
Abstract
High-quality, humic-acid-free pure DNA is a prerequisite for functional and sequence-based approaches of metagenomics. In the present investigation, an improved extraction buffer was developed by making a combination of powdered activated charcoal (2%; w/v), polyvinyl poly pyrrolidone (2%; w/v), and CaCl2 (2%; w/v). This trio significantly improved the purity and yield of the metagenomic DNA from the hot spring’s hot and alkaline soil. The quality of extracted metagenomic DNA was successfully validated by PCR amplification and restriction enzymes. Besides, the thermophilic amylase encoding genes were also retrieved from these soil DNA samples. Extreme habitats I harbour low microbial biomass and, therefore, demand in-situ lysis of the microbial cells to access their genomes. The protocol can potentially extract DNA from geothermal spring habitats where the count of microbial cells is low.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Traditional cultivation approaches lose a significant chunk of microorganisms of habitat due to their inability to simulate the environmental conditions in the laboratory (Stewart 2012). This count further reduces in extreme environments where the microbial biomass is comparatively low over the mesophilic habitats (Saxena et al. 2017). Geothermal hot springs represent one of the unique extreme habitats populated by microbial communities that are highly heterogeneous and have unique properties that can be exploited in the industrial sector (de León et al. 2013; Ward et al. 1998; Urbieta et al. 2014 2015). Several researchers have explored the hot and alkaline spring sites for analyzing microbial diversity and retrieving industrially relevant catalysts such as thermophilic carbohydrate degrading enzymes, lipolytic enzymes, hydrocarbon-degrading enzymes(López-López et al. 2015; Reichart et al. 2021; Saxena et al. 2017; Verma et al. 2021).
Traditional culturing techniques are insufficient to isolate microbes from low microbial load soil of alkaline hot springs; however, metagenomics have emerged as a robust tool for providing new insights into microbial communities harbouring in these extreme habitats. This non-conventional approach for analyzing microbes bypasses the tedious cultivation approaches of microbes on petri-dishes. It chiefly relies on cloning the direct community DNA of an environmental sample. Therefore, extraction of high molecular weight, humic-acid-free quality DNA from the habitat is the primary requisite of this technique. Soil DNA extraction is more challenging than other environmental materials due to the presence of humic acids (Solomon et al. 2016). Various approaches developed in the past encountered the common problem of humic acid co-precipitation with metagenomes throughout the isolation process. Humic acid is of concern because it interferes with downstream molecular biology processes such as PCR and restriction digestion by inhibiting the activity of Taq polymerase and restriction enzymes (Wilson 1997).
Previously, for metagenome extraction, many chemicals such as Cetyltrimethylammonium bromide (CTAB) (Zhou et al. 1996), Polyvinyl Poly Pyrrolidone (PVPP), Powdered Activated Charcoal (PAC) (Desai and Madamwar 2007; Verma and Satyanarayana 2011), Gel electrophoresis, and CaCl2, Aluminium Chloride, and CsCl2 density centrifugation were employed (Frostegård et al. 1999; Leff et al. 1995; Zhou et al. 1996). This paper aims to develop a procedure for extracting high-molecular-weight and quality metagenomic DNA from soil samples of hot and alkaline environments. The previous protocols and commercial kits on soil DNA isolation were assessed during this investigation on low biomass exhibiting soil samples of hot springs; however, most protocols compromise either DNA purity or its yield. Therefore, an improved protocol was developed by incorporating the CaCl2to extract soil DNA from hot and alkaline thermal springs. Thus, obtained metagenomic DNA significantly improved purity and yield from such extreme soil samples. The protocol was also successfully validated for downstream molecular biology processing, such as amplification and restriction digestion. The developed protocol can extract quality DNA from hot spring sediments and soils without compromising the DNA yield and purity.
Material and methods
Chemicals used in this study were of molecular biology grade and were obtained from Himedia, SRL, and Thermo-Fisher. HiPurA Soil DNA Purification Kit (Himedia) and Xpress DNA soil kit (Mag Genome) were also used.
