
Abstract
The gene (inuA) encoding exo-inulinase (EC 3.2.1.80) was cloned from the thermophilic Geobacillus stearothermophilus (Bacillus stearothermophilus) KP 1289 growing at between 41°C and 69°C. The inuA gene consisted of 1,482 bp encoding a protein of 493 amino acids. The deduced polypeptide of molecular mass (M) 56,744 Da showed strong sequence similarity to Pseudomonas mucidolens exo-inulinase, Bacillus subtilis levanase, Paenibacillus polymyxa (Bacillus polymyxa) fructosyltransferase, and so on, indicating that the enzyme belonged to glycosyl hydrolase family 32. The M of the purified exo-inulinase, expressed in Escherichia coli HB101, was estimated as approximately 54,000 Da by both SDS-PAGE and gel filtration. These results suggested that the active form of the enzyme is a monomer. The enzyme was active between 30 and 75°C with an optimum at 60°C. The properties were identical to those of the native enzyme. Additionally, for the first time for a prokaryotic GH32 protein, crystals of the recombinant enzyme were obtained.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inulin, a widespread polyfructan in plants, consists of linear β-2, 1-linked polyfructose chains that are attached to terminal sucrose molecules. This reserve carbohydrate is accumulated in the roots and tubers of plants such as Jerusalem artichoke, chicory, and dahlia, and is of great interest as a raw material for the production of fructose syrups or chemicals such as ethanol (Pandey et al. 1999; Vandamme and Derycke 1983). Inulin-hydrolyzing enzymes are classified into two types: endo-inulinase (2, 1-β-d-fructan fructanohydrolase, EC 3.2.1.7) and exo-inulinase (β-d-fructan fructanohydrolase, EC 3.2.1.80) according to the mode of action on inulin. Invertase (β-fructofuranosidase EC 3.2.1.26) is distinguished from the two inulinases by its lower, or complete lack of, affinity for inulin. Inulin is insoluble in cold water and only slightly (5%) soluble even in water at 55°C. The complete hydrolysis of inulin by inulinase is a single-step enzymatic reaction, and can attain yields of 95% fructose syrup. This enzyme reaction is attractive for the industrial production of ultrahigh-fructose syrups, because the conventional method with starch as the starting material needs at least three enzymatic steps and gives only 45% fructose syrups at best (Vandamme and Derycke 1983). Thus, a thermostable and inulinolytic enzyme would be expected to play an important role in food and chemical industries in which fructose syrup is widely applied.
Inulinases from plants, yeasts, fungi, and mesophilic bacteria have been extensively studied, and it is known that only a few thermophilic enzymes have an optimum temperature of 60°C or higher (Ferrira et al. 1991; Kato et al. 1999; Pandey et al. 1999; Uzunova et al. 2001). Recently, a β-fructofuranosidase gene of the hyperthermophile Thermotoga maritima has been expressed in Escherichia coli (Liebl et al. 1998). The purified recombinant enzyme is extremely thermostable and active on both sucrose and inulin, and is classified as a β-fructosidase (EC 3.2.1.26). We previously reported the purification and properties of a thermostable inulinase (EC 3.2.1.80) from Geobacillus stearothermophilus (Bacillus stearothermophilus) KP1289 (Kato et al. 1999). The thermophile grew between 41°C and 69°C and produced inulinase II, which was active at 30–75°C with an optimum at 60°C. The ratio of specificity constants (k cat/K m) for inulin, sucrose, and raffinose was 1.0:0.50:0.16. This enzyme is one of the most thermostable exo-inulinases known. In this paper, we describe the cloning of the exo-inulinase (inulinase II) gene from G. stearothermophilus KP1289 for molecular analysis. We have also characterized the enzyme expressed in E. coli in order to compare its enzymatic properties with those of the native enzyme. Additionally, crystals of the recombinant enzyme are described.
