
Abstract
The major histocompatibility complex (MHC) is critical to host-pathogen interactions. Class II MHC is a heterodimer, with α and β subunits encoded by different genes. The peptide-binding groove is formed by the first domain of both subunits (α1 and β1), but studies of class II variation or natural selection focus primarily on the β subunit and II B genes. We explored MHC II A in Leach’s storm-petrel, a seabird with two expressed, polymorphic II B genes. We found two II A genes, Ocle-DAA and Ocle-DBA, in contrast to the single II A gene in chicken and duck. In exon 2 which encodes the α1 domain, the storm-petrel II A genes differed strongly from each other but showed little within-gene polymorphism in 30 individuals: just one Ocle-DAA allele, and three Ocle-DBA alleles differing from each other by single non-synonymous substitutions. In a comparable sample, the two II B genes had nine markedly diverged alleles each. Differences between the α1 domains of Ocle-DAA and Ocle-DBA showed signatures of positive selection, but mainly at non-peptide-binding site (PBS) positions. In contrast, positive selection within and between the II B genes corresponded to putative PBS codons. Phylogenetic analysis of the conserved α2 domain did not reveal deep or well-supported lineages of II A genes in birds, in contrast to the pronounced differentiation of DQA, DPA, and DRA isotypes in mammals. This uncertain homology complicates efforts to compare levels of functional variation and modes of evolution of II A genes across taxa.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The major histocompatibility complex (MHC) is central to host-pathogen evolution because it encodes cell surface proteins that present pathogen peptides to the host immune system (Minias et al. 2018; Spurgin and Richardson 2010). The MHC is an ancient part of the adaptive immune system (Flajnik 2018) and is known for being extremely polymorphic, but the extent of polymorphism can differ between species or populations (Gómez et al. 2010; May et al. 2011). Thus, the MHC is an important functional gene target for considering the consequences of loss of genetic diversity, e.g., due to habitat fragmentation, population bottlenecks, or selective sweeps (De Groot et al. 2002; Kosch et al. 2016; Sutton et al. 2011; Ujvari and Belov 2011).
Among classical MHC genes, class II MHC is expressed by the immune system’s antigen-presenting cells and presents exogenously derived pathogen peptides to CD4+ T cells (Klein 1986). Class II proteins exist as heterodimers in which the alpha and beta subunits—encoded by different genes—both contribute to the peptide-binding groove (Brown et al. 1993). Because both subunits of class II MHC interact with antigens, both have the potential to be shaped by pathogen-mediated natural selection.
Despite the functional role of the alpha subunit of the protein, most studies of natural selection or sexual selection on MHC class II characterize II B without corresponding data on II A (Minias et al. 2019), because of a prevailing view that II A does not exhibit allelic polymorphism (Wilson et al. 2014). This perspective arose from compelling data on humans and several other primates (Bontrop et al. 1999) and was reinforced by data from two avian model systems, the chicken Gallus gallus domesticus (Salomonsen et al. 2003) and the duck Anas platyrhynchos (Ren et al. 2011). However, there are clear counter-examples that show extensive allelic variation in MHC II A, including house mouse Mus musculus (Číková et al. 2011), Xenopus (Liu et al. 2002), salmonids (Gómez et al. 2010), eel Anguilla anguilla (Bracamonte et al. 2015), bream Sparus aurata (Karaiskou et al. 2010), and others. This discrepancy is at least partly explained by genomic organization and homology of duplicated genes. Genes that are orthologs of DPA and DQA tend to exhibit at least modest allelic polymorphism and tight linkage with corresponding B genes. In contrast, orthologs of DRA tend to be monomorphic to achieve an “average best fit” with the diverse set of DRB alleles because frequent recombination occurs between DRA and DRB and prevents the formation of coevolved pairs of A and B alleles (Germain et al. 1985; Kaufman 1999; Traherne 2008). When studying MHC in a non-model species with an unknown number of genes and unknown homology of those genes, the extent of functional polymorphism in II A cannot be assumed and, instead, needs to be explored empirically.
In addition to the effects of A-B recombination rates on the extent of polymorphism in II A genes, there are other uncertainties about how II A genes evolve. Data from numerous studies of II B show signatures of positive selection in the exon that encodes the peptide-binding groove (Bernatchez and Landry 2003; Hughes and Nei 1989; Minias et al. 2018), but analogous data about patterns of selection on II A are scarce. Among the existing studies of exon 2 of II A, which encodes the peptide-binding region, there are cases of purifying selection (Aarnink et al. 2010; Jaratlerdsiri et al. 2014), which is consistent with the existence of very limited polymorphism. However, a number of studies have found signatures of positive selection at II A genes, similar to the pattern typical for II B genes (Bracamonte et al. 2015; Gómez et al. 2010; Liu et al. 2002; Liu et al. 2013; Wilson et al. 2014) and congruent with studies that have found associations between MHC II A genotypes and disease susceptibility (Liu et al. 2013; Xu et al. 2010; Yang et al. 2016). These contrasting results limit our ability to generalize about the variation and evolutionary dynamics of II A genes, and data on patterns of selection on II A genes are needed from additional species to expand our understanding of the class II heterodimer as a whole.
Here, we characterize MHC II A in Leach’s storm-petrel Oceanodroma leucorhoa, a long-lived seabird that is globally abundant but exhibiting population declines that have caused it to recently be classified by the IUCN as vulnerable (BirdLife International 2017; Hedd et al. 2018). Previously in this species, we have shown the presence of two class II B genes, both of which are expressed, show signatures of positive selection, and exhibit moderately high levels of polymorphism: in the II B 270-bp exon 2, which encodes the peptide-binding groove, we found 36 polymorphic sites and 11 alleles in Ocle-DAB1, and 39 polymorphic sites and 13 alleles in Ocle-DAB2 (Dearborn et al. 2016). Mated pairs of storm-petrels in our study population show no evidence of disassortative pairing at MHC, and a phylogenetic simulation suggests that this may be because random mating predictably yields functionally heterozygous offspring, due to the divergence in alleles of the two genes (Dearborn et al. 2016) (but see Hoover et al. (2018)). However, these II B genes encode only half of the MHC class II peptide-binding groove, with the other half encoded by II A gene(s) which to date are unidentified and uncharacterized.
