
Abstract
Bacterioplankton are the major driving force for biogeochemical cycles in estuarine ecosystems, but the communities that mediate these processes are largely unexplored. We sampled in the Pearl River Estuary (PRE) to examine potential differences in the taxonomic composition of resident (DNA-based) and active (RNA-based) bacterioplankton communities in free-living and particle-associated fractions. MiSeq sequencing data showed that the overall bacterial diversity in particle-associated fractions was higher than in free-living communities. Further in-depth analyses of the sequences revealed a positive correlation between resident and active bacterioplankton communities for the particle-associated fraction but not in the free-living fraction. However, a large overlapping of OTUs between free-living and particle-associated communities in PRE suggested that the two fractions may be actively exchanged. We also observed that the positive correlation between resident and active communities is more prominent among the abundant OTUs (relative abundance > 0.2%). Further, the results from the present study indicated that low-abundance bacterioplankton make an important contribution towards the metabolic activity in PRE.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Estuarine ecosystems are known for their high variations of salinity and nutrient supplies [1,2,3]. This heterogeneity is further complicated by an interplay between growing economy and high human population density [4], making estuaries one of the most complex and dynamic ecosystems [1]. Bacteria are major drivers for biogeochemical cycling in estuaries [5,6,7]. Examination of the structures and functions of estuarine bacterial community can, therefore, offer understandings on the ecology and health of estuaries.
Bacterioplankton in water column can be separated into free-living (FLB; fraction between 0.2 and 3 μm) and particle-associated communities (PAB; fraction > 3 μm), which appear to carry distinct taxonomic compositions and functions [8,9,10,11,12]. PAB are often associated with a higher metabolic potential than FLB in a number of oceanic and freshwater environments [8, 13]. While assessment of bacterial community structure has been done extensively in estuaries, only few studies reflect the importance of relationship between active and resident communities [14, 15]. Studies in bacterial communities of costal bay surface and coral sediments have indicated a positive relationship between DNA and RNA frequencies [16, 17], but not so in a few other estuary aquatic environments [18]. It is, however, unclear whether the same pattern holds in the Pearl River Estuary.
Pearl River Estuary (PRE) is located in Southern China within the subtropical zone, and its drainage basins cover an area of 39,380 km2. PRE watershed serves as home to more than 65 million people [1] and is one of the largest industrialized areas in China [19,20,21]. Consequently, PRE receives excessive nutrients and pollutants from agricultural and industrial activities [22], which shift local microbial community structures [23]. While biogeochemical studies on PRE have been predominantly focused on measuring the concentrations and fluxes of pollutants, particularly heavy metals, chlorinated hydrocarbons, and antibiotics [22, 24,25,26], a separate study on distribution of bloom-forming cyanobacteria-Microcystis determined that physicochemical parameters of the environment including total nitrogen to total phosphorus ratio, the ammonium to nitrate ratio, water temperature, and pH played an influencing role for microbial shift [23].
In this study, DNA-based techniques was employed together with RNA-based methods to characterize residential (DNA-based) and active (RNA-based) microbial community structure in PRE at both the PAB and FLB levels. Environmental factors that might influence the diversity, activity, and structure of PAB and FLB were also examined.
Materials and Methods
Sample Collection and Processing
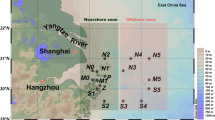
Surface water samples (~ 1 m depth) were collected from three sampling stations: upstream Zhongda (ZD, 23.1037° N, 113.3008° E), midstream Humen (HM, 22.7837° N, 113.5988° E), and downstream Zhuhai (ZH, 22.3408° N, 113.5953° E) (Fig. 1) along the course of Pearl River in May 2016. Environmental variables including temperature (T), salinity (S), conductivity (Cond), dissolved oxygen (DO), dissolved solid (DS), pH, turbidity (TU), and oxidation-reduction potential (ORP) were measured on site using a HQD Field Kit (HACH 58258; Loveland, Colorado, USA) and a portable Turbidimeter (HACH 2100Q; Loveland, Colorado, USA). Immediately after collection, 2 L of water samples were filtered through membrane filters (Pall Life Sciences, Ann Arbor, MI, USA) of pore sizes 3 and 0.2 μm sequentially. The filtrates were collected in 50-ml sterile centrifuge tubes and immediately kept on ice. These filtrates were, then, transported to the laboratory and stored at − 20 °C until analysis for dissolved organic carbon (DOC), dissolved nitrogen (DN), nitrate (NOx−), ammonium ion (NH4+), and soluble reactive phosphorus (SRP). Cells that were collected on 3 and 0.2-μm membrane filters were frozen immediately in liquid nitrogen on field sites for transportation to the laboratory and were processed within 24 h for the extraction of RNA and DNA (details provided below). Part of the filtrate (10 ml) that passed through 3-μm filter was preserved in 1% freshly prepared paraformaldehyde and stored on ice or at 4 °C before cell enumeration. Phytoplankton present in 500 ml whole water samples was collected on glass fiber filters (GF/F; Whatman International Ltd., Maidstone, England) and stored in the dark at − 20 °C for 1~2 weeks before measurement of chlorophyll a (Chl a) [27].

