
Abstract
Re-naturalized quarry lakes are important ecosystems, which support complex communities of flora and fauna. Microorganisms associated with sediment and water form the lowest trophic level in these ecosystems and drive biogeochemical cycles. A direct comparison of microbial taxa in water and sediment microbial communities is lacking, which limits our understanding of the dominant functions that are carried out by the water and sediment microbial communities in quarry lakes. In this study, using the 16S rDNA amplicon sequencing approach, we compared microbial communities in the water and sediment in two re-naturalized quarry lakes in Singapore and elucidated putative functions of the sediment and water microbial communities in driving major biogeochemical processes. The richness and diversity of microbial communities in sediments of the quarry lakes were higher than those in the water. The composition of the microbial communities in the sediments from the two quarries was highly similar to one another, while those in the water differed greatly. Although the microbial communities of the sediment and water samples shared some common members, a large number of microbial taxa (at the phylum and genus levels) were prevalent either in sediment or water alone. Our results provide valuable insights into the prevalent biogeochemical processes carried out by water and sediment microbial communities in tropical granite quarry lakes, highlighting distinct microbial processes in water and sediment that contribute to the natural purification of the resident water.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Quarry lakes form when quarrying activity ceases and the disused sites are naturally filled with rainwater, surface runoff, and/or groundwater. Re-naturalized quarry lakes are important ecosystems which support complex communities of flora and fauna. Microorganisms, the primary colonizers of the quarry lake ecosystem, are believed to play important roles in various environmental processes critical to the re-naturalization of quarry lake ecosystems [15]. In a quarry rich in acidic sulfate soil, microorganisms were found to promote the growth of grasses and shrubs [41]. Microorganisms are also involved in the degradation of anthropogenic chemicals in quarry lakes. For example, microorganisms in the water and sediments were reported to drive the degradation of anthropogenic contaminants such as acrylamide in a quarry lake [27]. In addition, through interacting with solid minerals, microorganisms in quarry lakes also contribute significantly to the biogeochemical cycles of elements (e.g., iron and manganese cycles), weathering, and biodeterioration of solid minerals [9, 25, 46, 63].
Microorganisms in quarry lakes are primarily associated with the sediments and the water. Sediment microbial communities in lakes and marine ecosystems have been frequently linked to methane production, phosphorus cycling, and metal transformation [22,23,24, 54]. Microorganisms in the water of lakes and other natural water bodies, for example, Actinobacteria and Cyanobacteria, are often heavily involved in the decomposition of organic matter, carbon fixation, and production of nutrients for heterotrophs [4, 55]. Although microbial communities in both the sediment and water have been reported to contribute greatly to the health of the lakes and other natural water bodies, they may have distinct community structures and functions. In particular, for the quarry lake ecosystem, a direct comparison of microbial taxa in water and sediment microbial communities is lacking, which limits our understanding of the dominant functions that are carried out by the water and sediment microbial communities, respectively, in the recycling of nutrients and purification of resident water in quarry lakes.
The objective of this study was to compare the microbial communities in the water and sediment in re-naturalized quarry lakes. Specifically, we conducted 16S rDNA amplicon sequencing using the water and sediment samples collected from two quarry lakes in Singapore and elucidated dominant potential functions of the sediment and water microbial communities in driving major biogeochemical processes in these ecosystems.
Materials and Methods
Sampling Sites
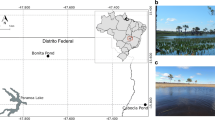
Water and sediment samples were collected from the lakes formed from the disused Singapore quarry (1° 21′ 23.7″ N, 103° 46′ 20.6″ E) and Ubin quarry (1° 24′ 29.2″ N, 103° 57′ 27.3″ E) in Singapore (Fig. 1a). Both quarry lakes were formed after the antecedent granite quarries ceased operating in the 1990s. The Singapore quarry lake is located near the central catchment of the main island of Singapore in a small (1.64 km2) nature reserve and is surrounded by a dense rainforest housing over 900 species of trees and ferns and nearly 500 species of fauna [14, 29]. The Ubin quarry lake is located on Pulau Ubin, an offshore island northeast to the main island of Singapore. Pulau Ubin is one of the last rural areas in Singapore and supports an abundance of plant and animal species. Unlike the Singapore quarry lake in which public access is restricted, the Ubin quarry lake is used for recreation frequently.

Geographic locations of the quarry lakes and details of the sediment and water samples. a The Singapore quarry lake is located in the mainland Singapore (1° 21′ 23.7″ N, 103° 46′ 20.6″ E) while the Ubin quarry lake (1° 24′ 29.2″ N, 103° 57′ 27.3″ E) is located on an offshore island, Pulau Ubin. b Sample, location, depth, and code of sediment and water samples collected. Sediment and water samples were collected from the surface of the Singapore quarry lake. Sediment and water samples were collected from the surface as well as at a depth of 25 m from the Ubin quarry lake
Sample Collection and Water Quality Measurement
The sediment samples from the two quarry lakes were collected using a Ponar sediment grab (Wildco Instruments, Wildlife Supply Company, USA), while the water samples were collected using a Van Dorn water sampler (Wildco Instruments, Wildlife Supply Company, USA). Sediment and water samples were collected from the surface (< 1 m below water level) for the Singapore quarry lake (S.00.SQ and W.00.SQ) and from the surface (< 1 m below water level) (S.00.UQ and W.00UQ) as well as 25 m below the surface for the Ubin quarry lake (S.25.UQ and W.25.UQ) (Fig. 1b). Samples were collected in triplicate. Samples from deeper locations in the Singapore quarry lake could not be collected as the access to those areas of the quarry lake is restricted. The collected water and sediment samples were immediately transferred to ice-packed clean autoclaved carboys and sterile plastic bags, respectively, and transported to the laboratory and stored at −80 °C for further use.