Collection of soil samples
Sediments and soil samples were taken from two locations in Tapovan hot springs; Uttarakhand (Longitude 30°29′27.2′′N and Latitude 79°38′48.0′′E) with temperatures of 88.8 °C and 55 °C, respectively, and pH was 8.0. Samples were collected with a sterile spatula and placed in sterile plastic bags and tubes before being stored at 4 °C in the laboratory and at − 20 °C for up to one year for further analysis.
Standardization of extraction protocol for metagenomic DNA from thermal hot-spring soil
For the isolation and purification of metagenomic DNA from sediments and soil samples, ten methods previously published and their modified forms were used (Bashir et al. 2015; Biver and Vandenbol 2013; Devi et al. 2015; Singh et al. 2014; Verma et al. 2021; Zhou et al. 1996). Three kits [Xpress soil DNA Kit, HiPurA soil DNA isolation kit (two alternate methods)] were also used in the current study for comparative analysis. The optimized protocol was also attempted to extract metagenomes from other soil samples of extreme environments (Chawalpani, Atri, and Manikaran).
Developed protocol for metagenome DNA extraction
For standardizing the DNA extraction protocol, all experiments were run in triplicates. Sediment/soil samples of 5.0 gm were taken in triplicates into sterile 50 ml falcon tubes and treated with 15 ml extraction buffer [100 mM Tris–HCl: pH-8.0, 100 mM sodium EDTA: pH-8.0, 1.5 M NaCl, 1% CTAB (w/v), 2% PVPP (w/v), 100 mM CaCl2, lysozyme (10 mg/ml), Proteinase K (10 mg/ml), and 100 mM sodium phosphate buffer: pH-8.0]. Two grams of poly-activated charcoal were added to the soil suspension. This mixture was vortexed and incubated at 37 °C for 45 min in an incubator shaker at 200 rpm. After that, 3.0 ml of 20% (w/v) SDS was mixed into the soil solution and maintained at 65 °C in a water bath for 1 h with intermittent mixing every 15 min. The lysed cell soup containing genomic DNA was centrifuged at 10,000 g for 20 min at 4 °C to collect the supernatant. An equal volume of supernatant and phenol, chloroform, and isoamyl alcohol (25:24:1; pH-8.0) solution was centrifuged at 12,000g for 20 min at 4 °C to obtain the aqueous phase. The DNA was precipitated by treating the aqueous phase with 0.1 V of 3 M sodium acetate (pH- 5.2) and 0.4 V of 30% (w/v) PEG-8000, followed by 20 min incubation at − 20 °C. The precipitated DNA was obtained by centrifuging the tube for 20 min at 14,000g at 4 °C, and it was washed with 70% (v/v) ethanol and air-dried. The pellet was dissolved in 1 ml of 1X TE buffer (pH 8.0), where an equal volume of chloroform and isoamyl alcohol (24:1) solution was further added. As mentioned above, the aqueous phase was obtained, and DNA was precipitated by adding 0.7 V of isopropanol and incubated at room temperature for 20 min. Finally, metagenomic DNA was pellet out by centrifugation at 14,000×g for 20 min at 4 °C. The DNA pellet was washed with 70% (v/v) ethanol and air-dried and dissolved in 50 µL 1X TE buffer (pH 8.0) and stored at low temperature (4 °C or − 20 °C) for further analysis.
Estimation of DNA purity and yield
Standard agarose gel electrophoresis was used to analyze the extracted metagenomic DNA, which involved loading equal amounts of DNA and a marker onto the agarose gel (Sambrook et al. 1989). The purity and concentration of DNA were determined using a Nano Vue plus spectrophotometer. The yield of isolated DNA was determined by measuring absorbance at 260 nm. The purity of DNA samples was determined using the A 260/A 280 (DNA/protein) and A 260/A 230 (DNA/humic acid) ratios to assess protein and humic acid contamination. A 260/A 280 ratio of less than 1.8 indicates protein contamination (Sambrook et al. 1989), whereas A 260/A 230 value of less than 2 indicates humic acid contamination (Ning et al. 2009). On a 0.8% agarose gel, DNA samples were visualized and electrophoresed at 70 V for 45 min. Ethidium bromide (10 mg/ml) was used to stain the gel, and DNA was identified using a UV trans illuminator (VilberLourmat).