Geographical location of the sampling stations in Pearl River Estuary
All samples were collected in triplicates. Data from the triplicate samples were combined to provide the standard error for each site. Glassware and GF/F filters used for sample collections were sterilized by heating at 500 °C for 4 h before use.
Analysis of Nutrient Content in the Water Samples
Nutrients in the whole water samples were measured according to standard procedures [27]. Concentration of DOC was determined using a total-organic-carbon (TOC) analyzer (TOC-VCPN; Shimadzu Corp., Tokyo, Japan) based on combustion-oxidation and combustion-detection methods. Concentrations of SRP and NOx− were measured spectrophotometrically based on molybdenum blue and hydrazine sulfate-NEDD [n-(1-napthyl)ethylene diamine dihydrochloride] methods, respectively. Concentrations of DN and NH4+ were determined spectrophotometry using flow injection protocols (Westco, Smart Chem 200 series flow injection analysis system, Scientific Instruments, Italy). Concentration of POM was determined using a TOC analyzer (TOC-VCPN; Shimadzu Corp., Tokyo, Japan), following fuming of GF/F filters with concentrated HCl for 4 h to remove any inorganic carbonate, oven-dried at 60 °C overnight as described earlier [13]. Chl a was extracted from the GF/F filters with 90% acetone and its concentration measured spectrophotometrically.
Bacterial Cell Enumeration
Bacterial cells were stained with SYBR Green II Nuclic Acid stain (Invitrogen™, Carlsbad, CA, USA) for 15 min in the dark. Stained bacterial cells were discriminated and enumerated using a Beckman flow cytometer (FC500; South Kraemer Blvd. Brea, CA, USA). Cell analysis was based on two parameters: green fluorescence emitted from SYBR Green II stain and side scatter, which is positively correlated with the size of the cells [28]. Quantitative PCR (qPCR) was performed as described by Kumar et al. [29]. Analyses of the real-time PCR data of all samples showed a single melting peak corresponding to the standard DNA. The R2 values for the standard curve were above 0.97 at the range of 1 × 102 to 1 × 1010, and the efficiency of each qPCR was between 95 and 99%.
Nucleic Acid Extraction and cDNA Synthesis
Nucleic acids (DNA and RNA) from each sample were extracted from the cells retained on 3 and 0.2-μm membrane filters. The frozen filters were cut into pieces and vortex with PowerSoil beads (MO BIO Laboratories, Inc., Carlsbad, CA, USA). Total RNA was extracted using PowerSoil® Total RNA Isolation kit (MO BIO Laboratories, Inc., Carlsbad, CA, USA), while reserving the RNA Capture Column for further extraction of DNA. Extracted RNA was reverse-transcribed into cDNA using M-MLV First-Strand cDNA Synthesis Kit (Omega Bio-Tek, Doraville, GA, USA). Community DNAs bound to RNA Capture Column on the previous experiment were extracted using RNA PowerSoil® DNA Elution Accessory kit (MO BIO Laboratories, Inc., Carlsbad, CA, USA).
PCR Amplification and Sequencing
Amplification of the V4~V5 region of 16S rRNA gene for both of the environmental DNAs and cDNAs was performed using the method described by Fierer et al. [30]. To permit multiplexing of samples, 12-bp barcode unique to each sample was linked to the 5′ end of the reverse primer using two bases CC. Following amplification, each set of PCR products were pooled together in equimolar concentrations. The final composite products were purified using E.Z.N.A.® Gel Extraction Kit (Omega BioTek, Doraville, GA, USA). Sequencing was performed on a 2 × 300 paired-end MiSeq platform (Illumina; San Diego, CA, USA).