Water quality parameters, including turbidity, pH, oxidation-reduction potential (ORP), nitrate, ammonium, and ammonia, were measured in situ using an EXO2 multiparameter Sonde fitted with water quality sensors (Xylem Analytics, Hemmant, Australia).
Sample Processing and Sequencing
DNA from the sediment samples was extracted using the FastDNA® SPIN Kit for soil (MP Biomedicals, Santa Ana, USA). To extract DNA from water samples, 5 L water was filtered through nitrocellulose filters (diameter 47 mm, pore size 0.2 μm) and the genomic DNA was isolated from the filters using the PowerWater® DNA Isolation Kit (Mo Bio Laboratories Inc., Carlsbad, USA) according to the manufacturer’s instructions. Degenerate primers 16SF (TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCAGCMGCCGCGGTAA) (M = A/C) and 16SR (GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTACNVGGGTATCTAATCC) (N = A/T/C/G, V = G/C/A) were used to amplify an approximately 300 bp amplicon of the hypervariable region four (HV4) of bacterial and archaeal 16S rDNA [36]. The primers were designed to include the Illumina-specific overhang adapter sequences for compatibility with the Illumina index and sequencing adapters. 16S rDNA amplicons were amplified using the 2X Kapa HiFi HotStart ReadyMix (Kapa Biosystems, Wilmington, MA). PCR cycling was carried out by using an initial denaturation step of 95 °C for 3 min, followed by 30 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 30 s. The final extension step was performed at 72 °C for 5 min. The amplicons were purified by cleaning with Agencourt AMPure XP beads (Beckman Coulter, Brea, CA). Amplified DNA was quantified by Invitrogen Qubit fluorometric quantitation (Thermo Fisher Scientific, Waltham, MA). Amplicons were sequenced using the PCR-free paired-end sequencing approach on an Illumina MiSeq sequencing by synthesis (SBS) platform.
Data Processing
The sequences were uploaded onto the MG-RAST server (version 3.0) [26, 65] under project ID “Quarry Amplicons” (MG-RAST IDs: mgm4706502.3–mgm4706519.3). Reads were preprocessed by using “SolexaQA” to trim low-quality regions. Artificial replication reads were analyzed and removed using “duplicate read inferred sequencing error estimation” (DRISEE). The sequences were screened for contamination using “Bowtie” against Homo sapiens NCBI v36 as a reference database [69]. An initial search using “vsearch” against a reduced RNA database, which is a 90% identity-clustered version of SILVA, Greengenes, and RDP databases, was used for rDNA detection. The reads were then clustered at 97% identity using cd-hit, and the longest sequence was picked as the cluster representative. A BLAT (https://genome.ucsc.edu/FAQ/FAQblat) similarity search for the longest cluster representative sequences was performed against SSU, M5RNA, Greengenes, and RDP databases with an E-value cut-off of 1E−10, minimum identity of 97%, and a minimum alignment of 50 bp. The RDP tool on the MG-RAST server produced the highest number of hits in comparison to the other three databases, and hence, it was used for further analysis. The operational taxonomic unit (OTU) table was generated and downloaded in a comma-separated value format for further downstream analysis. The OTU table was filtered to exclude eukaryotic and chloroplast sequences as well as sequences from unidentified domains, and only prokaryotic sequences were kept.
Data Analyses
Subsequent analyses were performed in R using “vegan” and “phyloseq” packages for the analysis of ecological data [42, 49]. All samples were rarefied to a sequencing depth of least-abundant sample for the estimation of diversity and clustering and ordination analyses. Rarefaction was performed using the “phyloseq” package, and the diversity indices (Shannon, Simpson, Chao, and Ace) were estimated from the rarefied data using the “vegan” package. Nonparametric statistical analyses on the rarefied data were performed using “adonis,” “anosim,” and “betadisper” functions in R [2, 12, 68]. Hierarchical cluster analysis was performed using “hclust” function by Ward’s minimum variance method [44, 64]. Nonmetric dimensional scaling (NMDS) ordination was performed using the weighted “UniFrac distance metric” in the “phyloseq” package. NMDS ordination was also performed using the “Bray-Curtis dissimilarity index” in the vegan package.
For the comparison of OTU abundances across the samples, normalized OTU tables were used instead of rarefied data. All the samples were normalized against the total OTU abundance of individual samples. To compare the OTU abundances among the groups of samples, Welch’s test statistics was used. Functional annotation of taxa was performed using the program “functional annotation of prokaryotic taxa” (FAPROTAX) on the normalized OTU table [40]. FAPROTAX is a manually constructed database that maps prokaryotic taxa (e.g., genera or species) to putative functions based on the literature on cultured representatives. Functions represented in FAPROTAX focus on marine and lake biogeochemistry. It comes with a Python script for converting OTU tables into putative functional tables based on the taxa identified in a sample and their functional annotations in the FAPROTAX database. The main limitation of applying this approach to our data is the implicit assumption of FAPROTAX that if all cultured members of a taxon can perform a particular function, then all members of the taxon (cultured and noncultured) can perform that function. Even considering this caveat, we believe that predicting putative functional groups using this approach is superior to genomic prediction approaches based on sequence homology. The predicted abundances of functions among the groups of samples were also compared using Welch’s test statistics. Significant differences in the mean values were calculated at the 95% confidence interval (p < 0.05). Bonferroni correction was applied, where appropriate, to control for the effect of testing multiple hypotheses simultaneously.
Results and Discussion
Microbial Communities Are Richer and More Diverse in Sediment Than in Water
After removing low-quality sequences and mismatches, a total of 15,886,002 16S rDNA reads were obtained from the 18 samples, which were binned into 3346 OTUs based on 97% or higher sequence similarity. Rarefaction curves of the individual samples were asymptotic, indicating that reasonable sequencing depth was attained (Fig. A.1).