Validation of the developed protocol
PCR amplification of 16S rRNA gene
To validate the methodology used in this study, the extracted metagenomic DNA was used as a template for amplification of 16S rRNA gene using universal bacterial specific 16S rRNA primers as Forward 5'-AGAGTTTGATCCTGGCTCAG-3' and reverse 5'-GGTTACCTTGTTACGACTT -3' (Srinivasan et al. 2015). The total reaction volume for all samples was 25 μL, which included the master mix (Himedia), DNA template-50–100 ng, forward and reverse primers in 10 pmol concentrations, and molecular grade water to balance the volume of the reactions. The PCR amplification was carried out using a thermal cycler (Eppendorf), and the optimal amplification conditions were as follows: initial denaturation: 95 °C for 5-min, denaturation: 94 °C for 30 secs, annealing: 58 °C for 30 secs, extension: 72 °C for 80 secs, final extension: 72 °C for 10 min, and hold at 4 °C. DNA from various published techniques was also used to amplify DNA from the same soil samples for further comparison.
Restriction digestion of metagenome
1 μg of isolated metagenomes from TP-B and TP-D soil samples were subjected to partial digestion with 1U of each EcoRI, BamHI, and Sau3AI restriction enzyme using the standardized procedure. The reaction mixture had a volume of 20 μL, and an equal amount of extracted DNA (1 μg) was digested with the respective enzymes at 37 °C for 1 h, followed by heat inactivation in a water bath at 65 °C for 20 min. The undigested environmental DNA (eDNA) was taken as a control.
Amplification of amylase gene using isolated metagenome
PCR was performed for amylase gene amplification utilising metagenome of TP-B and TP-D with chosen primers FP (5'-GGAGACAUATGAAACAACAAAAACGGCT-3') and RP (5'-GGGAAAGUGGGGCAAAATAA AAAAACGG-3') (Nathan and Nair 2013) and run-in triplicate reaction. The final reaction volume for two DNA samples was 25 µL, which included a master mix (Himedia), 50–100 ng of DNA template, forward and reverse primers at 10 pmol concentrations, and molecular grade water to balance the volume of the reactions. The amylase gene was amplified using a thermal cycler (Eppendorf), and the best conditions for amplification were initial denaturation at 95 °C for 5 min, denaturation at 94 °C for 30 s, annealing at 60 °C for 30 s, extension at 72 °C for 80 s, final extension at 72 °C for 10 min, and holding at 4 °C.
Effect of storage conditions on yield of DNA
The isolated metagenomic DNA is stored at – 20 °C (in triplicates), and the DNA concentration was measured for up to 6 months. The absorbance of the samples from different samples was taken at the wavelength of 260 nm (DNA), 230 nm (humic acid), and 280 nm (protein). The DNA concentration was calculated using the standard formula.
Statistical analysis
All extractions were done in triplicates and absorbance are average out for 260/230 and 260/280 ratio calculations. All the results were expressed as mean \(\pm\) standard deviation.
Results and discussion
Geothermal hot springs are one of those extreme ecosystems inhabited by extraordinarily varied microbial communities with unique features. Ward et al. (1990) used 16S rRNA sequencing data to reveal numerous uncultured microorganisms in hot springs and highlighted the problems with the traditional culture methods. Panda et al. (2016) reported the presence of many taxonomically novel unsolved 16S rRNA gene sequences from alkaline hot springs of north-eastern India (Panda et al. 2016). Hence, such sites have been proven as a reservoir of novel enzymes, antibiotics, and other biomolecules (López-López et al. 2015). However, the merit of this technique is marred by DNA yield due to low microbial load from such habitats and the presence of polyphenol/humic acid contamination which hamper the further use of isolated metagenomes in restriction digestion, PCR amplification and metagenomic library construction (Belkova et al. 2007; Singh et al. 2014; Robe et al. 2003; Whitehouse and Hottel 2007). Hence having a good quality inhibitor-free eDNA is imperative, and DNA isolation methods have high importance (Siddhapura et al. 2010).