Data Processing and Sequence Annotation
Raw read data containing bases with Phred quality score of less than 30 at 3′ end were trimmed to enhance the quality of the read. All pair-end reads were assembled using Mothur (version 1.36.1) [31] based on the overlapping regions. Assembled sequences with more than 10% mismatches were removed from the dataset. These resulting sequences were reassigned to their respective samples based on the 12-bp barcode. Finally, the primer sequences were trimmed using trim.seqs by applying the parameters (maxhomop = 12, qwindowsize = 50, qwindowaverage = 20, maxambig = 0, pdiffs = 2, bdiffs = 1, flip = T). Sequences that were longer than 360 bp were kept for further annotation.
Assembled raw sequence reads were clustered into operational taxonomic units (OTUs) using USEARCH software version 8.0 (USPARCE algorithm) at 97% similarity threshold, while removing the chimeric sequences. After applying the above quality control measures, a total of 357,699 reads were removed, leaving behind 199,086 high-quality sequences. Taxonomic affiliation of the OTUs was determined using RDP classifier [32] with a confidence threshold of 0.8. OTUs classified either as unknown, chloroplast, or mitochondria were filtered from further analysis.
Statistical Analysis
All statistical analyses based on normalized OTU table were performed with R software by applying the vegan package (R, Development Core Team, Vienna), unless otherwise mentioned. Diversity of the bacteria was calculated from the Chao1, Simpson, and Shannon diversity indices using QIIME [33]. Distances between different samples were analyzed using UPGMA algorithm. PCA plot of the abiotic characteristics was generated using the Bray-Curtis dissimilarity matrix. Venn diagrams were plotted using the online software “BioVenn” [34] to determine the number of exclusive or shared OTUs between different samples of the three stations.
Sequence Accession
Raw sequence reads were submitted to Sequence Read Archive (SRA), NCBI under the accession numbers SRR6173480–SRR6173503 (BioProject accession PRJNA413877; BioSample accessions SAMN07774240–SAMN07774251).
Results
Abiotic Characteristics of Sampling Sites
We observed significant differences in the physicochemical variables of the water samples collected from ZD, HM, and ZH (Fig. S1; Table S1). The first two axes explained 92.0% of the variation (PC1 56.5% and PC2 35.5%; Fig. 2). Variations (score > 1) were mainly resulted from Cond, T, DS, and S. A gradual increase in pH, TU, DO, and particulate organic matter (POM) was observed from the upstream site ZD to the downstream site ZH, while no particular gradient was maintained for the other parameters between the three sites during our sampling period (Table S1, p < 0.05). The upstream site ZD had the highest concentrations of DOC, DN, SPR, NO3−, NH4+, and POM, while the downstream site ZH displayed the highest values for Cond, DO, DS, ORP, POM, pH, S, T, and TU (Table S1, p < 0.05).

PCA plot indicating the relationship between sampling stations and the physicochemical variables in PRE
Relative Abundance of Bacterioplankton
As revealed by flow cytometry (FCM) analysis, the number of FLB cells was the highest (6.48 × 106 cells ml−1) in HM samples and the least (2.48 × 106 cells ml−1) in ZH samples (Table S1). Similar frequency ratio for FLB 16S rRNA gene copies was also observed in the qPCR analysis. However, a marked rise in the copy number for PAB communities was observed in ZH (6.54 × 108 copies ml−1) as compared to the other two sites (1.74 × 108 copies ml−1 and 2.73 × 108 copies ml−1).
Diversity of Bacterioplankton Communities
Following quality control steps, a total of 117,200 and 81,886 valid 16S rRNA gene sequence reads were recovered for the total resident (based on DNA) and active (based on cDNA) bacterioplankton communities, respectively. The sequences were organized between 389 and 1478 OTUs for each individual FLB and PAB libraries at 97% identity cutoff. Rarefaction curves were asymptotic for all sequence libraries (Fig. S1), indicating that the sequencing depths were sufficient to capture the species diversity of bacterioplankton communities.