We had three aims in this study: (1) find and characterize MHC class II A gene(s) in this species; (2) assess levels of polymorphism in comparison to II B genes, testing whether the α1 domain is highly polymorphic or relatively invariant, and (3) test whether II A shows signatures of positive selection similar to II B. These data will help broaden our understanding of selection pressures on class II MHC generally and, more specifically, will tell us whether studies of mate choice and pathogen-mediated selection in this storm-petrel system need to be considered from the perspective of the entire class II heterodimer.
Methods
Population and sampling
We studied Leach’s storm-petrels at the Bowdoin Scientific Station on Kent Island, New Brunswick, Canada (44 35′ N, 66 45′ W), which includes areas where storm-petrels have been monitored for more than 50 years (Mauck et al. 2018). For the present study, we collected blood samples in 2015, 2016, and 2017. Samples were collected following the Ornithological Council’s Guidelines for the Use of Wild Birds in Research (Fair et al. 2010) under permits from Bowdoin College and the Canadian Wildlife Service. Whole blood was collected by brachial venipuncture and stored in lysis buffer for subsequent DNA extraction. From a subset of birds, we also collected blood for RNA extraction, storing it in RNAlater (Qiagen, Valencia, CA) at ambient temperature for 48 h before freezing at − 20 °C.
We isolated DNA from lysis buffer using DNEasy (Qiagen, Valencia, CA). Total RNA was extracted from blood in RNAlater using a RiboPure Blood kit (Invitrogen, Vilnius, Lithuania) followed by digestion of any potential gDNA contamination with DNase I. We quantified nucleic acid concentrations with a NanoDrop 1000 (ThermoFisher, Wilmington, DE).
Because these birds are sexually monomorphic, we determined sex of individuals by amplifying a pair of differentiated sex chromosome genes (Fridolfsson and Ellegren 1999) as described in Dearborn et al. (2016).
Primer development
We began primer development by identifying conserved regions in GenBank class II A sequences from other bird species, including duck, Japanese crested ibis Nipponia nippon, oriental stork Ciconia boyciana, northern fulmar Fulmarus glacialis, and emperor penguin Aptenodytes forsteri. Based on these alignments and subsequent sequencing of storm-petrels, we developed initial primers 10F, 266F, 228F, 49R, and 291R (Fig. 1). Cloning and Sanger sequencing with multiple combinations of these primers revealed the presence of two highly diverged genes that became the basis for further primer design. We named these genes MhcOcle-DAA and MhcOcle-DBA following the conventions of Klein et al. (1990) and based on alignments with previously named II A genes in Japanese crested ibis and oriental stork (Chen et al. 2015; Tsuji et al. 2017). We assembled a consensus sequence from 10 individual storm-petrels for each gene, spanning from exon 1, which encodes the leader peptide, to the first few bases of exon 4, which encodes the membrane-spanning domain. We used these sequences for two purposes: to compare the α2 domain (exon 3) across species to determine orthology to other MHC genes, and to develop additional primers for focal work on the α1 domain (exon 2) that contributes to the peptide-binding groove (Fig. 1).

Primer locations. Primers in red italic font above the diagram are specific to Ocle-DAA, those in blue italic font below the diagram are specific to Ocle-DBA, and primers whose sequences are conserved across genes are given in plain black font in both locations. * denotes primers used to generate or verify exon 2 sequences in this paper. See text for reaction conditions and Appendix Table 2 for primer sequences
Exon 2 polymorphism
To assess variability in exon 2, which encodes the alpha subunit’s contribution to the peptide-binding groove, we sequenced 30 individuals using the primers indicated with asterisks in Fig. 1. To reduce biases in MHC characterization, we followed the suggestions of Burri et al. (2014) to use hot-start polymerase and long initial denaturing, and to repeat PCR reactions and sequencing to test for consistency of results. Reaction components for a 20-μl reaction were 2.0 μl 10× ABI AmpliTaq Gold 360 Buffer, 2.0 μl 25 mM MgCl2, 2.0 μl 2 mM dNTP, 0.8 μl 10 μM primer F, 0.8 μl 10 μM primer R, 1.0 μl ABI G-C Enhancer, 7.3 μl water, 0.1 μl AmpliTaq Gold 360 DNA Polymerase, and 4.0 μl 20 ng/μl template DNA. Cycling parameters were 95 °C for 10 min; 30 cycles of 95 °C for 30 s, TA for 30 s, and 72 °C for 60 s, ending with 72 °C for 5 min and a 4 °C hold; extension time for primer pairs with amplicons between 1 and 1.5 kb was increased to 90 s. Exon 2 of Ocle-DAA was amplified with 10F-1415R (TA = 62 °C; 1502 bp), 91F-1415R (TA = 61 °C; 1374 bp), 8F-291R (TA = 64 °C; 960 bp), 25F-291R (TA = 64 °C; 943 bp), and 25F-1415R (TA = 64 °C; 564 bp). Exon 2 of Ocle-DBA was amplified with 10F-49R (TA = 60 °C; 1504 bp), 391F-869R (TA = 61 °C; 480 bp), 800F-49R (TA = 61 °C; 672 bp), 800F-1298R (TA = 61 °C; 499 bp), 836F-1306R (TA = 61 °C; 471 bp), and 854F-1080R (TA = 61 °C; 227 bp).
PCR products were directly purified with Exo-SAP-IT (Thermo Fisher, Waltham, MA). Sanger sequencing was performed by Eurofins Genomics (Huntsville, AL) using their PowerRead protocol because of the very high GC content of our target sequences. When exon 2 sequences produced double peaks, indicative of heterozygotes, we cloned amplicons using a haploid cloning vector (TOPO TA Cloning Kit for Sequencing; Invitrogen, Carlsbad, CA) and sequenced the clones using our original primers.