The evenness indices were significantly higher for the microbial communities in the sediments (S.00.SQ, S.00.UQ, and S.25.UQ) than in the corresponding water (W.00.SQ, W.00.UQ, and W.25.UQ) samples (p < 0.05) for both the Singapore quarry lake and the Ubin quarry lake (Table 1). The evenness indices ranged from 0.50 ± 0.01 for the deep-sediment sample from the Ubin quarry lake to 0.55 ± 0.02 for the surface sediment collected from the Singapore quarry lake, while those for the water samples were estimated to be in the range of 0.45 ± 0.04 to 0.46 ± 0.01. The Ace and Chao indices, which extrapolate the data to estimate what the actual number of species would have been based on the occurrence of rare species in the samples [13], estimated significantly higher (p < 0.05) OTU richness for the deep sediment from the Ubin quarry lake as well as from the surface sediment of the Singapore quarry lake in comparison to the corresponding water samples. The richness estimates for the surface sediment and water from the Ubin quarry lake were comparable.
The γ-diversity, calculated based on Shannon’s index, of the microbial communities in the sediments appeared to be considerably higher than that in the water samples (Table 1). The γ-diversity ranged from 3.51 for the deep sediment from the Ubin quarry lake to 3.98 for the surface sediment from the Singapore quarry lake, while for the water samples, it ranged from 3.20 for the water from the Singapore quarry lake to 3.34 for the water from the Ubin quarry lake. Similarly, the means of α-diversity indices (based on Shannon’s index) were significantly higher (p < 0.05) for the sediment samples than for the water samples. The Simpson’s diversity indices (ranging from 0.84 ± 0.14 for the surface sediment in the Ubin quarry lake to 0.89 ± 0.05 for the water in the Singapore quarry lake) did not differ significantly across the samples.
Differences in microbial diversity between the sediment and water samples from lake ecosystems have been reported previously in a few studies [20, 21, 43, 52]. For example, Qu et al. [52] showed that, in a lake ecosystem in China, Shannon’s diversity index was higher for the sediment samples in comparison with the water samples. In the same study, the Ace and Chao estimates for the richness were also found to be higher for the sediment samples. Our results were consistent with those previously reported in literature that Shannon’s diversity of sediments is usually higher than that of the associated water [20, 52]. On the other hand, we observed no significant variation in Simpson’s diversity across all the six samples. Since Simpson’s diversity index focuses on major taxa, our results suggest that the differences in microbial diversity across the samples arose mainly from the differences in rare taxa.
Community Compositions of Sediments Are Similar while those of Water Differ Greatly
A nonparametric statistical test using “anosim” and “adonis2” showed that (i) the difference between microbial communities across the sediment and water samples among the six groups was significantly greater than the difference among replicates within the same group (p < 0.01) and (ii) there was a significant difference between the microbial communities in the Ubin and Singapore quarry lakes (p < 0.001), as well as between the microbial communities in water and sediment samples (p < 0.001). Analysis of variance using “betadisper” further indicated that the differences in the community composition of the samples were not merely due to the differences in their variance (p > 0.14). These tests showed that the maximum difference in the microbial community composition was between the samples from the sediment and the water group (Table A.2). While differences were also expected in the microbial community composition between the two quarry lakes, these differences were not as prominent as those between the sediment and the water.
The dendrogram generated using unsupervised hierarchical clustering of the 18 samples showed two very distinct clusters, one consisting of all the sediment samples and the other consisting of all the water samples (Fig. A.2). In addition, the distances between the samples in the sediment groups were lower compared to those between the samples in the water groups, suggesting a higher similarity among the sediment samples.
NMDS ordination plot produced based on the Bray-Curtis dissimilarity also showed that the sediment samples were clustered away from the water samples and there was more variability among the samples in the water group in comparison to the sediment group (Fig. 2a). The water samples from the Singapore quarry lake were clustered relatively far from the water samples from the Ubin quarry lake, suggesting a high dissimilarity in the community composition between the water in the two quarries. In contrast, the sediment samples from different groups were clustered close to one another, indicating similar community composition in the sediment samples from the two quarries as well as from the surface and the deep sediment in the Ubin quarry lake. In addition, the water and sediment samples from the two quarries were clustered on separate corners of the ordination plot. Hence, some differences in the overall community composition between the two quarries were expected as well. In addition, the water samples from different groups were clustered far away not only from the sediment samples but also from each other, suggesting that the water samples were highly dissimilar from the sediments as well as from each other. Similar observations were made with the NMDS ordination plot produced based on the UniFrac distance metric (Fig. 2b). Although the exact placements of the different samples on the ordination plot varied a little, there were no major discrepancies in the conclusions arising from the two ordination plots.

Ordination plots produced based on a Bray-Curtis dissimilarity and b UniFrac distance metric. The sediment samples were clustered together while the water samples were clustered far apart from each other, indicating that the community composition in the sediment samples was more similar than that in the water samples
Major Microbial Taxa in the Quarry Lake Samples
Less than 2% (60 OTUs) of the total 3346 OTUs were classified as the domain Archaea and the rest as bacteria. It should also be noted that less than 1% of the total reads were assigned to the domain Archaea. OTUs were further assigned to lower taxonomic ranks, and abundance for each rank was estimated across different groups. A total of 31 microbial phyla were identified which consisted of 61 classes, 130 orders, 297 families, 1004 genera, and 3346 species. Of these, 5 phyla consisting of 12 classes, 18 orders, 24 families, 39 genera, and 60 species belonged to the domain Archaea. A large fraction of the reads (~ 36%) could not be classified into any known phyla (unclassified sequences). Together with the unclassified sequences, Proteobacteria (17.2%), Actinobacteria (12.3%), Cyanobacteria (9.5%), Firmicutes (7.4%), and Bacteroidetes (6.8%) accounted for approximately 90% of the total reads. The other major phyla were Verrucomicrobia (2.3%), Planctomycetes (1.9%), Nitrospirae (1.6%), Chloroflexi (1.5%), Spirochaetes (0.6%), Acidobacteria (0.5%), Gemmatimonadetes (0.5%), and Fusobacteria (0.5%) (Fig. A.3). The major phyla belonging to the domain Archaea were Thaumarchaeota (0.4%), Crenarchaeota (0.1%), and Euryarchaeota (0.1%). Approximately 0.002% of the total reads were assigned to unclassified sequences derived from archaea.