Distribution of OTUs based on DNA sequencing library (Fig. 3a) indicated that 1088 OTUs were shared between the three sampling stations, while 1702 OTUs were unique to individual sites. On the basis of cDNA sequencing library, 828 OTUs were shared among the three sites and 1399 OTUs were unique (Fig. 3b). In both the libraries, HM samples yielded the most number of OTUs (both shared and unique).

Venn diagram representing distribution of OTUs based on DNA (a) and cDNA (b) sequencing libraries among Zhongda (ZD), Human (HM), and Zhuhai (ZH) stations. Number indicates the number of unique or shared OTUs among the different sites. Pie charts represent the proportion of each OTUs retrieved from either FLB or PAB fraction or determined in both the fraction
The OTUs were further classified as being retrieved from either PAB or FLB, or determined in both fractions by using pie charts. The number of unique OTUs obtained from PAB was higher than the corresponding FLB in ZD and HM (Fig. 3). In ZH, 228 unique OTUs were obtained from cDNA sequence library for either FLB or PAB communities, but more unique OTUs for FLB (172) were determined in the DNA sequence library than PAB (136). For the shared OTUs, majority of them were determined from both PAB and FLB fractions.
We calculated the α-diversity measures (Simpson, Shannon, and Chao1) on a randomized resampled dataset of 5755 reads to compare bacterial diversity among samples. For resident bacterial community, FLB and PAB of HM were more diverse than those of ZD and ZH. For active bacterioplankton, the highest α-diversity of FLB and PAB was determined in ZD followed by HM and ZH (Table S2). When the OTUs derived from each fraction were clustered based on the Bray-Curtis dissimilarities matrix, the resident bacterioplankton communities formed three separate groups based on sampling stations (Fig. 4a). However, the same analysis formed four groups for the active communities (Fig. 4b).

UPGMA dendrogram generated from the Bray-Curtis dissimilarities matrix of the total OTUs. a, b OTUs from DNA and cDNA sequence libraries, respectively
Relationship Between Active and Resident Bacterioplankton in the Water Samples, and in FLB and PAB Fractions
The relative frequency of each unique OTU in DNA and cDNA sequence libraries was used as an index to understand the relationships between resident and active bacterioplankton in the total bacterial communities and their FLB and PAB fractions (Fig. 5). Kendall’s nonparametric test revealed no correlation between the composition of active and resident communities (OTU distribution) in the total bacterial communities (τ = − 0.05, p < 0.01, n = 7251), or in the total FLB communities (τ = − 0.25, p < 0.01, n = 3823). However, a positive correlation was identified between active and resident communities in PAB fraction (τ = 0.18, p < 0.01, n = 3428). Both the abundant and rare bacterial groups contributed towards the relative abundance of active microbial community in PAB fraction (abundant τ = 0.32, p < 0.01, n = 160; rare τ = 0.10, p < 0.01, n = 3267). In contrast, active communities of the FLB fraction comprised mostly of the abundant bacterial group (abundant τ = 0.11, p < 0.01, n = 164; rare τ = − 0.32, p < 0.01, n = 3659).

Relative frequency of each unique OTU in DNA and cDNA sequence libraries. Each dot represents paired frequencies for each individual OTU in total (a–c), FLB (d–f), and PAB (g–i) fractions. Color of the dot indicates the sampling station from where the OTUs were derived
Composition of Bacterioplankton Community
Taxonomical classification of the OTUs based on the Ribosomal Database Project (RDP) classifier determined that PRE harbored a diverse bacterioplankton community (Fig. 6). These included taxa attributed to the phyla Proteobacteria, Bacteroidetes, Firmicutes, Chloroflexi, Cyanobacteria, Actinobacteria, Plantomycetes, and Acidobacteria (Fig. 6). At the phylum/class level, resident communities in HM station were predominantly represented by classes Betaproteobacteria and Sphingobacteria, while classes Bacteroidia (57.3%) and Clostridia (23.3%) formed the dominant active communities. At ZH sampling site, classes Flavobacteriia and Gammaproteobacteria occupied 84.9% of resident bacterioplankton communities, while active communities were dominated by phylum Cyanobacteria (40.1%) and class Betaproteobacteria (14.7%). Resident communities in ZD station were dominated by classes Betaproteobacteria (27.2%) and Actinobacteria (24.4%), followed by Sphingobacteria (7.2%), Alphaproteobacteria (4.5%), and Gammaproteobacteria (3.2%). These groups were observed as dominant active communities as well. Considering the PAB fraction, the compositions of resident and active communities at each site were relatively similar. At ZD station, Betaproteobacteria was one of the dominant taxa in both resident (23.8%) and active (24.9%) communities. In HM station, Cyanobacteria was the main dominant group in active community, and Actinobacteria in resident community. Betaproteabacteria was also present in relatively high proportion in both the communities. Classes Flavobacteriia and Gammaproteobacteria were the main dominant groups in ZH station.