Gene expression
To test for expression of these two genes, we selectively reverse transcribed total RNA to cDNA and then amplified cDNA using the Access RT-PCR kit (Promega, Madison, WI), with the following reaction conditions: 10 μl 5× AMV/Tfl buffer, 5 μl 2 mM dNTP, 5 μl 10 μM primer F, 5 μl 10 μM primer R, 2 μl 25 mM MgSO4, 20 μl water, 1 μl AMV reverse transcriptase, 1 μl Tfl polymerase, and 1 μl mRNA. Cycling parameters were 45 °C for 45 min; 94 °C for 2 min; 45 cycles of 94 °C for 30 s, TA for 30 s, and 68 °C for 2 min; ending with 68 °C for 7 min and a 4 °C hold. For Ocle-DAA, we amplified 274 bp of mRNA spanning exon 2 and exon 3, using primers 25F-1415R and a TA of 64 °C. For Ocle-DBA, we amplified 227 bp of mRNA within exon 2, using primers 854F-1080R and a TA of 61 °C.
Amplicons were gel purified with a Zymoclean Gel DNA Recovery kit (Zymo Research, Irvine, CA) and then Sanger sequenced in both directions by Eurofins Genomics (Huntsville, AL). Consensus of forward and reverse sequences of each allele from each bird was compared to gDNA sequences obtained from the same primers and from additional gDNA primers.
Last, we confirmed that our cDNA sequences were derived from mRNA rather than from any possible gDNA contamination that could have survived the DNase treatment. For Ocle-DAA, this was demonstrated by the absence of intron sequences in the mRNA data. For Ocle-DBA, the primers were within a single exon, so we conducted an additional PCR verification as follows: we used mRNA as template in a regular PCR (i.e., without reverse transcriptase) with exon 2 primers 832F-1080R, expecting no amplification; as a positive control, we also used gDNA from each bird as template, expecting that it would amplify.
Data analysis
Sequences were edited in Sequencher 5.4 (Gene Codes Corporation, Ann Arbor, MI).
We used phylogenetic analysis to explore whether Ocle-DAA and Ocle-DBA appear to be orthologs of the DPA, DQA, or DRA isotypes in humans. Because pathogen-mediated selection on the α1 domain would likely obscure evolutionary history in a tree of such distantly related taxa, we focused on the α2 domain which evolves more slowly. We used new storm-petrel sequences of Ocle-DAA (GenBank MN061395) and Ocle-DBA (GenBank MN061404) plus archived DNA sequences from HLA-DRA (NM_019111), HLA-DQA1 (NM_002122), and HLA-DPA1 (NM_033554); Rhesus macaque Macaca mulatta DRA (NM_001134298), DQA1 (NM_001134295), and DPA (NM_001048247); mouse M. musculus H2-Aa (NM_010378.3) and H2-Ea (BC106107.1); European badger Meles meles DQA (HQ908097) and DRA (HQ908099); California sea lion Zalophus californianus DQA (AF502560) and DRA (AY491450); African clawed frog Xenopus laevis DAA (AF454374) and DBA (AF454378); chicken G. gallus BLA (AY357253); duck A. platyrhynchos DRA (HM070250); Japanese crested ibis N. nippon DAA, DBA1, DBA2, and DBA3 (KP182408); oriental stork C. boyciana DAA and DBA (LC180358.1); Atlantic salmon Salmo salar (L77086); rainbow trout Oncorhynchus mykiss DAA (AJ251431); Japanese flounder Paralichthys olivaceus II A (AY997530); and miiuy croaker Miichthys miiuy DAA (GU936787). Boundaries of the α2 domain were verified from published alignments (Liu et al. 2002; Ren et al. 2011; Salomonsen et al. 2003; Sin et al. 2012; Stet et al. 2002; Taniguchi et al. 2014; Xu et al. 2010). The α2 DNA sequences were aligned as codons with MUSCLE in MEGA 7 (Tamura et al. 2013), resulting in an average amino acid p-distance of 0.420. We estimated the evolutionary relationships among genes with an unrooted maximum likelihood tree using Tamura’s 3-parameter model plus gamma, as determined by lowest BIC in model comparisons in MEGA.
The remaining analyses deal with exon 2, encoding the α1 domain which contributes to the peptide-binding groove. First, to test whether exon 2 genotype frequencies showed an excess or deficiency of heterozygotes, we used GenePop (Raymond and Rousset 1995). Second, to compare the level of exon 2 polymorphism in II A versus II B, we used our new II A data and prior data on Ocle-DAB1 and Ocle-DAB2 in this same population (Dearborn et al. 2016). Because our existing II B data was originally based on a larger sample, we used rarefaction analysis in ADZE (Szpiech et al. 2008) to calculate allelic richness for a sample of n = 30 diploid individuals (the size of our II A sample) and to identify which particular alleles would be likely to be found in that subsample. We then calculated the expected frequencies of these rarefied alleles in a sample of n = 30 animals by summing their original frequencies from the n = 188 sample and dividing each one by that sum. Using the resulting allele frequencies, we created haplotype networks for the II B genes and also for the II A genes, using HaploViewer (Salzburger et al. 2011) based on the dnapars module in PHYLIP 3.695 using jumbled outgroup selection (Felsenstein 2005). Nucleotide diversity for the II A alleles and for the rarefaction-reduced II B alleles was calculated with dnaSP (Librado and Rozas 2009).
Last, to compare the nature of selection on II A genes versus II B genes, we used CodeML in the PAML package (Yang 2007) with maximum likelihood trees generated in MEGA 7 (Tamura et al. 2013) to test for the presence and location of sites showing a signature of positive selection. We implemented site models in CodeML using the preset mode of EasyCodeML (Gao et al. 2019), to test null model M1a (nearly neutral) against alternate model M2a (nearly neutral and positively selected sites) and also to test null model M7 (beta) against alternate model M8 (beta and ω > 1). If the likelihood ratio tests rejected the null model in favor of the alternate model with positively selected sites, the specific sites hypothesized to be under positive selection were identified as those with posterior probabilities > 0.95 using the Bayes empirical Bayes (Yang et al. 2005) approach. Separately from the selection analyses, putative peptide-binding site codons were identified by alignment with chicken sequences for II A genes (Salomonsen et al. 2003) and by alignment with human and seabird sequences for II B genes as previously described (Dearborn et al. 2016).