Materials and methods
Bacterial strains, culture conditions, and DNA manipulations
The G. stearothermophilus KP1289 used in this study was a wild strain from our culture collection. Slant and liquid cultures of the bacterial strain were incubated at 55 or 60°C as previously described (Kato et al. 1999). All DNA manipulations were performed essentially as described by Sambrook et al (1989). E. coli JM109 was used for library construction, overexpression, and preparation of subclones for DNA sequencing. E. coli DH5 α, HB101, BL21, MV1184, and AD494 were also used for overexpression. pUC vectors were used as a routine cloning vectors. E. coli transformants were cultured at 37°C in LB medium supplemented with 100 μg/ml ampicillin (LA). Restriction endonucleases and DNA modifying enzymes were purchased from New England Biolabs (Beverly, Mass.), TOYOBO (Osaka, Japan) and TAKARA (Kyoto, Japan), and were used as recommended by their respective suppliers. Polymerase chain reaction (PCR) was performed with rTaq DNA polymerase (TOYOBO) for 30 cycles according to the manufacturer's directions.
Cloning of the thermostable exo-inulinase gene and its flanking regions
A genomic library was used for colony hybridization with a digoxigenin (DIG)-labeled probe. DNA labeling, Southern blotting and DNA hybridization were performed as recommended by the supplier of the DIG reagents (Boehringer Mannheim, Germany).
Two sets of degenerate PCR primers were derived, one from the N-terminal protein sequence (MKTHNSE) the other from a conserved sequence (FRDPKVFW) in glycosyl hydrolase family 32. The PCR fragment was labeled with DIG, and this labeled fragment was used as the probe for Southern hybridization.
Genomic DNA isolated from the thermophile was digested with restriction enzymes, separated according to size by agarose gel electrophoresis, and then subjected to the Southern blotting procedure using the DIG-labeled fragment. A single hybridizing band was observed with various restriction enzymes. The genomic DNA was digested with HindIII and XbaI, and DNA fragments of approximately 6–7 kb were collected. The collected fragments, including those that would contain the thermostable exo-inulinase gene, were cloned into the HindIII and XbaI sites of pUC119. The ligation mixture was used to transform E. coli JM109. Transformants bearing recombinant plasmid were selected on LA plates. Plasmid DNA, pYKA-2, isolated from a positive clone was digested with a series of restriction enzymes. The resulting fragments were subcloned into appropriate restriction sites of pUC vectors for further analysis. A 4.0-kb PstI-EcoRI fragment on pYKA-25, the positive clone, was used for sequence analysis.
Nucleotide sequencing
DNA fragments were sequenced on both strands by the dideoxy chain termination method of Sanger (1981), using a DYEnamic™ ET terminator sequencing kit (Perkin Elmer, Applied Biosystems, Foster City, Calif.). Oligonucleotide primers were purchased from TAKARA, Sigma, and Invitrogen.
Nucleotide sequence accession number
The nucleotide sequence reported here has been deposited in the DDBJ/GenBank/EMBL database under accession number AB086444.
Protein analysis
Inulinase activity was determined by measuring the initial rate of inulin hydrolysis as previously described (Kato et al. 1999). Protein was assayed by the method of Lowry et al. (1951) with bovine serum albumin as the standard. SDS- and native-polyacrylamide gel electrophoresis (PAGE) were performed by the method of Laemmli (1970). Western blotting analysis was performed according to standard methods. Rabbit antisera raised against G. stearothermophilus inulinase II were prepared as previously described (Kato et al. 1999). Polyvinylidene difluoride (PVDF) membrane, Immobilon-P, was obtained from Millipore (Bedford, Mass.). Goat anti-rabbit IgG conjugated with horseradish peroxidase was from Jackson ImmunoResearch Laboratories (West Grove, Pa.). N-terminal sequencing of proteins was carried out by automated Edman degradation with an Applied Biosystems 477A gas/liquid-phase protein sequencer with an on-line phenylthiohydantoin 120A analyzer.
Purification of the recombinant thermostable exo-inulinase
E. coli HB101 bearing pYKA-25 grown aerobically for 8 h at 37°C in LA medium was harvested, washed and suspended in 50 mM phosphate buffer/5 mM EDTA (pH 7.0, Buffer A). The cells were sonicated in Buffer A. The sonicate was centrifuged. The supernatant was treated for 10 min at 55°C and then centrifuged. The enzyme in the supernatant was purified by four-column chromatography, DEAE-cellulose SH, Sephadex CL6B, Ethyl-agarose, DEAE-Sepharose CL6B, and Hydroxylapatite HTP according to methods described previously (Kato et al. 1999) with appreciable modification. The purity was judged by both SDS-PAGE and gel filtration.