Dominant bacterioplankton composition (relative abundance of sequences ≥ 2%) at phylum/class level for each sample used in the study
Heat map with accompanying clustering analysis of major bacterial families (relative abundance > 2% of total sequences) (Fig. S2) showed dominant FLB structures among samples. The resident FLB communities of ZD and HM were predominantly occupied by members of the families Comamonadaceae and Sporichthyaceae. The resident communities of the two sites, however, differed in the number of OTUs attributed to families Cytophagaceae, Chitiniophagaceae, and LD12 freshwater group. ZH samples were dominated by OTUs of families Moraxellaceae and Flavobacteriaceae. For the active bacterioplankton, composition of the dominant FLB communities in ZH and ZD was mostly similar and comprised of OTUs attributed to family Comamonadaceae and Family I Cyanobacteria. The active FLB taxa of HM was, however, dominantly represented by OTUs of family Rikenellaceae, and a lower proportion of OTUs for families Porphyromonadaceae, Bacteroidales_UCH-001, Syntrophomonadaceae, and Ruminococcaceae.
At family hierarchy level, the dominant PAB communities were comparatively more similar among samples than FLB (Fig. S2). Family I Cyanobacteria was detected as one of the dominant active taxa in PAB communities in all the three sites. The active families in the three sites, however, differ in the proportion of OTUs of families Moraxellaceae and Comamonadaceae. For the resident communities, families Comamonadaceae and Sporichthyaceae constituted the dominant PAB taxa in ZD and HM samples, while that of ZH was dominated by OTUs of families Moraxellaceae and Flavobacteriaceae.
Potential Activities and Distribution of Active Bacterioplankton Communities
Potential activity level of each classified taxon was established from the ratio (R) of relative abundance of all OTUs in cDNA and DNA sequence libraries, and was interpreted as either high (R > 20), medium-high (5 < R ≤ 20), medium (1 < R ≤ 5), or low (0 < R < 1). OTUs which were determined only in cDNA sequence library but not on the DNA sequence library were labeled as “only found in cDNA.” It was determined that majority of the OTUs in FLB fractions of HM and ZH samples belonged to the “only found in cDNA” category (Fig. 7). Few FLB OTUs from ZH showed ratio above 1 [high (22 OTUs), medium-high (42), medium (54), and low (63)]. Comparatively, OTUs from ZD showed different levels of potential activities (high, 7 OTUs; medium-high, 40; medium, 161; low, 150). For the PAB fractions, maximum OTUs showed either medium or low level of activities, or belonged to “only found in cDNA” category (Fig. 7).

Level of potential activities of the bacterioplankton communities in the three sampling stations
Analysis of the relative abundance of the major active taxa in all the sites indicated that they were widely distributed, but the level of activities was not necessarily dependent on the relative abundance of the sequences (Fig. 8, Table S3). For example, OTU175 (family Synergistaceae) were retrieved from FLB fraction of ZH and PAB fraction of ZD in high abundance, but the highest level of potential activity (R = 178.0) was exhibited by the OTUs determined from ZH fraction only. Similarly, OTU187 (family Burkholderiaceae) from the FLB fraction of HM exhibited high level of potential activity (R = 113.7), while its sequences from other fractions showed lower level of potential activities. The phylum Cyanobacteria was another highly active taxon, and its OTUs were determined from two or more of the samples. OTU37, affiliated with cyanobacteria Microcystis, was obtained from all the three sampling sites, and that of OTU11168, Family I Cyanobacteria, from HM and ZH stations (Fig. 8). Another active taxon OTU10 (affiliated with the marine cyanobacteria Synechococcus) was obtained from all samples with high abundance, but the highest level of activity was determined only from the ZH station.