Actinobacteria, Bacteroidetes, Cyanobacteria, Chloroflexi, and Verrucomicrobia Dominate in Water
Actinobacteria, Bacteroidetes, Cyanobacteria, Chloroflexi, and Verrucomicrobia were mainly associated with the water samples in the two quarry lakes (Fig. A.4). Actinobacteria and Verrucomicrobia were in significantly higher proportion (p < 0.01) in the water from the Singapore quarry lake in comparison to the other sample groups (Fig. 3). This is consistent with previous studies where bacteria from these two phyla have been reported to be dominating in the water of a freshwater lake [45]. The dominant genera in these two phyla were represented in significantly higher proportion in the water samples in comparison to the sediment samples (p < 0.01). For example, the top five dominant genera identified in the phylum Actinobacteria, including Tetrasphaera and Streptomyces, were relatively more abundant in the water samples as compared to the sediment samples (Fig. A.5). Similarly, the dominant genera identified in the phylum Verrucomicrobia (Chthoniobacter, Rubritalea, and Prosthecobacter) were more abundant in the water samples as compared to the sediment samples.

Heatmap representing major differences in prokaryotic phyla among different samples. Actinobacteria, Firmicutes, Verrucomicrobia, Cyanobacteria, Chloroflexi, and Bacteroidetes were more abundant in water whereas Nitrospirae, Aquificae, and the phyla of Archaea were dominant in sediments. Proteobacteria were well represented in water as well as in sediments. The color code represents the row z-score; the number of standard deviations of a value differs from the mean. The numeric values represent relative abundance in a column in arbitrary units. The line profile in the color key is the histogram of all the values
In addition, the phylum Cyanobacteria was in higher proportion in the water samples as compared to the sediment samples. Nonetheless, some genera in this phylum were also prevalent in sediments (Fig. A.6). The most dominant genus in this phylum, Synechococcus, was in much higher proportion in the water samples as compared to the sediment samples, while Microcystis was more abundant in the deep-water samples compared to the surface water. Many taxa in the phylum Cyanobacteria, such as Synechococcus, have an affinity for the bright sunlight [1], while other genera, such as Microcystis, are known to remain in the sediments for a prolonged period of time for reasons not clearly understood [37]. Oscillatoria and Anabaena, along with other benthic Cyanobacteria genera such as Pleurocapsa, Arthrospira, Symploca, and Lyngbya, were represented in higher proportion in the sediments in comparison to water. These observations are consistent with previous reports where these cyanobacteria were found to form mats associated with aquatic sediments instead of freely floating in water [16, 18, 60].
Other phyla such as Bacteroidetes and Chloroflexi were found to be well represented in water as well as in sediments. In the phylum Chloroflexi, the dominant genus Chloroflexus was more abundant in the water samples while the other genus Herpetosiphon was more abundant in the sediment samples. In the phylum Bacteroidetes, the dominant genera Saprospira and Alistipes were more abundant in the water samples, while other genera such as Prolixibacter, Cytophaga, and Flexibacter were more abundant in the sediment samples (Fig. A.7). Prolixibacter was reported to be prevalent in marine sediments in a previous study [32]. Here, we found that these microorganisms were relatively more represented in the water of the quarry lakes rather than in the sediments. The reasons why these microorganisms appeared in higher proportion in the water samples rather than in the sediment samples remain elusive. Nonetheless, it has been reported that the turbulent mixing in the ecosystem may cause the upwelling of these microorganisms from sediment into the water [62], which could possibly be a reason for the relatively higher proportion of these microorganisms in the water samples rather than in the sediment samples.
Proteobacteria Are Well Represented in Water as well as in Sediments
Proteobacteria, the phylum with maximum number of reads, was well represented in both the water and the sediment samples from the two quarry lakes (Fig. 3). Most Proteobacteria identified in this study are commonly found in water and sediments of aquatic environments and are the dominant bacteria in a variety of freshwater biofilms such as river and streams and drinking water biofilms [3, 5, 19, 30, 71]. The Alphaproteobacteria class was in higher proportion in the water samples, while Gammaproteobacteria and Deltaproteobacteria were in higher proportion in the sediment samples (Fig. A.8). The abundances of other genera from this phylum also differed between the water and sediment samples. For example, Hyphomicrobium, Methylocystis, and Vibrio were in higher proportion in water, while Rhodovulum, Geobacter, Thioalkalivibrio, Gluconacetobacter, and Desulfococcus were more abundant in sediments.
Nitrospirae, Firmicutes, Aquificae, and the Phyla of Archaea Dominate in Sediments
Planctomycetes, Firmicutes, Nitrospirae, Chloroflexi, Spirochaetes, Acidobacteria, Aquificae, and Fibrobacteres were dominant in the sediment samples but scarce in the water samples for both quarry lakes. In addition, all phyla in the domain Archaea were in much higher proportion in the sediment samples than in the water.
Overall, the phylum Firmicutes was abundant in sediments (Fig. 3). The major genera in this phylum, e.g., Desulfotomaculum, Paenibacillus, and Sporanaerobacter, were dominant in sediments and scarce in water, although a few other genera in this phylum such as, Seinonella and Fusibacter, were more abundant in water (Fig. A.9).
All the three genera in the phylum Nitrospirae, i.e., Leptospirillum, Nitrospira, and Thermodesulfovibrio, were in significantly higher proportion in the sediment samples as compared to the water samples. Similarly, in the phylum Aquificae, the four dominant genera Sulfurihydrogenibium, Persephonella, Hydrogenobacter, and Hydrogenobaculum were more abundant in the sediment samples (Fig. A.10).