Various groups have developed several protocols in the past two decades for the isolation of metagenome from alkaline hot springs (Bashir et al. 2015; Biver and Vandenbol, 2013; Devi et al. 2015; Singh et al. 2014; Verma and Satyanarayana 2011; Verma et al. 2017; Zhou et al. 1996). In this investigation, seven previously reported protocols and their modified forms, and kit-based protocols, were used to compare the yield and purity of metagenomes obtained from two different soil samples (TP-B-88.8 °C and TP-D-55.5 °C) of Tapovan hot spring. These soil samples differed from each other due to their chemical elemental composition at different soil depths and varying temperatures that may represent diverse microbiota (Belkova et al. 2007; Miller et al. 2009). Three chemicals (PAC, PVPP, and CaCl2) were incorporated into the extraction buffer to remove humic acids. PAC is a highly porous material with a large surface area and has been successfully used to purify soil-based metagenomic DNA (Arvanitoyannis et al. 2008). It exhibits a solid physical adsorption force and is an adsorber of contaminants/soil impurities on its surface. At the same time, PVPP removes the phenolic group containing humic acid by forming hydrogen bonds with phenolic substances and forming a PVPP-phenol complex (Frostegård et al. 1999). Calcium ions of CaCl2, being cation, bind to anionic functional groups of polyphenols like- COO− and OH−, thus preventing oxidation of phenol groups containing humic acid to quinones. These quinones form a covalent bond with the precipitated DNA and cause a hindrance in the activity of restriction enzymes and DNA polymerases (Verma et al. 2017). Verma et al. (2017) incorporated CaCl2 into the extraction buffer and obtained quality DNA. However, the extraction buffer used by Verma and colleagues was devoid of PAC and PVPP. Therefore, to take advantage of the characteristics of PAC and PVPP, these chemicals were added to the present investigation's extraction buffer. This way, a three-fold improvement may be achieved to obtain the quality metagenomic DNA from soil samples.
Comparative analysis of metagenomic DNA extraction protocols for assessment of DNA purity and yield
The quality and quantity of extracted metagenomic DNA using the developed protocol of this investigation were compared with the other reported methods of metagenome isolation (Table 1; Fig. 1a, b). The developed protocol yielded 62.3 ± 1.52 ng/μL and 70.6 ± 2.08 ng/μL of DNA from the respective soil samples. The purity ratios in both the samples were satisfactory as TP-B and TP-D showed the value of A260/280 nm-1.65 and 1.67, respectively, while values of A260/230 nm were 2.04 and 2.24, respectively. The values of purity ratios and DNA yield are summarized in Table 1.

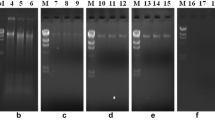
Purity and yield assessment of metagenome isolated from soil samples TP-B (A) and TP-D (B), using various reported protocols and current protocol on agarose gel electrophoresis (0.8%). A and B: L1-Zhou et al. 1996, L2-Biver and Vandenbol (2013), L3-Singh et al. 2014, L4- Himedia kit alternative protocol, L5-Current protocol, L6- Xpress soil DNA kit, L7- Verma et al. 2017, L8- 1 Kb DNA Ladder, L9- Devi et al. 2015, L10- Verma and Satyanarayana (2011), L11- Bashir et al. 2015, L12-Himedia kit protocol-1
In comparison with the protocol of Verma et al. (2017), better purity ratios (A260/280–1.65, 1.67 and A260/230–2.04, 2.24) and DNA yield (62.3 ng/µL, 70.6 ng/µL) were obtained from the developed protocol during this investigation. Similarly, the present findings were significantly better than the protocol published by Verma and Satyanarayana (2011), which does not consider CaCl2 in their extraction buffer. These DNA were from hot environmental soil samples (Table 1). The maximum eDNA yield was 62.3 ng/µL (TP-B) and 70.6 ng/µL (TP-D) through the current protocol, which was almost double the DNA yield (36.9 ng/µL (TP-B) and 30.4 ng/µL (TP-D) from the respective samples using the protocol of Verma and Satyanarayana (2011). The maximum yield of eDNA for both samples was given 249 ng/µL (TP-B) and 232.5 ng/µL (TP-D) by Biver and Vandenbol (2013); however, the A260/230 ratio was low 0.48–0.6. The current protocol gave a moderate DNA yield, and Himedia kit protocols gave a low yield. However, the Express soil DNA kit protocol gives moderate yield but low purity, so it cannot be used for metagenomic library preparation. The protocol developed by Zhou et al. (1996) and Biver and Vandenbol (2013) provided high DNA yield;however, all these protocols could not achieve the required purity of soil DNA. The yield in these published protocols was high due to the harsh detergents used in the study and did not follow the double precipitation of DNA. However, such obtained DNA is of no use and needs to be further purified either using gel extraction or passing through silica-based columns that further reduce the DNA yield significantly to achieve quality DNA. The purity and quality of DNA by processing soil DNA with commercial kits were poor compared to manual methods. Thus obtained DNA cannot be processed for metagenomic library construction due to further loss of DNA in various steps involved (Singh et al. 2014; Verma et al. 2017). Other protocols used during the investigation to compare extracted DNA are presented in Table 1. These show the significance of the PAC, PVPP, and CaCl2. The developed protocol was reproducible on other soil samples of extreme environments at the level of DNA yield and purity (Table 2).