Results
Overall structure, divergence, expression, and orthology
Using multiple primers, we sequenced approximately 1750 bp of each gene from the start of exon 1, encoding the leader peptide, to the start of exon 4, encoding the transmembrane domain. We sequenced these regions in 10 individuals (5 males, 5 females) and found two clearly differentiated II A genes in all individuals that we examined (GenBank MN061391-MN061399 for MhcOcle-DAA and MN061400-408 for MhcOcle-DBA). Two of the 10 individuals were homozygous and identical to each other, both for Ocle-DAA (GenBank MN061395) and Ocle-DBA (GenBank MN061404). Our quantitative descriptions of differences between the genes (Fig. 2) are based on this common haplotype. In brief, the two genes are fairly similar in exons 1 and 3, moderately diverged in exon 2 and intron 3, and strikingly diverged in introns 1 and 2 (Fig. 2). Intron 1 was different length in the two genes and after the first 80 bp was so diverged as to be impossible to align. The intron 2 sequence was identical between the two genes for the first 156 bp but then the two genes diverged so much as to be impossible to align; in Ocle-DAA only, intron 2 contained a 9-bp indel, with 7 individuals homozygous for the short haplotype and 3 individuals heterozygous for the indel.

Percent sequence identity between duplicate II A genes, Ocle-DAA and Ocle-DBA. Values for each exon and intron are given as % sequence identity, number of segregating sites S, and total length of region. Exon 1 = leader peptide, exon 2 = peptide-binding groove (α1 domain), exon 3 = non-peptide-binding extracellular domain (α2 domain), exon 4 = transmembrane and cytoplasmic domain. Sequence of exon 4 is only partial because of primer location
As expected of genes that are expressed and functionally important, all exon sequences lacked stop codons or indels. We established the expression of both genes by amplifying cDNA in four individuals, finding consensus between the mRNA and gDNA sequences; controls confirmed a lack of gDNA contamination.
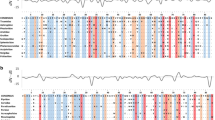
Multispecies alignment and phylogenetic analysis of the more conserved α2 domain (i.e., exon 3) showed that II A genes in birds exist as a weakly supported clan, separate from fish, and separate from the well-differentiated mammalian lineages of DQA, DRA, and DPA (Figs. 3 and 4). Within birds, Ocle-DBA is adjacent to DBA in oriental stork, while Ocle-DAA is adjacent to DAA in oriental stork and Japanese crested ibis; however, both clans are weakly supported with short branch lengths. The chicken BLA, which is thought to be orthologous to mammalian DRA (Salomonsen et al. 2003), appears to be quite diverged from II A genes in other birds (Fig. 4). Similar results (not shown) were obtained with neighbor joining.

Amino acid alignment of the α2 domain of MHC II A genes from mammals, birds, fish, and an amphibian. Identities and gaps are indicated by – and / respectively. Species names and GenBank accession numbers are given in “Methods”

Unrooted maximum likelihood tree of the α2 domain of MHC II A genes in a selection of mammals (blue circles), frog (green diamonds), birds (red triangles), and fish (yellow crosses). Branch supports are based on 1000 bootstraps. The clustering of mammalian isotypes DQA, DRA, and DPA is well supported, but the relationships of avian genes are less strongly resolved and show no clear affinity to a particular mammalian isotype
α1 polymorphism within genes
The pronounced differences between the two storm-petrel II A genes allowed us to use gene-specific primers to characterize allelic diversity in exon 2 for 30 individuals (12 males, 18 females). At Ocle-DAA, all 30 birds were homozygous for a single allele, Ocle-DAA*01 (Figs. 5 and 6; GenBank MK981896). At Ocle-DBA, we found three alleles which differed from each other by single non-synonymous substitutions (Figs. 5 and 6; GenBank MK981897-MK981899). Genotype frequencies of these Ocle-DBA alleles did not differ from Hardy-Weinberg expectations (Weir and Cockerham FIS = 0.1672, p = 0.1822; Ocle-DBA*01/01 36.7%, Ocle-DBA*01/02 36.7%, Ocle-DBA*01/03 3.3%, Ocle-DBA*02/02 13.3%, Ocle-DBA*02/03 6.7%, Ocle-DBA*03/03 3.3%).

Exon 2 nucleotide alignment of two MHC class II A genes in Leach’s storm-petrel. Ocle-DBA exhibited two polymorphic sites, both non-synonymous, at positions 61 and 87. Ocle-DAA exhibited no polymorphic sites, with all 30 individuals homozygous for Ocle-DAA*01. Reading frame is + 3. Putative peptide contact codons (Salomonsen et al. 2003) are marked above with a p

Exon 2 amino acid alignment of two MHC class II A genes in Leach’s storm-petrel. Ocle-DBA exhibited two polymorphic codons, at positions 20 and 29. Ocle-DAA exhibited no polymorphic sites, with all 30 individuals homozygous for Ocle-DAA*01. Putative peptide contact codons (Salomonsen et al. 2003) are marked above with a p. Symbols below alignment reflect similarity of properties in the Gonnet PAM 250 matrix, with * indicating a single, fully conserved residue, : indicating strongly similar properties scoring > 0.5, and . indicating weakly similar properties scoring = < 0.5
Comparing α1 diversity of II A and II B genes
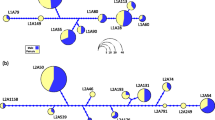
In both II A and II B in our population of Leach’s storm-petrels, the duplicate gene copies were markedly diverged from each other in the α1 domain (Ocle-DAA vs. Ocle-DBA, and Ocle-DAB1 vs. Ocle-DAB2; Fig. 7). However, the pattern in the II A genes differed from that in the II B genes in two respects. First, we observed low exon 2 allelic richness in the two II A genes (1 and 3 alleles, respectively, in 30 individuals), whereas the II B genes were much more polymorphic even in our rarefaction analysis: AR for n = 30 birds was 8.6 for Ocle-DAB1 and 8.7 for Ocle-DAB2. Second, the II A exon 2 alleles within the polymorphic locus differed from each other by only a single substitution, which contrasts with the pronounced level of divergence seen within each of the II B genes (Fig. 7). These differences were captured in the respective values of nucleotide diversity, π, which were roughly an order of magnitude greater for the II B genes: 0.0441 ± 0.0026 SD for Ocle-DAB1 and 0.0393 ± 0.0043 for Ocle-DAB2, versus 0 for Ocle-DAA and 0.0024 ± 0.00025 for Ocle-DBA.