In the phylum Thaumarchaeota, all the three genera Nitrososphaera, Cenarcheum, and Nitrosopumilus were in higher proportion in the Ubin quarry lake sediments as compared to the Singapore quarry lake sediments (Fig. A.11). Nitrosocladus, the dominant genera in the phylum Crenarchaeota, was equally represented in the sediments of both quarry lakes. In the phylum Euryarchaeota, the dominant genera (Methanobacterium, Methanococcus, Methanosaeta, Methanolinea, and Methanoregula) were more abundant in the sediment samples from the Singapore quarry lake, while the subordinate genera (Methanothermus, Methanosarcina, and Methanomethylovorans) were more dominant in the sediment samples from the Ubin quarry lake.
Bacteria genera such as Geobacter, Desulfotomaculum, and Sporanaerobacter which were in higher proportion in the sediments in the current study have also been reported to be found mainly in sludge, soils, and sediments in previous studies [53, 58]. Similarly, gliding bacteria like Saprospira and Cytophaga, which were dominant in the sediment samples in this study, are abundant in epilithic biofilms in freshwater aquatic habitats like rivers and lakes [48]. A few other examples of bacteria that were abundant in sediments included Desulfotomaculum and Sporanerobacter which are anaerobic spore-forming bacteria commonly found in sediments and sludge [17, 31, 50, 51].
Potential Roles of Key Microbial Players in the Sediment and Water Samples
A number of microorganisms identified are known to be involved in crucial biogeochemical processes and interspecies interactions which occur primarily in sediments. For example, Geobacter, a metal reducer in soils and sediments, may play an important role in direct electron transfer to iron oxide as well as in interspecies electron transfer to methane-producing archaea [53, 58]. Similarly, major taxa in the domain Archaea, Thaumarchaeota and Crenarchaeota, may be involved in the oxidation of ammonia to nitrite, while Euryarchaeota is involved in methanogenesis [11, 34]. Bacteria in the phylum Spirochaetes (Leptospira, Treponema, and Aminobacterium) may contribute to the degradation of organic matter in sludge and sediments [6, 28]. To identify the potential biogeochemical processes in the water and sediment of these quarry lakes, a comprehensive assignment of microbial taxa to function was performed (Fig. 4).

Heatmap representing major differences in predicted functions among different sample groups. Methanogenesis, methylotrophy, degradation of aromatic compounds and hydrocarbons, and metal and sulfur transformation as well as nitrification were associated with sediments. Phototrophy, aerobic chemoheterotrophy, and denitrification were abundant in water. The color code represents the row z-score; the number of standard deviations of a value differs from the mean. The numeric values represent relative abundance in a column in arbitrary units. The line profile in the color key is the histogram of all the values
Among the putative functions, aerobic chemoheterotrophy and phototrophy were more abundant in water as compared to sediments. On the other hand, degradation of biomass, aromatic compounds, and hydrocarbons, as well as transformation of metals and sulfur, was more abundant in sediments as compared to water. Aerobic chemoheterotrophy in the water of the Ubin and Singapore quarry lakes was mainly attributed to the abundance of bacteria such as Tetrasphaera, Saprospira, Microbacterium, and Streptomyces, to name a few (Fig. A.12). Synechococcus was the most abundant genus in the phylum Cyanobacteria and was associated with phototrophy (Fig. A.13). Bacillus, Cytophaga, Fibrobacter, Chitinophaga, and Rhodothermus were the predominant genera associated with extracellular hydrolysis in the sediments (Fig. A.14). Bacillus and Cytophaga are soil bacteria which are known to degrade cellulosic materials [38, 66, 67]. Degradation of aromatic compounds in the sediments was attributed mainly to Clostridia (Fig. A.15). Clostridium has been previously found in sludge and predicted to metabolize aromatic compounds to methane in anaerobic conditions [70]. Other major genera, such as Rhodococcus, capable of degrading both aromatic and nonaromatic hydrocarbons [7, 35], were found primarily in the sediments of the two quarries. Metal transformation was attributed to Geobacter, Desulfobacterium, Desulfomonas, Shewanella, and Leptospirillum in the sediments. Transformation of sulfur compounds could be attributed mainly to Desulfotomaculum, Desulfobacterium, Desulfococcus, Sporanaerobacter, and Thioalkalivibrio (Fig. A.16). Most of the nitrifiers were found primarily in sediments, and hence, nitrification was predicted to occur primarily in the sediments (Fig. A.17). This is counterintuitive as nitrification is an aerobic process and the oxygen concentration in the sediments is expected to be lower than that in the water. However, it should be noted that many of these microorganisms have often been found in sludge, sediments, or the transition zone at the oxic-anoxic interface [8, 33, 39, 56, 61]. Previous studies have shown that nitrification is inhibited by sunlight [10], which could be a possible reason for the localization of these nitrifying bacteria in the sediments during mid-day, when the samples were collected from the quarry lakes. In oxygenated waters, nitrifiers are believed to be prevalent at the sediment-water interface, and nitrification takes place primarily in the upper layers of sediments [54, 57, 59].
It should be noted that the functional assignment of the quarry lake ecosystems based on microbial taxonomy is putative. Nevertheless, these predictions provide insights into the potential functions of water and sediment microbial communities in quarry lakes. Additionally, the temporal microbial dynamics of the quarry lake ecosystems are not explored in this study as the quarry lakes in Singapore are protected areas with restricted access. Nonetheless, it is likely that there are no significant temporal variations in these quarry lakes as there is little seasonal variation in tropical regions. For example, a recent study on bacterial communities in reservoir water in Singapore showed a weak temporal variation in the overall bacterial community composition [47].