Validation of the developed protocol
PCR amplification of bacterial specific 16S rRNA gene
After the quality check, it was crucial to evaluate the metagenomic DNA for its suitability in restriction digestion and PCR amplification to validate the quality. Therefore, the obtained DNA was processed for16S rRNA amplification and restriction digestion. The extracted metagenomic DNA from the existing protocol, as well as the previous procedures stated, were employed for the amplification of bacterial-specific the16S rRNA genes. Amplification of 16S rRNA gene using universal bacterial specific primer showed an amplicon of size ̴1.5 kb using metagenome isolated through optimized protocol from TP-B and TP-D soil samples (Fig. 2). PCR amplification of the 16S rRNA gene was also performed using metagenomes extracted from previously published protocols and commercial kits. Thick bands were observed from metagenomes isolated through the developed protocol; however, no amplification was found from DNA extracted through Bashir et al. (2015) and Devi et al. (2015). Faint and thin bands were observed from metagenomes recovered from Singh et al. (2014), Verma et al. (2017) and Verma and Satyanarayana (2011) because of the presence of a low concentration of humic acid impurities along with the metagenome. Various researchers have adopted similar approaches to validate the metagenome quality (Singh et al. 2014; Verma et al. 2017). DNA isolated using protocols of Singh et al. (2014), Verma and Satyanarayana (2011), and Verma et al. (2017) have also been used as a template for PCR amplification; however, the titres/intensities of the amplicons were comparatively less due to the presence of humic acid substances. Metagenomes obtained from Bashir et al. (2015) and Devi et al. (2015) exhibited a significantly high content of humic impurities, hence 16SrRNA genes were not amplified even after dilution (Fig. 2). The humic acid chelates the required metal ions. It thus inhibits the optimum activity of various enzymes (Sidstedt et al. 2015).

PCR amplification of bacterial specific 16S rRNA gene using universal primers. A and B: PCR amplification of 16S rRNA gene products of soil samples TP-B and TP-D isolated using various protocols- L1- Current protocol, L2- Verma et al. 2017, L3- Verma and Satyanarayana 2011, L4- 100 bp DNA ladder, L5- Devi et al. 2015, L6-Bashir et al. 2015, L7-. Singh et al. 2014
Assessment of quality of extracted eDNA for molecular techniques
Restriction digestion of eDNA for the construction of a metagenomic library
The extracted eDNA quality was also validated using restriction digestion. Metagenome isolated from other protocols from the same soil samples was hard to digest and showed hindrance in the activity of the respective restriction enzymes (Fig. 3a, b). Because the improved methodology yields a metagenome free of humic acid content, restriction digestion of metagenome from soil sample TP-D yielded a smear of digested small DNA fragments using three different enzymes EcoRI, BamHI, and Sau3AI and was found to be suitable for the construction of a metagenomic library (Fig. 3c). Hence, the developed protocol can potentially remove the humic acid substance to a significant level that does not hinder the activity of restriction enzymes. Humic acid chelates the various divalent metal ions, thus trapping the activity (Sidstedt et al. 2015; Tebbe and Vahjen 1993). Several metagenome-based protocols follow such validation to check the quality of DNA (Singh et al. 2014; Verma and Satyanarayana 2011).