Dot plot indicating distribution of bacterioplankton with high level of potential activities. Size of the dot represents the relative abundance of each OTU. Taxonomic affiliations (up to classified hierarchy level) for each OTU are indicated on the right side of the plot
Discussion
It is vital to understand the community structure to determine the dynamics of bacterioplankton communities in Pearl River. Despite a high taxonomic diversity, assessment of their distribution pattern indicated that bacterioplankton in Pearl River ecosystem are preferentially particle-associated (Fig. 3). Large distribution of PAB in aquatic environment is generally associated with high concentration of particulate carbon, since attachment to particles is the first step towards utilization of organic matter [35, 36]. But, interestingly, PAB’s exclusive OTUs in PRE were more dominant in sampling stations with lower POM content (ZD and HM), in contrast to findings in earlier studies at coastal areas around Mackenzie River [8, 37, 38]. This result may probably be related with a poor quality of particulate material, as also observed along Southern California coast [39]. More importantly, a high dispersal potential of bacterioplankton communities may essentially be a part of ecology of PRE [8, 40]. This is evident from the large overlapping OTUs between two or more sampling stations (Fig. 3). Under such condition, the bacterioplankton communities do not exclusively occur as either free-living or particle-associated but are conjunct between the two; since the physicochemical conditions of PRE do not remain static during seasonal variation [24, 26], the co-occurrence of bacterioplankton as both free-living and particle-associated may potentially help them to bear and utilize the fluctuant nutrients influx [38, 41].
During our study, we determined the relationships between the resident and active bacterioplankton within the free-living and particle-associated communities to assess the contribution of the different bacterioplankton communities in the ecology of PRE (Fig. 5). In the Pearl River system, we observed a major inconformity in the abundant taxa between active and resident communities in the FLB fraction, but less in the PAB fraction. For instance, the predominant active communities in FLB fraction of HM comprised of unclassified bacteria of the classes Bacteroidia and Clostridia, which was unique from either the resident communities in FLB fraction of HM or any of the other remaining sites (Fig. 6). It is also evident from our study that active bacterioplankton communities in PRE were preferentially found to be associated with the PAB fraction (Fig. 5). It may be due to the fact that particles (or substrates) provide a stable microenvironment in the fluctuant and dynamic ecosystem, whereby particle-associated communities can attach and utilize these substrates [8, 37]. This is particularly true for members of the phylum Cyanobacteria who exist in the photic zones of PRE, particularly in ZD and ZH station (Fig. 8), utilizing the organic nutrients from the substrate. They have the added advantages of establishing symbiotic relationship with different groups of bacteria [42, 43], though a deep investigation will be necessary to understand the biotic interaction between Cyanobacteria and other groups of bacteria in PRE.
Physicochemical parameters may act as a major driver for the distribution of the bacterioplankton communities in PRE (Fig. 2). For instance, OTUs of class Betaproteobacteria were predominantly detected in stations with less salinity (ZD and HM), while that of Gammaproteobacteria in station located closer to the South-China Sea (ZH), and having a salinity concentration much higher than the other two stations (Table S1). This finding is in congruence with the review on the distribution of Proteobacteria by Nold and Zwart [44] whereby members of the classes Alphaproteobacteria and Gammaproteobacteria were found to be more dominant in saline environments (seawater), and that of Betaproteobacteria in freshwater. Similarly, the distribution of phylum Cyanobacteria has often been related with the influx in dissolved organic nutrients [20, 23]. In our case, the abundance of OTUs affiliated with the phylum Cyanobacteria at all the sites may be related with high content of dissolved nitrogen and phosphorus compounds (Table S1). While cultures belonging to family Sporichthayaceae (class Actinobacteria) have been found to be obligate anaerobes of soil [45], detection of OTUs related to this family in the upstream station ZD may probably be related with low DO content. The oxygen-rich condition of ZH station, supplemented by high concentration of POM and abundant presence of Chl a, seems to support the chemoorganotrophic bacteria, particularly member of the class Flavobacteria (family Flavobacteriaceae) [46,47,48]. The physiological implication for detection of a large number of sequences affiliated with family Moraxellaceae (class Gammaproteobacteria) in ZH, however, requires further investigation, as members of this family, excepting the genus Psychrobacter, have not been reported from any aquatic environments [49].