Haplotype networks showing divergence and allelic diversity at exon 2 of II A (Ocle-DAA and Ocle-DBA; top panel) compared to exon 2 of II B (Ocle-DAB1 and Ocle-DAB2; bottom panel). Data are based on n = 30 individuals, sampled directly for II A and subsampled by rarefaction analysis of a larger published sample for II B (Dearborn et al. 2016). Area of circle is proportional to allele frequency, and each line segment represents one nucleotide change; within each class, alleles of the two genes are differentiated by color
Comparing signatures of selection on α1 domain of II A and II B genes
The low polymorphism in exon 2 of the II A genes limited our scope to test for selection, as Ocle-DAA was invariant and Ocle-DBA had only three alleles that differed by single non-synonymous substitutions. In analysis of just the three Ocle-DBA alleles, we failed to reject the null model M1a in favor of M2a (2ΔLnL = 0.88, df = 2, p = 0.643) or the null model M7 in favor of M8 (2ΔLnL = 0.88, df = 2, p = 0.643).
As a complementary approach, we asked whether the divergence between the duplicated II A genes showed evidence of positive selection. To do this, we combined the Ocle-DAA sequence and the three Ocle-DBA sequences into a single alignment. This analysis rejected the null model M1a in favor of M2a (2ΔLnL = 12.16, df = 2, p = 0.0023; Table 1) and likewise rejected M7 in favor of M8 (2ΔLnL = 12.7, df = 2, p < 0.0017). Bayes empirical Bayes analysis suggested that 11 sites were under positive selection (Table 1); only 2 of these 11 sites are putative peptide-binding sites based on alignment with chicken sequences.
To compare the evolutionary processes acting on II A genes and II B genes, we conducted the same analyses on the rarified set of alleles from the II B genes. Here, we found strong evidence of positively selected sites in both Ocle-DAB1 and Ocle-DAB2. Specifically, among the 9 alleles of Ocle-DAB1, we rejected the null model M1a in favor of M2a (2ΔLnL = 48.2, df = 2, p < 0.0001; Table 1) and likewise rejected M7 in favor of M8 (2ΔLnL = 48.0, df = 2, p < 0.0001). Bayes empirical Bayes analysis suggested that 10 sites were under positive selection (Table 1); 9 of these 10 sites were previously identified as putative peptide-binding sites based on alignment with human MHC data (see Dearborn et al. 2016). At the other II B gene, Ocle-DAB2, we also rejected the null model M1a in favor of M2a (2ΔLnL = 35.6, df = 2, p < 0.0001; Table 1) and rejected M7 in favor of M8 (2ΔLnL = 31.6, df = 2, p < 0.0001). Bayes empirical Bayes analysis suggested that 11 sites were under positive selection (Table 1), 10 of which were previously identified as putative peptide-binding sites based on alignment with human MHC data (see Dearborn et al. 2016). Thus, exon 2 alleles belonging to each of the II B genes showed strong evidence of positively selected sites, almost exclusively in putative PBS codons.
For comparison to the II A multigene analysis, we ran an equivalent analysis using the combined set of sequences from the II B genes. This analysis rejected the null model M1a in favor of M2a (2ΔLnL = 89.9, df = 2, p < 0.0001; Table 1) and likewise rejected M7 in favor of M8 (2ΔLnL = 90.9, df = 2, p < 0.0001). Bayes empirical Bayes analysis suggested that 10 sites were under positive selection (Table 1), 9 of which were previously identified as putative peptide-binding sites based on alignment with human MHC data (see Dearborn et al. 2016). Similar results were obtained if using a smaller number of alleles from each II B gene to better mimic the II A data.
Overall, then, we found significant evidence for positively selected sites within each of the II B genes, between the II B genes, and between the II A genes, but not within the II A genes. For the II B genes, but not the II A genes, these selected sites strongly coincided with sites independently identified as putative PBS sites.
Data availability
GenBank Accession #s MN061391-MN061399: population set for MhcOcle-DAA, spanning from the start of exon 1 to part of exon 4; 1786 bp for the short haplotype of the intron 2 indel, and 1795 bp for the long haplotype of the intron 2 indel
GenBank Accession #s MN061400-MN061408: population set for MhcOcle-DBA, spanning from the start of exon 1 to part of exon 4; 1751 bp
GenBank Accession # MK981896: 258 bp sequence of the only exon 2 allele found in n = 30 birds for MhcOcle-DAA
GenBank Accession #s MK981897-MK981899: 258 bp sequences of the three exon 2 alleles found in n = 30 birds for MhcOcle-DBA
Discussion
We found evidence of two distinct MHC class II A genes in Leach’s storm-petrels. Both genes were expressed in blood samples and neither showed indels or stop codons in any of the exon sequences, consistent with them being functionally important. There was minimal variation within genes at exon 2 which encodes the α1 domain, i.e., the α subunit’s contribution to the peptide-binding groove. However, the two II A genes are quite different from each other. At exon 2 in particular, there was only 81% identity between the two genes’ nucleotide sequences, and 15 of 85 positions in the amino acid alignment had scores ≤ 0.5 in the Gonnet PAM 250 matrix, suggestive of functional differences between the genes. This divergence between Ocle-DAA and Ocle-DBA appears to be due to positive selection rather than a nearly neutral process, based on site model comparisons in CodeML. In this respect, II A is similar to II B in this species. However, the specific II A exon 2 codons that appear to have experienced positive selection, based on BEB analysis, do not coincide with those that were identified as PBS codons from alignment with chicken sequences. This runs counter to the II B genes, where the positively selected sites were almost exclusively putative PBS codons.