Quality assessment of extracted metagenomic DNA using BamHI restriction digestion on 1.2% of Agarose gel electrophoresis. A and B: Undigested and digested metagenome from soil sample TP-B isolated using different protocols. L1 and L2- Singh et al. 2014, L3 and L4-Verma and Satyanarayana 2011, L5- 1 Kb lambda DNA marker, L6 and L7- Verma et al. 2017.C: Undigested and digested metagenome from soil sample TP-D using Current protocol. L1- 100 bp DNA ladder, L2- Undigested metagenome, L3- EcoRI digested DNA, L4-BamHI digested DNA, L5-Sau3AI digested DNA
PCR amplification of α-amylase gene
To further check the significance of the extracted metagenomic DNA applicability in retrieving industrially important enzyme coding genes, we successfully amplified the thermo-alkali-stable amylase encoding genes from both the eDNA sample (TP-B and TP-D). The amplicon of 1.5 kb was visualized on an agarose gel (Fig. 4). A thermostable alpha amylase gene was also amplified from eDNA of Atri, Odisha hot spring and further cloned and sequenced (Chauhan et al. 2023). Impure environmental DNA is prone to degradation over time due to humic substances and other polyphenolics. It thus needs to be processed at the earliest after their extraction. DNA yield of extracted metagenomic DNA of TP-D and TP-B showed an insignificant loss in DNA on their storage at low temperatures (− 20 °C). The TP-B and TP-D samples retained 100% DNA content up to the fifth month and 96.8% and 98.5% of the total DNA after the storage of 6 months at − 20 °C, respectively (data not shown here). Thus, the developed protocol is convincing enough and provides a way to achieve high-quality metagenomic DNA of extreme habitats like thermal geysers and hot springs without compromising its DNA yield and purity.

Validation of purity of extracted metagenomic DNA by PCR amplification of α-amylase gene for molecular cloning. L1- Amylase gene amplified using metagenome TP-B, L2- Amylase gene amplified using metagenome TP-D, L3-100 bp DNA Ladder
Conclusion
The PAC, PVPP, and CaCl2 developed in the present investigation offer an alternative to extracting humic acid-free metagenomic DNA from extreme environmental samples, especially soil. The protocol has significance because we were simultaneously achieving high DNA yield along with their purity, which is usually missing in most of the previously published protocols and commercial soil DNA kits. As the protocol relies on direct lysis of the samples, the extracted metagenomic DNA could be a good representative of various microbial communities, which is highly required in the metagenome-based study to avoid sample biasing. The protocol was successfully validated by various restriction enzymes and DNA polymerases and thus can be explored for various molecular downstream processing such as shotgun sequencing and metagenomic library construction.
References
Arvanitoyannis I, Kassaveti A, Ladas D (2008) Food waste treatment methodologies. In: Food science and technology, waste management for the food industries, Arvanitoyannis I (ed) 1st edn. Academic Press, USA, pp 345–410.https://doi.org/10.1016/B978-012373654-3.50009-2
Bashir Y, Rather IA, Konwar BK (2015) Rapid and simple DNA extraction protocol from goat rumen digesta for metagenomic analysis. Pak J Pharm Sci 28:2305–2309
Belkova NL, Tazaki K, Zakharova JR, Parfenova VV (2007) Activity of bacteria in water of hot springs from Southern and Central Kamchatskaya geothermal provinces, Kamchatka Peninsula, Russia. Microbiol Res 162:99–107. https://doi.org/10.1016/j.micres.2006.01.006