In the present study, a major portion of the OTUs was detected to be exclusive to the cDNA sequence library (Fig. 7). This data could mean that a significant and functionally relevant portion of the microbial communities is missed by our DNA sequencing approaches [50]. This inference is further illustrated by the fact that the relative abundance of OTUs with high level of potential activity was generally very low (Table S3). In other words, estuarine bacterioplankton with low abundance can be highly active.
References
Mao Q, Shi P, Yin K, Gan J, Qi Y (2004) Tides and tidal currents in the Pearl River Estuary. Cont Shelf Res 24:1797–1808. https://doi.org/10.1016/j.csr.2004.06.008
Fontanez KM, Eppley JM, Samo TJ, Karl DM, DeLong EF (2015) Microbial community structure and function on sinking particles in the North Pacific Subtropical Gyre. Front Microbiol 6:469. https://doi.org/10.3389/fmicb.2015.00469
Leorri E, Cearreta A, Irabien MJ, Yusta I (2008) Geochemical and microfaunal proxies to assess environmental quality conditions during the recovery process of a heavily polluted estuary: the Bilbao estuary case (N. Spain). Sci Total Environ 396:12–27. https://doi.org/10.1016/j.scitotenv.2008.02.009
Zhang L, Wang L, Yin K, Lü Y, Zhang D, Yang Y, Huang X (2013) Pore water nutrient characteristics and the fluxes across the sediment in the Pearl River estuary and adjacent waters, China. Estuar Coast Shelf Sci 133:182–192. https://doi.org/10.1016/j.ecss.2013.08.028
Reed HE, Martiny JBH (2012) Microbial composition affects the functioning of estuarine sediments. ISME J 7:868–879. https://doi.org/10.1038/ismej.2012.154
Bernhard AE, Tucker J, Giblin AE, Stahl DA (2007) Functionally distinct communities of ammonia-oxidizing bacteria along an estuarine salinity gradient. Environ. Microbiol. 9:1439–1447. https://doi.org/10.1111/j.1462-2920.2007.01260.x
Hawkins RJ, Purdy KJ (2007) Genotypic distribution of an indigenous model microorganism along an estuarine gradient. FEMS Microbiol. Ecol. 62:187–194. https://doi.org/10.1111/j.1574-6941.2007.00376.x
Garneau MÈ, Vincent WF, Terrado R, Lovejoy C (2009) Importance of particle-associated bacterial heterotrophy in a coastal Arctic ecosystem. J. Mar. Syst. 75:185–197. https://doi.org/10.1016/j.jmarsys.2008.09.002
Eloe EA, Shulse CN, Fadrosh DW, Williamson SJ, Allen EE, Bartlett DH (2011) Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment. Environ. Microbiol. Rep. 3:449–458. https://doi.org/10.1111/j.1758-2229.2010.00223.x
Ortega-Retuerta E, Joux F, Jeffrey WH, Ghiglione JF (2013) Spatial variability of particle-attached and free-living bacterial diversity in surface waters from the Mackenzie River to the Beaufort Sea (Canadian Arctic). Biogeosciences 10:2747–2759. https://doi.org/10.5194/bg-10-2747-2013
D'Ambrosio L, Ziervogel K, MacGregor B, Teske A, Arnosti C (2014) Composition and enzymatic function of particle-associated and free-living bacteria: a coastal/offshore comparison. ISME J 8:2167–2179. https://doi.org/10.1038/ismej.2014.67
Rieck A, Herlemann DPR, Jürgens K, Grossart HP (2015) Particle-associated differ from free-living bacteria in surface waters of the Baltic Sea. Front Microbiol 6:1297. https://doi.org/10.3389/fmicb.2015.01297
Crump BC, Hopkinson CS, Sogin ML, Hobbie JE (2004) Microbial biogeography along an estuarine salinity gradient: combined influences of bacterial growth and residence time. Appl Environ Microbiol 70:1494–1505. https://doi.org/10.1128/aem.70.3.1494-1505.2004