Conclusions
The richness and diversity of microbial communities in sediments of the quarry lakes were higher than those in the water. In addition, the compositions of the microbial communities in the sediments from the two quarries were highly similar to one another, while those in the water differed greatly. Although the microbial communities of the sediment and water samples shared some common members, a large number of microbial taxa (at the phylum and genus levels) were predominantly found either in sediment or in water. Bacteria in the phyla Actinobacteria, Verrucomicrobia, Cyanobacteria, Chloroflexi, and Bacteroidetes were more abundant in water whereas Nitrospirae, Aquificae, and Firmicutes along with Archaea were abundant in sediments. Proteobacteria was well represented in water as well as in sediments. Phototrophy and aerobic chemoheterotrophy were expected to be the most prevalent microbial processes in water, while degradation of organic matter and transformation of metals and sulfur compounds were expected to occur mainly in sediments. Intriguingly, nitrification, which is traditionally expected to occur in water, was implied to occur primarily in the sediments. Our results provide valuable insights into the putative microbial processes in water and sediment that potentially contribute to the biogeochemical processes carried out by water and sediment microbial communities in tropical granite quarry lakes.
References
Allewalt JP, Bateson MM, Revsbech NP, Slack K, Ward DM (2006) Effect of temperature and light on growth of and photosynthesis by Synechococcus isolates typical of those predominating in the octopus spring microbial mat community of Yellowstone National Park. Appl Environ Microbiol 72:544–550
Anderson MJ (2006) Distance-based tests for homogeneity of multivariate dispersions. Biometrics 62:245–253
Anderson-Glenna MJ, Bakkestuen V, Clipson NJ (2008) Spatial and temporal variability in epilithic biofilm bacterial communities along an upland river gradient. FEMS Microbiol Ecol 64:407–418
Bai L, Cao C, Wang C, Xu H, Zhang H, Slaveykova VI, Jiang H (2017) Toward quantitative understanding of the bioavailability of dissolved organic matter in freshwater lake during Cyanobacteria blooming. Environ Sci Technol 51:6018–6026
Battin TJ, Besemer K, Bengtsson MM, Romani AM, Packmann AI (2016) The ecology and biogeochemistry of stream biofilms. Nat. Rev. Microbiol. 14:251–263
Benacer D, Woh PY, Zain SNM, Amran F, Thong KL (2013) Pathogenic and saprophytic Leptospira species in water and soils from selected urban sites in peninsular Malaysia. Microbes Environ. 28:135–140
Bicca FC, Fleck LC, Ayub MAZ (1999) Production of biosurfactant by hydrocarbon degrading Rhodococcus ruber and Rhodococcus erythropolis. Rev. Microbiol. 30:231–236
Bollmann A, Bullerjahn GS, McKay RM (2014) Abundance and diversity of ammonia-oxidizing archaea and bacteria in sediments of trophic end members of the Laurentian Great Lakes, Erie and Superior. PLoS One 9(5):e97068
Cámara B, De los Ríos A, Urizal M, De Buergo MÁ, Varas MJ, Fort R, Ascaso C (2011) Characterizing the microbial colonization of a dolostone quarry: implications for stone biodeterioration and response to biocide treatments. Microb Ecol 62:299–313
Capone DG, Bronk DA, Mulholland MR, Carpenter EJ (2008) Nitrogen in the marine environment. Academic Press, Cambridge
Chin K-J, Lukow T, Conrad R (1999) Effect of temperature on structure and function of the methanogenic archaeal community in an anoxic rice field soil. Appl Environ Microbiol 65:2341–2349
Clarke K (1993) Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 18:117–143
Colwell RK, Chao A, Gotelli NJ, Lin S-Y, Mao CX, Chazdon RL, Longino JT (2012) Models and estimators linking individual-based and sample-based rarefaction, extrapolation and comparison of assemblages. J Plant Ecol 5:3–21
Corlett RT (1988) Bukit Timah: the history and significance of a small rain-forest reserve. Environ Conserv 15:37–44
Cotner JB, Biddanda BA (2002) Small players, large role: microbial influence on biogeochemical processes in pelagic aquatic ecosystems. Ecosystems 5:105–121
Dadheech PK, Glöckner G, Casper P, Kotut K, Mazzoni CJ, Mbedi S, Krienitz L (2013) Cyanobacterial diversity in the hot spring, pelagic and benthic habitats of a tropical soda lake. FEMS Microbiol Ecol 85:389–401
De Rezende JR, Kjeldsen KU, Hubert CR, Finster K, Loy A, Jørgensen BB (2013) Dispersal of thermophilic Desulfotomaculum endospores into Baltic Sea sediments over thousands of years. ISME J 7:72–84
Domaizon I, Savichtcheva O, Debroas D, Arnaud F, Villar C, Pignol C, Alric B, Perga ME (2013) DNA from lake sediments reveals the long-term dynamics and diversity of Synechococcus assemblages. Biogeosciences 10:3817–3838
Douterelo I, Jackson M, Solomon C, Boxall J (2016) Microbial analysis of in situ biofilm formation in drinking water distribution systems: implications for monitoring and control of drinking water quality. Appl Microbiol Biotechnol 100:3301–3311
Fang L, Chen L, Liu Y, Tao W, Zhang Z, Liu H, Tang Y (2015) Planktonic and sedimentary bacterial diversity of Lake Sayram in summer. Microbiologyopen 4:814–825
Feng B-W, Li X-R, Wang J-H, Hu Z-Y, Meng H, Xiang L-Y, Quan Z-X (2009) Bacterial diversity of water and sediment in the Changjiang estuary and coastal area of the East China Sea. FEMS Microbiol Ecol 70:236–248