Gene copy number varies widely across avian taxa for MHC class I and class II B, but comparable data are generally lacking for class II A (Minias et al. 2019). Although we used a large number of primers in our sequencing efforts (Fig. 1), we cannot exclude the possibility that additional II A genes or pseudogenes exist, particularly given cases such as the Japanese crested ibis which has a set of extremely similar MhcNini-DBA genes that appear to be the product of recent duplication (Chen et al. 2015). In our storm-petrel population, though, our finding of two II A genes matches the number of II B genes that have been found to date, which could be the result of a tandem duplication as is common in mammals (Bontrop et al. 1999; Hughes and Nei 1990). The presence of tandem A B pairs is seen in some other birds including Japanese crested ibis (Chen et al. 2015) and oriental stork (Tsuji et al. 2017) but is different from two of the best-studied avian models: the chicken has one II A and two II B genes (Salomonsen et al. 2003) and the duck has one II A and five II B genes (Ren et al. 2011). This raises the question of homology of storm-petrels’ MHC genes. The two II B genes in storm-petrels appear to correspond to two ancient lineages of II B genes in birds (Burri et al. 2010; Goebel et al. 2017), but II A data are not yet available from enough avian species to reconstruct the lineage history and assess whether that history is consistent with a preserved tandem duplication event in the storm-petrel clade.
We also looked more deeply at the evolutionary history of II A, using data from birds, mammals, fishes, and amphibians. Phylogenetic analysis of the conserved α2 domain (Fig. 4) did not reveal a clear evolutionary relationship between the storm-petrel II A genes and the three mammalian isotypes, DQA, DRA, and DPA—despite the fact that each isotype formed its own clan within mammals. The avian part of the tree was poorly resolved, with short branches and low bootstrap support, suggestive of some combination of more recent gene duplications, a lesser extent of diversifying selection, or a greater extent of concerted evolution.
In the peptide-binding region encoded by exon 2, polymorphism within the two II A genes in storm-petrels was limited, with a single exon 2 allele at Ocle-DAA and three alleles at Ocle-DBA in 30 individuals. This oligomorphism is consistent with data from chickens (Salomonsen et al. 2003) and some primates (Bontrop et al. 1999) but differs from a growing number of studies which have found a large number of II A alleles (Bracamonte et al. 2015; Číková et al. 2011; Karaiskou et al. 2010; Xu et al. 2016; Yang et al. 2016). Oligomorphism in Ocle-DAA and Ocle-DBA might be expected if the storm-petrel II A genes are orthologs of mammalian DRA but would be more unusual if these genes are orthologs of the more polymorphic DPA or DQA isotypes. Although prior work has determined that the lone II A gene in chicken and in duck is orthologous to DRA (Ren et al. 2011; Salomonsen et al. 2003), the evolutionary origin of the two storm-petrel II A genes is not evident from our phylogenetic analysis.
The limited II A polymorphism observed in our population is not because of a bottleneck or other mechanism of genetic drift. The breeding population on this 80-ha island has been estimated at 15,000 breeding pairs (L. Minich and R. Mauck, unpubl. data), there is a general lack of genetic differentiation in this species across the Atlantic basin (Bicknell et al. 2012), and this particular population has a typical level of variability at microsatellite loci (Dearborn et al. 2016). More crucially, the limited number of alleles within the two II A genes departs markedly from the allelic richness of the two II B genes in this same population. In addition, the II A polymorphism that does exist is limited in scope: the three α1 alleles of Ocle-DBA are minimally different from each other, which contrasts with alleles of either of the II B genes. This contrast is reflected in nucleotide diversity, π, being an order of magnitude lower for Ocle-DBA than for either of the II B genes.
The evolution of allelic polymorphism within a class II gene can be affected by the recombination rate between the A and B gene. That is, frequent recombination between A and B prevents the development of tightly coevolved A-B haplotypes, with the possible result that selection favors the existence of a single II A allele characterized by an “average best fit” with the diverse set of II B alleles (Germain et al. 1985; Kaufman 1999). The genomic organization of class II MHC in storm-petrels, including the extent of recombination hotspots, is not currently known. If the lack of allelic polymorphism seen in II A is the result of selection for average best fit with the numerous II B alleles, then we would predict the eventual discovery of high rates of recombination between each A and B gene—because of either a large physical distance or a recombination hotspot.
We also do not currently know if heterodimer formation in storm-petrels—or in other birds with duplicated II A genes—is restricted to the pairing of particular A and B genes, e.g., Ocle-DAA with Ocle-DAB1 and Ocle-DBA with Ocle-DAB2, versus all four possible combinations. In mice, which have two isotypes of II A and II B genes, the most common A-B pairings are within isotype, but there are exceptions (Lotteau et al. 1987; Ruberti et al. 1992). Furthermore, the two II A storm-petrel genes appear to be less differentiated from each other than are the isotypes of mice or humans, such that cross-gene heterodimer formation perhaps could occur more readily.
A final area of uncertainty is the pattern of gene expression for Ocle-DAA and Ocle-DBA. We showed that transcripts of both genes are present in whole blood, but we do not know if the two genes are expressed at equal rates overall, nor whether their relative expression differs in different parts of the body. In chickens, for example, the two II B genes are differentially expressed overall, resulting in a “major” gene and a “minor” gene (Jacob et al. 2000), but relative expression patterns of the two genes differ further by tissue type (Parker and Kaufman 2017). There is also evidence from class I MHC in chickens that expression can differ between promiscuous (i.e., generalist) alleles and fastidious (i.e., specialist) alleles (Kaufman 2018). It is interesting to speculate whether one of the storm-petrel II A genes (perhaps the invariant Ocle-DAA) could encode a generalist allele while the other encodes one or more specialist versions.
Overall, we found evidence that this species has two II A genes and two II B genes, which differs from the best-studied avian systems and is more consistent with the tandem duplications seen in mammals and in some non-model bird species. II A polymorphism was low in comparison to II B, consistent with data from chickens and from DRA in primates but in contrast to DQA and DPA in mammals and II A genes in many fish species. Positive selection appears to have played a role in the divergence of the II A genes but was not as clearly related to peptide-binding sites compared with II B. Divergence between the α1 domains of the II A genes raises the unanswered question of whether these α and β subunits assemble in a coevolved, gene-specific manner.
