et al. (2002) are rather similar. Furthermore, another comparison of different DNA extraction methods (Martin-Laurent et al. 2001) clearly shows that the soil DNA extraction method used can affect both phylotype abundance and the composition of the indigenous bacterial community. In that work, DNA extraction methods based on mechanical treatment by bead-beating were compared. Thus, it may be possible that, by altering the composition of the glass beads used with the method according to Moré, the bias can further be lowered and DNA from a greater number of microorganisms can be obtained.
Modification of the Moré method
A further problem that is faced when DNA is isolated directly from soil is its contamination with humic compounds, inhibiting for example PCR reactions (Tsai and Olson 1992). As the DNA isolated by both methods was not pure enough for PCR reactions and restriction digestion, ethanol precipitation was replaced by a direct purification step, using silica and guanidine thiocyanate. This procedure resulted in the same DNA yield, compared with ethanol precipitation in the method of Moré. The purity of the isolated DNA, however, was much higher when it was purified with silica. The DNA obtained by ethanol precipitation had to be purified up to three times with the Wizard DNA clean-up system to be suitable for PCR analysis, whereas the silica-purified DNA could be used without further treatment for PCR (data not shown).
Combination of methods
As DNA from Pseudomonas was most effectively extracted using the method of Zhou, while the method of Moré facilitated the extraction of DNA from Arthrobacter and Rhodococcus, the two methods for DNA isolation were combined to isolate DNA from a mixture of Pseudomonas, Arthrobacter and Rhodococcus in autoclaved soil. The selective isolation of DNA from Gram-positive and Gram-negative cells was confirmed by PCR analysis, using 23S rDNA-specific primers. Applying the method of Zhou to a mixture of all three strains in autoclaved soil led exclusively to the recovery of DNA from Pseudomonas, as demonstrated by the main PCR product being 250 bp in size. DNA from actinomycetes possesses a 100-bp insertion at this site in the 23S rDNA, resulting in a PCR product of 350 bp (Roller et al. 1992). After further lysis of the remaining cells by the method of Moré and subsequent PCR analysis, using the same primers, the main PCR product contained the actinomycete-characteristic 100-bp insertion. Thus, we were able to show that, by the combination of freeze-thaw cycles, SDS and proteinase K, Pseudomonas cells can be lysed and DNA extracted, whereas DNA from Arthrobacter and Rhodococcus cannot be obtained by this process, probably due to inefficient cell lysis. By further treatment of the samples, using glass beads, the remaining Arthrobacter and Rhodococcus cells in the sample are disrupted. The combination of both methods is therefore suitable to selectively isolate DNA from different microorganisms.
To test whether this selective cell lysis by a combination of the two methods can also be obtained with environmental samples, DNA was isolated from 0.5-g soil samples. Initial PCR analysis of 23S rDNA did not result in a 350-bp fragment from templates isolated by either method. This could be due to a strong bias of non-actinomycetes in the soil sample. To test this, the sample was spiked with 200 μl of Rhodococcus cells. PCR analysis with 23S rDNA-specific primers resulted in a 250-bp fragment, using the DNA extracted from soil by the method of Zhou as template. When the DNA that was isolated subsequently by the Moré method from the remaining microorganisms in the sample was used for PCR analysis, both a 250-bp fragment and a 350-bp fragment were visible after agarose gel electrophoresis. This shows that it is possible to avoid the isolation of actinomycete DNA from soil samples using the method of Zhou. Subsequently applying the method according to Moré allows the isolation of both actinomycete DNA and DNA from other Gram-positive and Gram-negative bacteria that could escape lysis by the method of Zhou.
Conclusions
E. coli expresses actinomycete genes very inefficiently, as only 20% of actinomycete promoters can be recognized by E. coli (Strohl 1992) and because of the high G+C content of actinomycete genes. For the construction of libraries, the DNA isolated by the method of Zhou is suitable for cloning in E. coli as host, because DNA from actinomycetes can hardly be extracted. This reduces the number of colonies carrying genes from actinomycetes that cannot be expressed in E. coli and therefore reduces the number of clones identified as negative. However, if a library is to be constructed in Streptomyces, it is useful to take DNA from a combination of both the Zhou and Moré methods. This reduces the amount of DNA obtained from Gram-negative organisms and allows the extraction of DNA from actinomycetes that are hard to lyse.