Ferris FG, Fyfe W, Beveridge T (1987) Bacteria as nucleation sites for authigenic minerals in a metal-contaminated lake sediment. Chem Geol 63:225–232
Frenzel P, Thebrath B, Conrad R (1990) Oxidation of methane in the oxic surface layer of a deep lake sediment (Lake Constance). FEMS Microbiol Ecol 6:149–158
Gächter R, Meyer JS, Mares A (1988) Contribution of bacteria to release and fixation of phosphorus in lake sediments. Limnol Oceanogr 33:1542–1558
Garcia-Valles M, Urzì C, Vendrell-Saz M (2002) Weathering processes on the rock surface in natural outcrops: the case of an ancient marble quarry (Belevi, Turkey). Environ Geol 41:889–897
Glass EM, Wilkening J, Wilke A, Antonopoulos D, Meyer F (2010) Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb Protoc. https://doi.org/10.1101/pdb.prot5368
Guezennec A-G, Michel C, Ozturk S, Togola A, Guzzo J, Desroche N (2015) Microbial aerobic and anaerobic degradation of acrylamide in sludge and water under environmental conditions—case study in a sand and gravel quarry. Environ Sci Pollut Res 22:6440–6451
Hamdi O, Hania WB, Postec A, Bouallagui H, Hamdi M, Bonin P, Ollivier B, Fardeau M-L (2015) Aminobacterium thunnarium sp. nov., a mesophilic, amino acid-degrading bacterium isolated from an anaerobic sludge digester, pertaining to the phylum Synergistetes. Int J Syst Evol Microbiol 65:609–614
Henderson JC (2000) The survival of a forest fragment: Bukit Timah Nature Reserve, Singapore. Forest tourism and recreation: case studies in environmental management. New York: Cabi Pub, pp 23–39
Henne K, Kahlisch L, Brettar I, Höfle MG (2012) Analysis of structure and composition of bacterial core communities in mature drinking water biofilms and bulk water of a citywide network in Germany. Appl Environ Microbiol 78:3530–3538
Hernandez-Eugenio G, Fardeau M-L, Cayol J-L, Patel BK, Thomas P, Macarie H, Garcia J-L, Ollivier B (2002) Sporanaerobacter acetigenes gen. nov., sp. nov., a novel acetogenic, facultatively sulfur-reducing bacterium. Int J Syst Evol Microbiol 52:1217–1223
Holmes DE, Nevin KP, Woodard TL, Peacock AD, Lovley DR (2007) Prolixibacter bellariivorans gen. nov., sp. nov., a sugar-fermenting, psychrotolerant anaerobe of the phylum Bacteroidetes, isolated from a marine-sediment fuel cell. Int J Syst Evol Microbiol 57(4):701–707
Iguchi A, Terada T, Narihiro T, Yamaguchi T, Kamagata Y, Sekiguchi Y (2009) In situ detection and quantification of uncultured members of the phylum Nitrospirae abundant in methanogenic wastewater treatment systems. Microbes Environ 24(2):97–104
Kerou M, Offre P, Valledor L, Abby SS, Melcher M, Nagler M, Weckwerth W, Schleper C (2016) Proteomics and comparative genomics of Nitrososphaera viennensis reveal the core genome and adaptations of archaeal ammonia oxidizers. Proc Natl Acad Sci 113:E7937–E7946
Kim D, Kim Y-S, Kim S-K, Kim SW, Zylstra GJ, Kim YM, Kim E (2002) Monocyclic aromatic hydrocarbon degradation by Rhodococcus sp. strain DK17. Appl Environ Microbiol 68:3270–3278
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e1–e1
Latour D, Sabido O, Salençon M-J, Giraudet H (2004) Dynamics and metabolic activity of the benthic cyanobacterium Microcystis aeruginosa in the Grangent reservoir (France). J Plankton Res 26:719–726
Lin C-C, San Yan CJ, Kan S-C, Hsueh N-C, Yang L-Y, Shieh C-J, Huang C-C, Liu Y-C (2017) Deciphering characteristics of the designer cellulosome from Bacillus subtilis WB800N via enzymatic analysis. Biochem Eng J 117:147–155
Liu B, Li Y, Zhang J, Zhou X, Wu C (2014) Abundance and diversity of ammonia-oxidizing microorganisms in the sediments of Jinshan Lake. Curr Microbiol 69(5):751–757
Louca S, Parfrey LW, Doebeli M (2016) Decoupling function and taxonomy in the global ocean microbiome. Science 353:1272–1277
Maki T, Nomachi M, Yoshida S, Ezawa T (2008) Plant symbiotic microorganisms in acid sulfate soil: significance in the growth of pioneer plants. Plant Soil 310:55–65
McMurdie PJ, Holmes S (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217
Mesbah NM, Abou-El-Ela SH, Wiegel J (2008) Novel and unexpected prokaryotic diversity in water and sediments of the alkaline, hypersaline lakes of the Wadi An Natrun, Egypt. Microb Ecol 55:369–369
Murtagh F, Legendre P (2014) Ward’s hierarchical agglomerative clustering method: which algorithms implement ward’s criterion? J Classif 31:274–295
Newton RJ, Jones SE, Eiler A, McMahon KD, Bertilsson S (2011) A guide to the natural history of freshwater lake bacteria. Microbiol Mol Biol Rev 75:14–49
Ng DHP, Kumar A, Cao B (2016) Microorganisms meet solid minerals: interactions and biotechnological applications. Appl Microbiol Biotechnol 100:6935–6946
Nshimyimana JP, Freedman AJ, Shanahan P, Chua LC, Thompson JR (2017) Variation of bacterial communities with water quality in an urban tropical catchment. Environ Sci Technol 51(10):5591–5601
O’Sullivan LA, Weightman AJ, Fry JC (2002) New degenerate Cytophaga-Flexibacter-Bacteroides-specific 16S ribosomal DNA-targeted oligonucleotide probes reveal high bacterial diversity in River Taff epilithon. Appl Environ Microbiol 68:201–210
Oksanen J, Blanchet F, Kindt R, Legendre P, O’Hara R (2016) Vegan: community ecology package. R Packag. 2.3-3