References
Aarnink A, Estrade L, Apoil PA, Kita YF, Saitou N, Shiina T, Blancher A (2010) Study of cynomolgus monkey (Macaca fascicularis) DRA polymorphism in four populations. Immunogenetics 62:123–136
Bernatchez L, Landry C (2003) MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol 16:363–377
Bicknell AWJ, Knight ME, Bilton D, Reid JB, Burke T, Votier SC (2012) Population genetic structure and long-distance dispersal among seabird populations: implications for colony persistence. Mol Ecol 21:2863–2876
BirdLife International I (2017) Hydrobates leucorhous (amended version of 2016 assessment). The IUCN Red List of Threatened Species 2017
Bontrop RE, Otting N, De Groot NG, Doxiadis GGM (1999) Major histocompatibility complex class II polymorphisms in primates. Immunol Rev 167:339–350
Bracamonte SE, Baltazar-Soares M, Eizaguirre C (2015) Characterization of MHC class II genes in the critically endangered European eel (Anguilla anguilla). Conserv Genet Resour 7:859–870
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39
Burri R, Promerová M, Goebel J, Fumagalli L (2014) PCR-based isolation of multigene families: lessons from the avian MHC class IIB. Mol Ecol Resour 14:778–788
Burri R, Salamin N, Studer RA, Roulin A, Fumagalli L (2010) Adaptive divergence of ancient gene duplicates in the avian MHC class II β. Mol Biol Evol 27:2360–2374
Chen LC, Lan H, Sun L, Deng YL, Tang KY, Wan QH (2015) Genomic organization of the crested ibis MHC provides new insight into ancestral avian MHC structure. Sci Rep 5:7963
Číková D, De Bellocq JG, Baird SJE, Piálek J, Bryja J (2011) Genetic structure and contrasting selection pattern at two major histocompatibility complex genes in wild house mouse populations. Heredity 106:727–740
De Groot NG, Otting N, Doxiadis GGM, Balla-Jhagjhoorsingh SS, Heeney JL, Van Rood JJ, Gagneux P, Bontrop RE (2002) Evidence for an ancient selective sweep in the MHC class I gene repertoire of chimpanzees. Proc Natl Acad Sci U S A 99:11748–11753
Dearborn DC, Gager AB, McArthur AG, Gilmour ME, Mandzhukova E, Mauck RA (2016) Gene duplication and divergence produce divergent MHC genotypes without disassortative mating. Mol Ecol 25:4355–4367
Fair J, Paul E, Jones J (2010) Guidelines to the use of wild birds in research. Ornithological Council, Washington
Felsenstein J (2005) PHYLIP (phylogeny inference package). 3.6.5 edn. Distributed by the author, Department of Genome Sciences, University of Washington, Seattle
Flajnik MF (2018) A cold-blooded view of adaptive immunity. Nat Rev Immunol 18:438–453
Fridolfsson A-K, Ellegren H (1999) A simple and universal method for molecular sexing of non-ratite birds. J Avian Biol 30:116–121
Gao F, Chen C, Arab DA, Du Z, He Y, Ho SYW (2019) EasyCodeML: a visual tool for analysis of selection using CodeML. Ecol Evol 9:3891–3898
Germain RN, Bentley DM, Quill H (1985) Influence of allelic polymorphism on the assembly and surface expression of class II MHC (Ia) molecules. Cell 43:233–242
Goebel J, Promerová M, Bonadonna F, McCoy KD, Serbielle C, Strandh M, Yannic G, Burri R, Fumagalli L (2017) 100 million years of multigene family evolution: origin and evolution of the avian MHC class IIB. BMC Genomics 18:460
Gómez D, Conejeros P, Marshall SH, Consuegra S (2010) MHC evolution in three salmonid species: a comparison between class II alpha and beta genes. Immunogenetics 62:531–542
Hedd A, Pollet IL, Mauck RA, Burke CM, Mallory ML, McFarlane Tranquilla LA, Montevecchi WA, Robertson GJ, Ronconi RA, Shutler D, Wilhelm SI, Burgess NM (2018) Foraging areas, offshore habitat use, and colony overlap by incubating Leach’s storm-petrels Oceanodroma leucorhoa in the northwest Atlantic. PLoS One 13:e0194389
Hoover B, Alcaide M, Jennings S, Sin SYW, Edwards SV, Nevitt GA (2018) Ecology can inform genetics: disassortative mating contributes to MHC polymorphism in Leach’s storm-petrels (Oceanodroma leucorhoa). Mol Ecol 27:3371–3385
Hughes AL, Nei M (1989) Nucleotide substitution at major histocompatibility complex class II loci: evidence for overdominant selection. Proc Natl Acad Sci U S A 86:958–962
Hughes AL, Nei M (1990) Evolutionary relationships of class II major-histocompatibility-complex genes in mammals. Mol Biol Evol 7:491–514
Jacob JP, Milne S, Beck S, Kaufman J (2000) The major and a minor class II β chain (B-LB) gene flank the Tapasin gene in the B-F/B-L region of the chicken major histocompatibility complex. Immunogenetics 51:138–147
Jaratlerdsiri W, Isberg SR, Higgins DP, Miles LG, Gongora J (2014) Selection and trans-species polymorphism of major histocompatibility complex class II genes in the order Crocodylia. PLoS One 9:e87534
Karaiskou N, Moran P, Georgitsakis G, Abatzopoulos TJ, Triantafyllidis A (2010) High allelic variation of MHC class II alpha antigen and the role of selection in wild and cultured Sparus aurata populations. Hydrobiologia 638:11–20
Kaufman J (1999) Co-evolving genes in MHC haplotypes: the ‘rule’ for nonmammalian vertebrates? Immunogenetics 50:228–236
Kaufman J (2018) Generalists and specialists: a new view of how MHC class I molecules fight infectious pathogens. Trends Immunol 39:367–379
Klein J (1986) Natural history of the major histocompatability complex. Wiley, New York
Klein J, Bontrop RE, Dawkins RL, Erlich HA, Gyllensten UB, Heise ER, Jones PP, Parham P, Wakeland EK, Watkins DI (1990) Nomenclature for the major histocompatibility complexes of different species: a proposal. Immunogenetics 31:217–219
Kosch TA, Bataille A, Didinger C, Eimes JA, Rodríguez-Brenes S, Ryan MJ, Waldman B (2016) Major histocompatibility complex selection dynamics in pathogen-infected túngara frog (Physalaemus pustulosus) populations. Biol Lett 12