Biver S, Vandenbol M (2013) Characterization of three new carboxylic ester hydrolases isolated by functional screening of a forest soil metagenomic library. J Ind Microbiol Biotechno l40:191–200. https://doi.org/10.1007/s10295-012-1217-7
Chauhan G, Kumar V, Arya M, Kumari A, Srivastava A, Khanna P, Sharma M (2023) Mining of thermostable alpha-amylase gene from geothermal springs using a metagenomics approach. J Pure Appl Microbiol 17:362–370. https://doi.org/10.22207/JPAM.17.1.26
de León KB, Gerlach R, Peyton BM, Fields MW (2013) Archaeal and bacterial communities in three alkaline hot springs in heart lake geyser basin, yellowstone national park. Front Microbiol 12:330. https://doi.org/10.3389/fmicb.2013.00330
Desai C, Madamwar D (2007) Extraction of inhibitor-free metagenomic DNA from polluted sediments, compatible with molecular diversity analysis using adsorption and ion-exchange treatments. Bioresour Technol 98:761–768. https://doi.org/10.1016/j.biortech.2006.04.004
Devi SG, Fathima AA, Radha S, Arunraj R, Curtis WR, Ramya M (2015) A rapid and economical method for efficient DNA extraction from diverse soils suitable for metagenomic applications. PLoS ONE 10: e0132442. https://doi.org/10.1371/journal.pone.0132441
Frostegård Å, Courtois S, Ramisse V, Clerc S, Bernillon D, le Gall F, Jeannin P, Nesme X, Simonet P (1999) Quantification of bias related to the extraction of DNA directly from soils. Appl Environ Microbiol 65:5409–5420. https://doi.org/10.1128/aem.65.12.5409-5420.1999
Leff LG, Dana JR, McArthur JV, Shimket LJ (1995) Comparison of methods of DNA extraction from stream sediments. Appl Environ Microbiol 61:1141–1143. https://doi.org/10.1128/aem.61.3.1141-1143.1995
López-López O, Knapik K, Cerdán ME, González-Siso MI (2015) Metagenomics of an alkaline hot spring in Galicia (Spain): Microbial diversity analysis and screening for novel lipolytic enzymes. Front Microbiol 6:1291. https://doi.org/10.3389/fmicb.2015.01291
Miller SR, Williams C, Strong AL, Carvey D (2009) Ecological specialization in a spatially structured population of the thermophilic cyanobacterium mastigocladuslaminosus. Appl Environ Microbiol 75:729–734. https://doi.org/10.1128/AEM.01901-08
Nathan S, Nair M (2013) Engineering a repression-free catabolite-enhanced expression system for a thermophilic alpha-amylase from Bacillus licheniformis MSG. J Biotechnol 168:394–402. https://doi.org/10.1016/j.jbiotec.2013.09.016
Ning J, Liebich J, Kästner M, Zhou J, Schäffer A, Burauel P (2009) Different influences of DNA purity indices and quantity on PCR-based DGGE and functional gene microarray in soil microbial community study. Appl MicrobiolBiotechnol82, 983–993.https://doi.org/10.1007/s00253-009-1912-0
Panda AK, Bisht SS, de Mandal S, Kumar NS (2016) Bacterial and archeal community composition in hot springs from Indo-Burma region. North-East India AMB Express 6:111. https://doi.org/10.1186/s13568-016-0284-y
Reichart NJ, Bowers RM, Woyke T, Hatzenpichler R (2021) High potential for biomass-degrading enzymes revealed by hot spring metagenomics. FrontMicrobiol 12:914. https://doi.org/10.3389/fmicb.2021.668238
Robe P, Nalin R, Capellano C, Vogel TM, Simonet P (2003) Extraction of DNA from soil. Eur J Soil Biol 39:183–190. https://doi.org/10.1016/S1164-5563(03)00033-5
Sambrook J, Fritsch EF, Maniatis T (1989). In: Molecular cloning: a laboratory manual, cold spring harbor laboratory, Cold Spring Harbor, New York.