Protein crystallization
The protein solution of the purified recombinant enzyme was concentrated to 8.5 mg/ml by ultrafiltration with an Amicon PM-10 membrane (Millipore), and 1.4 μl of the concentrate was mixed with 2.6 μl 16% poly(ethyleneglycol) 4000 (PEG/4000) in 0.1 M Tris buffer/0.2 M magnesium chloride (pH 8.5). Hanging drops were equilibrated against an appropriate volume of the same PEG/4000 solution at 18°C.
Results
N-Terminal sequence of the native exo-inulinase from G. stearothermophilus KP1289 and construction of the enzyme-specific DNA probe
The N-terminal sequence of the native exo-inulinase, which was previously purified from cultures of the thermophile, was determined to be MKTHNSEKYRPTFHFSPKKN by automated Edman degradation. Homology analysis of the N-terminal sequence using BLAST (Pearson and Lipman 1988) and FASTA (Altschul et al. 1990) showed that the exo-inulinase belonged to glycosyl hydrolase family 32. Thus, two sets of degenerate PCR primers were derived, one from the N-terminal protein sequence (MKTHNSE) and the other from a conserved sequence (FRDPKVFW) from glycosyl hydrolase family 32 (see below). PCR amplification of the G. stearothermophilus genomic DNA yielded a DNA fragment of approximately 0.5 kb. The fragment was labeled with DIG, and the labeled fragment was used as a probe for Southern hybridization. Genomic Southern analysis indicated that the gene encoding exo-inulinase exists as a single copy in the G. stearothermophilus KP1289 genome.
Isolation and sequence analysis of the thermostable exo-inulinase gene (inuA)
As described in Materials and methods, genomic DNA fragments of approximately 6–7 kb digested with HindIII and XbaI were used in the construction of a genomic library. The library was screened for the thermostable exo-inulinase gene (inuA) by colony hybridization; one positive clone was obtained among 900 colonies screened. Cell extracts of this clone hydrolyzed inulin at 55°C. The HindIII-XbaI fragment was digested with several restriction enzymes. The resulting fragments were subcloned into appropriate restriction sites of pUC vectors for further analysis.
The 4.0-kb PstI-EcoRI fragment on pYKA-25, the positive clone, was used in sequence analysis. It comprised 3,969 bp and contained an open reading frame (ORF) of 1,482 bp. This ORF, which commenced with a TTG (837–839) start codon and terminated with a TGA (2,316–2,318) stop codon, corresponded to a protein of 493 amino acid residues with molecular mass (M) of 56,744 Da (Fig. 1). The 5′-flanking region of the ORF contained the AGGGGG (822–827) assumed to be the Shine-Dalgarno ribosome-binding sites. Two sequences in the 5′-flanking region, TTGAAA (778–783) and CAGATT (803–808), seemed to be the −35 and −10 promoter sequences. The G+C content of the ORF sequence was 34.9 mol%.

Nucleotide sequence of the Geobacillus stearothermophilus inuA region. The deduced amino acid sequence of the open reading frame is shown. Nucleotides of a 3,969-bp PstI-EcoRI DNA fragment are numbered from the 5′ terminus. Deduced amino acids are numbered from the initiator methionine. The putative Shine-Dalgarno sequence is boxed. The possible −35 and −10 sequences are underlined
The classification of glycosyl hydrolases into families depends on amino acid sequence similarities (Davies and Henrissat 1995; Henrissat 1991; Henrissat and Bairoch 1993, 1996; http://afmb.cnrs-mrs.fr/CAZY/). The β-fructofuranosidases deposited in protein databases have been classified into the glycosyl hydrolase families 32 and 68 (GH32 and GH68) (Naumoff 2001). The deduced polypeptide (inuA) showed strong sequence similarity to Pseudomonas mucidolens exo-inulinase (50% identity), Bacillus subtilis levanase (47% identity), Paenibacillus polymyxa (Bacillus polymyxa) fructosyltransferase (44% identity), Saccharomyces cerevisiae invertase (37% identity), and so on, indicating that the enzyme belonged to GH32. Models for the catalytic mechanism of GH32 have been proposed, although they have not been completely proven (Pons et al. 1998; Reddy and Maley 1990, 1996).