O’sullivan LA, Roussel EG, Weightman AJ, Webster G, Hubert CR, Bell E, Head I, Sass H, Parkes RJ (2015) Survival of Desulfotomaculum spores from estuarine sediments after serial autoclaving and high-temperature exposure. ISME J 9:922–933
Otwell AE, Callister SJ, Zink EM, Smith RD & Richardson RE (2016) Comparative proteomic analysis of Desulfotomaculum reducens MI-1: insights into the metabolic versatility of a gram-positive sulfate- and metal-reducing bacterium. Front Microbiol 7:191
Qu J, Zhang Q, Zhang N, Shen L, Liu P (2015) Microbial community diversity in water and sediment of an eutrophic lake during harmful algal bloom using MiSeq illumina technology. Int Congr Adv Environ Res. https://doi.org/10.7763/IPCBEE
Rotaru A-E, Shrestha PM, Liu F, Markovaite B, Chen S, Nevin KP, Lovley DR (2014) Direct interspecies electron transfer between Geobacter metallireducens and Methanosarcina barkeri. Appl. Environ Microbiol 80:4599–4605
Rothfuss F, Bender M, Conrad R (1997) Survival and activity of bacteria in a deep, aged lake sediment (Lake Constance). Microb Ecol 33:69–77
Savage DF, Afonso B, Chen AH, Silver PA (2010) Spatially ordered dynamics of the bacterial carbon fixation machinery. Science 327:1258–1261
Schramm A, De Beer D, Gieseke A, Amann R (2000) Microenvironments and distribution of nitrifying bacteria in a membrane-bound biofilm. Environ Microbiol 2(6):680–686
Seitzinger S, Nixon S (1987) Eutrophication and the rate of denitrification and N2O production in nearshore marine sediments. Limnol Oceanogr 30:1332–1339
Smith JA, Tremblay P-L, Shrestha PM, Snoeyenbos-West OL, Franks AE, Nevin KP, Lovley DR (2014) Going wireless: Fe (III) oxide reduction without pili by Geobacter sulfurreducens strain JS-1. Appl Environ Microbiol 80:4331–4340
Sørensen J, Jørgensen BB, Revsbech NP (1979) A comparison of oxygen, nitrate, and sulfate respiration in coastal marine sediments. Microb Ecol 5:105–115
Stal LJ, Severin I, Bolhuis H (2010) The ecology of nitrogen fixation in cyanobacterial mats. In: Hallenbeck PC (ed) Recent advances in phototrophic prokaryotes. Springer, Berlin/Heidelberg, pp 31–45
Stieglmeier M, Klingl A, Alves RJ, Simon KM, Melcher M, Leisch N, Schleper C (2014) Nitrososphaera viennensis gen. nov., sp. nov., an aerobic and mesophilic, ammonia-oxidizing archaeon from soil and a member of the archaeal phylum Thaumarchaeota. Int J Syst Evol Microbiol 64(8):2738–2752
Tsujimura S, Okubo T (2003) Development of Anabaena blooms in a small reservoir with dense sediment akinete population, with special reference to temperature and irradiance. J Plankton Res 25:1059–1067
Urzí C, Realini M (1998) Colour changes of Notos calcareous sandstone as related to its colonisation by microorganisms. Int Biodeterior Biodegrad 42:45–54
Ward Jr JH (1963) Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 58:236–244
Wilke A, Gerlach W, Harrison T, Paczian T, Trimble WL, Meyer F (2016) MG-RAST Manual for version 4, revision
Wrótniak-Drzewiecka W, Brzezińska AJ, Dahm H, Ingle AP, Rai M (2016) Current trends in myxobacteria research. Ann Microbiol 66:17–33
Xie G, Bruce DC, Challacombe JF, Chertkov O, Detter JC, Gilna P, Han CS, Lucas S, Misra M, Myers GL (2007) Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii. Appl Environ Microbiol 73:3536–3546
Zapala MA, Schork NJ (2006) Multivariate regression analysis of distance matrices for testing associations between gene expression patterns and related variables. Proc Natl Acad Sci 103:19430–19435
Keegan KP, Glass EM, Meyer F (2016) MG-RAST, a metagenomics service for analysis of microbial community structure and function. In: Martin F, Uroz S (eds) Microbial environmental genomics. Springer, Berlin/Heidelberg, pp 207–233
Shcherbakova VA, Laurinavichyus KS, Chuvil’skaya NA, Ryzhmanova YV, Akimenko VK (2015) Anaerobic bacteria involved in the degradation of aromatic sulfonates to methane. Appl Biochem Microbiol 51(2):209–214
Liu G, Ling FQ, van der Mark EJ, Zhang XD, Knezev A, Verberk JQJC, van der Meer WGJ, Medema GJ, Liu WT, van Dijk JC (2016) Comparison of particle-associated bacteria from a drinking water treatment plant and distribution reservoirs with different water sources. Sci Rep 6:20367
Acknowledgements
Sequencing was carried out with the help of Dr. Daniela Moses and Professor Stephan Schuster using the sequencing facilities at the Singapore Centre for Environmental Life Sciences Engineering (SCELSE), Nanyang Technological University, Singapore. We thank the Singapore National Parks Board (NParks), in particular, Ms. Samantha Lai and Ms. Joanna Yeo, for their assistance in sample collection. This material is based on research/work supported by the Singapore Ministry of National Development and National Research Foundation under L2 NIC Award No. L2NICCFP1-2013-3.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Disclaimer
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the L2 NIC.
Electronic Supplementary Material
ESM 1
(PDF 3659 kb)
Rights and permissions
About this article

Cite this article
Kumar, A., Ng, D.H.P., Wu, Y. et al. Microbial Community Composition and Putative Biogeochemical Functions in the Sediment and Water of Tropical Granite Quarry Lakes. Microb Ecol 77, 1–11 (2019). https://doi.org/10.1007/s00248-018-1204-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-018-1204-2