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Liu Y, Kasahara M, Rumfelt LL, Flajnik MF (2002) Xenopus class II a genes: studies of genetics, polymorphism, and expression. Dev Comp Immunol 26:735–750
Liu Z, Hu DD, Shao SJ, Huang JQ, Wang JF, Yang J (2013) Polymorphisms in major histocompatibility complex class IIα genes are associated with resistance to infectious hematopoietic necrosis in rainbow trout, Oncorhynchus mykiss (Walbaum, 1792). J Appl Ichthyol 29:1234–1240
Lotteau V, Teyton L, Burroughs D, Charron D (1987) A novel HLA class II molecule (DRα-sDQβ) created by mismatched isotype pairing. Nature 329:339–341
Mauck RA, Dearborn DC, Huntington CE (2018) Annual global mean temperature explains reproductive success in a marine vertebrate from 1955 to 2010. Glob Chang Biol 24:1599–1613
May S, Zeisset I, Beebee TJC (2011) Larval fitness and immunogenetic diversity in chytrid-infected and uninfected natterjack toad (Bufo calamita) populations. Conserv Genet 12:805–811
Minias P, Pikus E, Whittingham LA, Dunn PO (2018) A global analysis of selection at the avian MHC. Evolution 72:1278–1293
Minias P, Pikus E, Whittingham LA, Dunn PO (2019) Evolution of copy number at the MHC varies across the avian tree of life. Genome Biol Evol 11:17–28
Parker A, Kaufman J (2017) What chickens might tell us about the MHC class II system. Curr Opin Immunol 46:23–29
Raymond M, Rousset F (1995) Genepop (version-1.2)—population-genetics software for exact tests and ecumenicism. J Hered 86:248–249
Ren L, Yang Z, Wang T, Sun Y, Guo Y, Zhang Z, Fei J, Bao Y, Qin T, Wang J, Huang Y, Hu X, Zhao Y, Li N (2011) Characterization of the MHC class II α-chain gene in ducks. Immunogenetics 63:667–678
Ruberti G, Sellins KS, Hill CM, Germain RN, Fathman CG, Livingstone A (1992) Presentation of antigen by mixed isotype class II molecules in normal H-2d mice. J Exp Med 175:157–162
Salomonsen J, Marston D, Avila D, Bumstead N, Johansson B, Juul-Madsen H, Olesen GD, Riegert P, Skjødt K, Vainio O, Wiles MV, Kaufman J (2003) The properties of the single chicken MHC classical class II α chain (B-LA) gene indicate an ancient origin for the DR/E-like isotype of class II molecules. Immunogenetics 55:605–614
Salzburger W, Ewing GB, Von Haeseler A (2011) The performance of phylogenetic algorithms in estimating haplotype genealogies with migration. Mol Ecol 20:1952–1963
Sin YW, Dugdale HL, Newman C, MacDonald DW, Burke T (2012) MHC class II genes in the European badger (Meles meles): characterization, patterns of variation, and transcription analysis. Immunogenetics 64:313–327
Spurgin LG, Richardson DS (2010) How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc R Soc B Biol Sci 277:979–988
Stet RJ, De Vries B, Mudde K, Hermsen T, Van Heerwaarden J, Shum BP, Grimholt U (2002) Unique haplotypes of co-segregating major histocompatibility class II A and class II B alleles in Atlantic salmon (Salmo salar) give rise to diverse class II genotypes. Immunogenetics 54:320–331
Sutton JT, Nakagawa S, Robertson BC, Jamieson IG (2011) Disentangling the roles of natural selection and genetic drift in shaping variation at MHC immunity genes. Mol Ecol 20:4408–4420
Szpiech ZA, Jakobsson M, Rosenberg NA (2008) ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24:2498–2504
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Taniguchi Y, Matsumoto K, Matsuda H, Yamada T, Sugiyama T, Homma K, Kaneko Y, Yamagishi S, Iwaisaki H (2014) Structure and polymorphism of the major histocompatibility complex class II region in the Japanese crested Ibis, Nipponia nippon. PLoS One 9:e108506
Traherne JA (2008) Human MHC architecture and evolution: implications for disease association studies. Int J Immunogen 35:179–192
Tsuji H, Taniguchi Y, Ishizuka S, Matsuda H, Yamada T, Naito K, Iwaisaki H (2017) Structure and polymorphisms of the major histocompatibility complex in the Oriental stork, Ciconia boyciana. Sci Rep 7:42864
Ujvari B, Belov K (2011) Major histocompatibility complex (MHC) markers in conservation biology. Int J Mol Sci 12:5168–5186
Wilson AB, Whittington CM, Bahr A (2014) High intralocus variability and interlocus recombination promote immunological diversity in a minimal major histocompatibility system. BMC Evol Biol 14:273
Xu T, Liu J, Sun Y, Zhu Z, Liu T (2016) Characterization of 40 full-length MHC class IIA functional alleles in miiuy croaker: polymorphism and positive selection. Dev Comp Immunol 55:138–143
Xu TJ, Chen SL, Zhang YX (2010) MHC class IIα gene polymorphism and its association with resistance/susceptibility to Vibrio anguillarum in Japanese flounder (Paralichthys olivaceus). Dev Comp Immunol 34:1042–1050
Yang M, Wei J, Li P, Wei S, Huang Y, Qin Q (2016) MHC class IIα polymorphisms and their association with resistance/susceptibility to Singapore grouper iridovirus (SGIV) in orange-spotted grouper, Epinephelus coioides. Aquaculture 462:10–16
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang Z, Wong WSW, Nielsen R (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Acknowledgments
We thank Bob Mauck, Frank Hailer, Erin Voss, Sabine Berzins, and Hillary Chavez for help with this project. Funding and equipment were provided by the Arthur Vining Davis Foundations and Institutional Development Awards (P20GM0103423) from the National Institute of General Medical Sciences of the National Institutes of Health. This is contribution number 277 from the Bowdoin Scientific Station on Kent Island.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendix
Appendix
Rights and permissions
About this article

Cite this article
Rand, L.M., Woodward, C., May, R. et al. Divergence between genes but limited allelic polymorphism in two MHC class II A genes in Leach’s storm-petrels Oceanodroma leucorhoa. Immunogenetics 71, 561–573 (2019). https://doi.org/10.1007/s00251-019-01130-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-019-01130-z