Expression of the thermostable exo-inulinase in E. coli
E. coli HB101 bearing pYKA-25 was grown aerobically for 8 h at 37°C in LA medium. The recombinant exo-inulinase produced without IPTG induction was not an extracellular but an intracellular enzyme in the E. coli expression system. These results indicated that the gene was most probably transcribed under the control of its own promoter present in the 4.0-kb PstI-EcoRI fragment. Additionally, five other E. coli strains (JM109, AD494, MV1184, DH5α, and BL21) were used for overexpression. Each transformant was aerobically grown for 24 h in 5 ml LA medium and used in an enzyme assay. The total inulinase activities in each crude extract, with the host strains shown in parentheses, were: 2.45 U (JM109), 1.11 U (AD494), 0.70 U (MV1184), 1.22 U (DH5α), 0.03 U (BL21), and 8.95 U (HB101). These results showed that HB101 bearing pYKA-25 produced the enzyme most effectively. These results might be attributed to different plasmid and/or protein stability in each of the host cells.
Purification, size, N-terminal sequence, and characterization of the recombinant exo-inulinase
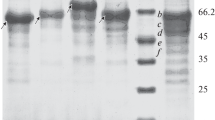
E. coli HB101 bearing pYKA-25 was used for overexpression of the recombinant exo-inulinase. The enzyme was purified 88.1-fold compared with the cell extract. The specific activity was 133 U/mg. The final preparation migrated as a single protein band on SDS-PAGE (Fig. 2). The M of the recombinant enzyme was estimated to be approximately 54,000 Da by both SDS-PAGE and gel filtration on Sephadex G-100.

SDS-PAGE of native and recombinant exo-inulinases. Electrophoresis was carried out on 10% polyacrylamide gels containing SDS. Proteins on the gels after SDS-PAGE were stained with Coomassie Brilliant Blue R-250. Lanes: M M r marker, 1 purified native exo-inulinase, 2 recombinant exo-inulinase
The N-terminal sequence of the recombinant enzyme was determined to be MKTHNSEKYR. The sequence was identical to that of the native enzyme. Analysis of native-PAGE suggested that there were slightly different structures between the two preparations, suggesting slight differences in their folding structures and/or modifications (data not shown).
The recombinant and native exo-inulinases were active between pH 4.5 and pH 8.6, with an optimum at approximately pH 6.0 (data not shown). They were reconfirmed to be thermostable and most active at 60°C (Fig. 3). These profiles agreed well with the previous results (Kato et al. 1999).

Effect of temperature on the activity (a) and stability (b) of native and recombinant G. stearothermophilus exo-inulinase. ▲ Activity of native enzyme, ○ activity of recombinant enzyme. a The activity of each enzyme was assayed at 35–80°C. The activity found at 60°C was taken to be 100%. b Each enzyme was heated for 10 min at different temperatures up to 80°C, and assayed at 60°C for the residual activity. The activity of the enzyme kept at 4°C was taken to be 100%
Crystallization of the recombinant and purified exo-inulinase
We examined the crystallization of the exo-inulinase from G. stearothermophilus and obtained regular octahedral crystals (Fig. 4). The present report is the third to characterize the crystals of GH32 proteins, and the first as far as prokaryotic proteins are concerned (Arand et al. 2002; Nakamura et al. 1978). The crystals started to grow in 1 day, and reached a size of approximately 0.1 mm ×0.1 mm ×0.2 mm after 3 days. We also obtained larger crystals and are now collecting X-ray data. The three-dimensional structure of exo-inulinase will be described elsewhere.

Crystals of a thermostable exo-inulinase from G. stearothermophilus KP1289
Discussion
Genomic Southern analysis indicated that the gene encoding exo-inulinase exists as a single copy in the G. stearothermophilus KP1289 genome, while it has been reported that the thermophile produces two inulinases. These two inulinases have extremely similar properties and slightly different mobility in native-PAGE (Kato et al. 1999). The results of that study suggested that a small difference in folding structure and/or modification would result in different mobilities in native-PAGE. Thus, both enzymes are certainly derived from the same gene (inuA) product.