References
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169
Berthelet M, Whyte LG, Greer CW (1996) Rapid, direct extraction of DNA from soils for PCR analysis using polyvinylpolypyrrolidone spin columns. FEMS Microbiol Lett 138:17–22
Borneman J, Skroch PW, O’Sullivan KM, Palus JA, Rumjanek NG, Jansen JL, Nienhuis J, Triplett EW (1996) Molecular microbial diversity of an agricultural soil in Wisconsin. Appl Environ Microbiol 62:1935–1943
Degrange V, Bardin R (1995) Detection and counting of Nitrobacter populations in soil by PCR. Appl Environ Microbiol 61:2093–2098
Entcheva P, Liebl W, Johann A, Hartsch T, Streit WR (2001) Direct cloning from enrichment cultures, a reliable strategy for isolation of complete operons and genes from microbial consortia. Appl Environ Microbiol 67:89–99
Höss M, Pääbo S (1993) DNA extraction from pleistocene bones by a silica-based purification method. Nucleic Acids Res 21:3913–3914
Jacobsen CS, Rasmussen OF (1992) Development and application of a new method to extract bacterial DNA from soil based on separation of bacteria from soil with cation exchange resin. Appl Environ Microbiol 58:2458–2462
Johnston CG, Aust SD (1994) Detection of Phanerochaete chrysosporium in soil by PCR and restriction enzyme analysis. Appl Environ Microbiol 60:2350–2354
La Montagne MG, Michel FC, Holden PA, Reddy CA (2002) Evaluation of extraction and purification methods for obtaining PCR-amplifiable DNA from compost for microbial community analysis. J Microbiol Methods 49:255–264
Lee SY, Bollinger J, Bezdicek D, Ogram A (1996) Estimation of the abundance of an uncultured soil bacterial strain by a competitive quantitative PCR method. Appl Environ Microbiol 62:3787–3793
Martin-Laurent F, Philippot L, Hallet S, Chaussod R, Germon JC, Soulas G, Catroux G (2001) DNA extraction from soils: old bias for new microbial diversity analysis methods. Appl Environ Microbiol 67:2354–2359
Moré MI, Herrick JB, Silva MC, Ghiorse WC, Madsen EL (1994) Quantitative cell lysis of indigenous microorganisms and rapid extraction of microbial DNA from sediment. Appl Environ Microbiol 60:1572–1580
Picard C, Ponsonnet C, Paget E, Nesme X, Simonet P (1992) Detection and enumeration of bacteria in soil by direct DNA extraction and polymerase chain reaction. Appl Environ Microbiol 58:2717–2722
Porteous LA, Armstrong JL, Seidler RJ, Watrud LS (1994) An effective method to extract DNA from environmental samples for polymerase chain reaction amplification and DNA fingerprint analysis. Curr Microbiol 29:301–307
Purdy KJ, Embley TM, Takii S, Nedwell DB (1996) Rapid extraction of DNA and rRNA from sediments by a novel hydroxyapatite spin-column method. Appl Environ Microbiol 62
Roller C, Ludwig W, Schleifer KH (1992) Gram-positive bacteria with a high DNA G+C content are characterized by a common insertion within their 23S rRNA genes. J Gen Microbiol 138:1167–1175
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Selenska S, Klingmüller W (1991) DNA recovery and direct detection of tn5 sequences from soil. Lett Appl Microbiol 13:21–24
Steffan RJ, Goksoyr J, Bej AK, Atlas RM (1988) Recovery of DNA from soils and sediments. Appl Environ Microbiol 54:2908–2915
Strohl WR (1992) Compilation and analysis of DNA sequences associated with apparent streptomycete promoters. Nucleic Acids Res 20:961–974
Tebbe CC, Vahjen W (1993) Interference of humic acids and DNA extracted directly from soil in detection and transformation of recombinant DNA from bacteria and a yeast. Appl Environ Microbiol 59:2657–2665
Torsvik V, Goksoyr J, Daae FL (1990) High diversity in DNA of soil bacteria. Appl Environ Microbiol 56:782–787
Tsai YL, Olson BH (1991) Rapid method for direct extraction of DNA from soil and sediments. Appl Environ Microbiol 57:1070–1074
Tsai YL, Olson BH (1992) Rapid method for separation of bacterial DNA from humic substances in sediments for polymerase chain reaction. Appl Environ Microbiol 58:2292–2295
Volossiouk T, Robb EJ, Nazar RN (1995) Direct DNA extraction for PCR-mediated assays of soil organisms. Appl Environ Microbiol 61:3972–3976
Wikstrom P, Wiklund A, Andersson AC, Forsman M (1996) DNA recovery and PCR quantification of catechol 2,3-dioxygenase genes from different soil types. J Biotechnol 52:107–120
Wintzingerode F von, Gobel UB, Stackebrandt E (1997) Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev 21:213–229
Zhou J, Bruns MA, Tiedje JM (1996) DNA recovery from soils of diverse composition. Appl Environ Microbiol 62:316–322
Acknowledgements
This work was supported by BASF AG, Ludwigshafen and the Bundesministerium für Bildung und Forschung. We thank Ms Marian Turner for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kauffmann, I.M., Schmitt, J. & Schmid, R.D. DNA isolation from soil samples for cloning in different hosts. Appl Microbiol Biotechnol 64, 665–670 (2004). https://doi.org/10.1007/s00253-003-1528-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-003-1528-8