Saxena R, Dhakan DB, Mittal P, Waiker P, Chowdhury A, GhatakA SVK (2017) Metagenomic analysis of hot springs in central india reveals hydrocarbon degrading thermophiles and pathways essential for survival in extreme environments. Front Microbiol 7:2123. https://doi.org/10.3389/fmicb.2016.02123
Siddhapura PK, Vanparia S, Purohit MK, Singh SP (2010) Comparative studies on the extraction of metagenomic DNA from the saline habitats of Coastal Gujarat and Sambhar Lake, Rajasthan (India) in prospect of molecular diversity and search for novel biocatalysts. Int J BiolMacromol47:375–379. https://doi.org/10.1016/j.ijbiomac.2010.06.004
Sidstedt M, Jansson L, Nilsson E, Noppa L, Forsman M, Rådström P, Hedman J (2015) Humic substances cause fluorescence inhibition in real-time polymerase chain reaction. Anal Biochem 487:30–37. https://doi.org/10.1016/j.ab.2015.07.002
Singh R, Devi T, Verma V, Rasool S (2014) Comparative studies on the extraction of metagenomic DNA from various soil and sediment samples of jammu and kashmir region in prospect for novel biocatalysts. IOSR J Environ Sci Toxicol Food Technol 8(4):46–56. https://doi.org/10.9790/2402-08414656
Solomon S, Kachiprath B, Jayanath G, Sajeevan TP, Bright Singh IS, Philip R (2016) High-quality metagenomic DNA from marine sediment samples for genomic studies through a preprocessing approach. 3 Biotech 6:160. https://doi.org/10.1007/s13205-016-0482-y
Srinivasan R, Karaoz U, Volegova M, MacKichan J, Kato-Maeda M, Miller S, Nadarajan R, Brodie EL, Lynch SV (2015) Use of 16S rRNA gene for identification of a broad range of clinically relevant bacterial pathogens. PLoS ONE 10:e0117617. https://doi.org/10.1371/journal.pone.0117617
Stewart EJ (2012) Growing unculturable bacteria. J Bacteriol 16:4151–4160. https://doi.org/10.1128/JB.00345-12
Tebbe CC, Vahjen W (1993) Interference of humic acids and DNA extracted directly from soil in detection and transformation of recombinant DNA from bacteria and a yeast. Appl Environ Microbiol 59:2657–2665. https://doi.org/10.1128/aem.59.8.2657-2665.1993
Urbieta MS, Donati ER, Chan KG, Shahar S, Sin LL, Goh KM (2015) Thermophiles in the genomic era: Biodiversity, science, and applications. BiotechnolAdv 33:633–647. https://doi.org/10.1016/j.biotechadv.2015.04.007
Urbieta MS, Toril EG, Alejandra Giaveno M, Bazán ÁA, Donati ER (2014)Archaeal and bacterial diversity in five different hydrothermal ponds in the Copahue region in Argentina. SystApplMicrobiol37:429–441. https://doi.org/10.1016/j.syapm.2014.05.012
Verma S, Meghwanshi GK, Kumar R (2021) Current perspectives for microbial lipases from extremophiles and metagenomics. Biochimie 7:171. https://doi.org/10.1016/j.biochi.2020.12.027
Verma D, Satyanarayana T (2011) An improved protocol for DNA extraction from alkaline soil and sediment samples for constructing metagenomic libraries. ApplBiochemBiotechnol165:454–464. https://doi.org/10.1007/s12010-011-9264-5
Verma SK, Singh H, Sharma PC (2017) An improved method suitable for isolation of high-quality metagenomic DNA from diverse soils. 3 Biotech 182:23–36. https://doi.org/10.1007/s13205-017-0847-x
WardDM FMJ, Nold SC, Bateson MM (1998) A natural view of microbial biodiversity within hot spring cyanobacterial mat communities. MicrobiolMolBiol Rev 345:63–65. https://doi.org/10.1128/mmbr.62.4.1353-1370.1998
Whitehouse CA, Hottel HE (2007) Comparison of five commercial DNA extraction kits for the recovery of Francisella tularensis DNA from spiked soil samples. Mol Cell Probes 21:92–96. https://doi.org/10.1016/j.mcp.2006.08.003
Wilson IG (1997) Inhibition and facilitation of nucleic acid amplification. Appl Environ Microbiol 63:3741–3751. https://doi.org/10.1128/aem.63.10.3741-3751.1997
Zhou J, Bruns MA, Tiedje JM (1996) DNA recovery from soils of diverse composition. Appl Environ Microbiol 62:316–322. https://doi.org/10.1128/aem.62.2.316-322.1996
Acknowledgements
The UGC-Senior Research Fellowship given to Ms Garima Chauhan, UGC-non-NET PhD Fellowship given to Ms Meghna Arya from BBAU and Project Assistant Fellowship from CST-UP to Mr Vikas Kumar are duly acknowledged. The funding for this research work was given to Dr Monica Sharma by the Uttar Pradesh Council of Science & Technology (Grant number CST/8275).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We declare that there is no conflict of interest.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chauhan, G., Arya, M., Kumar, V. et al. An improved protocol for metagenomic DNA isolation from low microbial biomass alkaline hot-spring sediments and soil samples. 3 Biotech 14, 34 (2024). https://doi.org/10.1007/s13205-023-03824-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-023-03824-4