The G+C content of the inuA ORF sequence (34.9 mol%) was lower than the G+C content of the KP1289 genome DNA (48.1 mol%). The G+C content is not likely to be related to the thermostability of exo-inulinase.
Analysis of multiple alignments showed that the N-terminal region of GH32 is highly conserved. The inuA gene product contains three conserved regions, 21-WMNDANGLVY-30, 149-DFRDPKVFWH-158, and 202-WECP-205 (putative catalytic residues italicized).
Computer analysis using a PSORT program (Prediction of Protein Sorting Signals) suggested that the enzyme would be an intracellular protein without a signal peptide in both Gram-positive and Gram-negative bacteria (http://psort.ims.u-tokyo.ac.jp/). In fact, the recombinant enzyme was produced in the cytosol. In addition, the observed N-terminal sequence of the purified recombinant enzyme was in complete agreement with the theoretical results (Fig. 1). These results suggest that the inuA gene product does not have a signal peptide. We previously reported that G. stearothermophilus KP1289 produced extracellular inulinases. Western blot analysis using anti-inulinase II (extracellular inulinase) antisera, however, showed that the extracellular enzyme would be identical to the intracellular one, and that the extracellular one would not be subjected to post-translational modification (data not shown). The native enzyme in the thermophile culture appeared to be derived from the cell lysis and/or the leakage from cytosol. These considerations were strongly supported by the fact that the stop codon was in the −4 to −6 nt upstream of the predicted start codon (Fig. 1).
Analysis of the sequence homology shows that the exo-inulinase belongs to GH32. Many enzymes of this family have been isolated from various organisms, and their nucleotide and deduced amino acid sequences have been reported. Pons et al. (1998, 2000) predicted that the structure of GH32 proteins might have a β-propeller catalytic domain. Until now, however, their 3-D structures based on X-ray analysis have remained obscure. We obtained regular octahedral crystals of the exo-inulinase from G. stearothermophilus. Enzymes from thermophilic microorganisms developed unique structure-function properties of thermostability and optimal activity at higher temperatures. They can be used in several industrial processes. The main advantages of higher temperatures are reduced risk of microbial contamination, lower viscosity, improved transfer rates, and improved solubility of substrates. Thus, we have focused on the molecular determinants of protein thermostability. We previously found a strong correlation between the increase in the number of proline residues and the rise in thermostability of bacillary oligo-1,6-glucosidases. Based on this correlation, we proposed the proline rule, a general principle for increasing thermostability (Suzuki 1989, 1999). This proposal was verified by protein engineering and 3-D structure analysis (Watanabe et al. 1990, 1991, 1994, 1996, 1997). On the other hand, protein engineering studies of substrate specificities have also been initiated, although without much success. Recently, we have succeeded in proving the substrate specificities on α-glucosidases (unpublished data). The present research is expected to add a new aspect to the relationship between structure and activity in the β-fructosidase superfamily, and to contribute to our understanding of strategies leading to protein thermal stabilization.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Arand M, Golubev AM, Nedo JRB, Polikarpov I, Wattiez R, Korneeva OS, Eneyskaya EV, Kulminskaya AA, Shebalin KA, Shishliannikov SM, Chepurnaya OV, Neustroev KN (2002) Purification, characterization, gene cloning and preliminary X-ray data of the exo-inulinase from Aspergillus awamori. Biochem J 362:131–135
Davies G, Henrissat B (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3:853–859
Ferrira MSS, De Andrade AVM, Kenndey JF (1991) Properties of a nonspecific fructofuranosidase produced by Cladosporium cladosporioides cells for hydrolysis of Jerusalem artichoke extract. Appl Biochem Biotechnol 31:1–9
Henrissat B (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280:309–316
Henrissat B, Bairoch A (1993) New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 293:781–788
Henrissat B, Bairoch A (1996) Updating the sequence-based classification of glycosyl hydrolases. Biochem J 316:695–696
Kato K, Araki T, Kitamura T, Morita N, Moori M, Suzuki Y (1999) Purification and properties of a thermostable inulinase (β-d-fructan fructohydrolase) from Bacillus stearothermophilus KP1289. Starch 51:253–258
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Liebl W, Brem D, Gotschlich A (1998) Analysis of the gene for β-fructosidase (invertase, inulinase) of the hyperthermophilic bacterium Thermotoga maritima, and characterization of the enzyme expressed in Escherichia coli. Appl Microbiol Biotechnol 50:55–64
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Nakamura T, Kurokawa T, Nakatsu S, Ueda S (1978) Crystallization and general properties of an extracellular inulase from Aspergillus sp. (in Japanese). Nippon Nogeikagaku Kaishi 52:159–166
Naumoff DG (2001) β-Fructosidase superfamily: homology with some α-d-arabinases and β-d–xylosidases. Proteins 42:66–76
Pandey A, Soccol CR, Selvakumar P, Soccol VT, Krieger N, Fontana JD (1999) Recent developments in microbial inulinases: its production, properties, and industrial applications. Appl Biochem Biotechnol 81:35–52
Pearson WR, Lipman DJ (1988) Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 85:2444–2448
Pons T, Olmea O, Chinea G, Beldarra A, Marquez G, Acosta N, Rodriguez L, Valencia A (1998) Structural model for family 32 of glycosyl-hydrolase enzymes. Proteins 33:383–395
Pons T, Hernadez L, Batista FR, Chinea G (2000) Prediction of a common β-propeller catalytic domain for fructosyltransferase of different origin and substrate specificity. Protein Sci 9:2285–2291
Reddy VA, Maley F (1990) Identification of an active-site residue in yeast invertase by affinity labeling and site-directed mutagenesis. J Biol Chem 265:10817–10820
Reddy VA, Maley F (1996) Studies on identifying the catalytic role of Glu-204 in the active site of yeast invertase. J Biol Chem 271:13953–13958
Sanger F (1981) Determination of nucleotide sequence in DNA. Science 214:1205–1210
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Suzuki Y (1989) A general principle of increasing protein thermostability. Proc Jpn Acad B 65:146–148
Suzuki Y (1999) The proline rule-A strategy for protein thermal stabilization. Proc Jpn Acad B 75:133-137
Uzunova K, Vesileva A, Kambourova M, Ivanova V, Spasova D, Mandeva R, Derekova A, Tonkova A (2001) Production and properties of a bacterial thermostable exo-inulinase. Z Naturforsch 56c:1022–1028
Vandamme EJ, Derycke DG (1983) Microbial inulinases: fermentation process, properties, and application. Adv Appl Microbiol 29:139–176
Watanabe K, Kitamura K, Iha H, Suzuki Y (1990) Primary structure of the oligo-1,6-glucosidase of Bacillus cereus ATCC7064 deduced from the nucleotide sequence of the cloned gene. Eur J Biochem 192:609–620
Watanabe K, Chishiro K, Kitamura K, Suzuki Y (1991) Proline residues responsible for thermostability occur with high frequency in the loop regions of an extremely thermostable oligo-1,6-glucosidase from Bacillus thermoglucosidasius KP1006. J Biol Chem 266:24287–24294
Watanabe K, Masuda T, Ohashi H, Mihara H, Suzuki Y (1994) Multiple proline substitutions cumulatively thermostabilize Bacillus cereus ATCC7064 oligo-1,6-glucosidase. Irrefragable proof supporting the proline rule. Eur J Biochem 226:277–283
Watanabe K, Kitamura K, Suzuki Y (1996) Analysis of the critical sites for protein thermostabilization by proline substitution in oligo-1,6-glucosidase from Bacillus coagulans ATCC 7050 and the evolutionary consideration of proline residues. Appl Environ Microbiol 62:2066–2073
Watanabe K, Hata Y, Kizaki H, Katsube Y, Suzuki Y (1997) The refined crystal structure of Bacillus cereus oligo-1,6-glucosidase at 2.0 Åresolution: structural characterization of proline-substitution sites for protein thermostabilization. J Mol Biol 269:142–153
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tsujimoto, Y., Watanabe, A., Nakano, K. et al. Gene cloning, expression, and crystallization of a thermostable exo-inulinase from Geobacillus stearothermophilus KP1289. Appl Microbiol Biotechnol 62, 180–185 (2003). https://doi.org/10.1007/s00253-003-1261-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-003-1261-3