Zhang Y, Zhao Z, Dai M, Jiao N, Herndl GJ (2014) Drivers shaping the diversity and biogeography of total and active bacterial communities in the South China Sea. Mol Ecol 23:2260–2274. https://doi.org/10.1111/mec.12739
Cardoso DC, Sandionigi A, Cretoiu MS, Casiraghi M, Stal L, Bolhuis H (2017) Comparison of the active and resident community of a coastal microbial mat. Sci Rep 7:2969. https://doi.org/10.1038/s41598-017-03095-z
Gaidos E, Rusch A, Ilardo M (2011) Ribosomal tag pyrosequencing of DNA and RNA from benthic coral reef microbiota: community spatial structure, rare members and nitrogen-cycling guilds. Environ. Microbiol. 13:1138–1152. https://doi.org/10.1111/j.1462-2920.2010.02392.x
Campbell BJ, Yu L, Heidelberg JF, Kirchman DL (2011) Activity of abundant and rare bacteria in a coastal ocean. Proc Natl Acad Sci U S A. 108:12776–12781. https://doi.org/10.1073/pnas.1101405108
Campbell BJ, Kirchman DL (2013) Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J 7:210–220. https://doi.org/10.1038/ismej.2012.93
Yang Y, Chen F, Zhang L, Liu J, Wu S, Kang M (2012) Comprehensive assessment of heavy metal contamination in sediment of the Pearl River Estuary and adjacent shelf. Mar Pollut Bull 64:1947–1955. https://doi.org/10.1016/j.marpolbul.2012.04.024
Dai M, Zhai W, Cai W-J, Callahan J, Huang B, Shang S, Huang T, Li X, Lu Z, Chen W, Chen Z (2008) Effects of an estuarine plume-associated bloom on the carbonate system in the lower reaches of the Pearl River estuary and the coastal zone of the northern South China Sea. Cont Shelf Res 28:1416–1423. https://doi.org/10.1016/j.csr.2007.04.018
Harrison PJ, Yin K, Lee JHW, Gan J, Liu H (2008) Physical–biological coupling in the Pearl River Estuary. Cont Shelf Res 28:1405–1415. https://doi.org/10.1016/j.csr.2007.02.011
Strokal M, Kroeze C, Li L, Luan S, Wang H, Yang S, Zhang Y (2015) Increasing dissolved nitrogen and phosphorus export by the Pearl River (Zhujiang): a modeling approach at the sub-basin scale to assess effective nutrient management. Biogeochemistry 125:221–242. https://doi.org/10.1007/s10533-015-0124-1
Liu X, Lu X, Chen Y (2011) The effects of temperature and nutrient ratios on Microcystis blooms in Lake Taihu, China: an 11-year investigation. Harmful Algae 10:337–343. https://doi.org/10.1016/j.hal.2010.12.002
Cao JJ, Lee SC, Ho KF, Zou SC, Fung K, Li Y, Watson JG, Chow JC (2004) Spatial and seasonal variations of atmospheric organic carbon and elemental carbon in Pearl River Delta Region, China. Atmos Environ 38:4447–4456. https://doi.org/10.1016/j.atmosenv.2004.05.016
Huang S, He S, Xu H, Wu P, Jiang R, Zhu F, Luan T, Ouyang G (2015) Monitoring of persistent organic pollutants in seawater of the Pearl River Estuary with rapid on-site active SPME sampling technique. Environ Pollut 200:149–158. https://doi.org/10.1016/j.envpol.2015.02.016
Cai WJ, Dai M, Wang Y, Zhai W, Huang T, Chen S, Zhang F, Chen Z, Wang Z (2004) The biogeochemistry of inorganic carbon and nutrients in the Pearl River estuary and the adjacent Northern South China Sea. Cont Shelf Res 24:1301–1319. https://doi.org/10.1016/j.csr.2004.04.005
Lu X, Sun S, Zhang YQ, Hollibaugh JT, Mou X (2015) Temporal and vertical distributions of bacterioplankton at the Gray’s Reef National Marine Sanctuary. Appl Environ Microbiol 81:910–917. https://doi.org/10.1128/aem.02802-14
Lebaron P, Servais P, Agogué H, Courties C, Joux F (2001) Does the high nucleic acid content of individual bacterial cells allow us to discriminate between active cells and inactive cells in aquatic systems? Appl Environ Microbiol 67:1775–1782. https://doi.org/10.1128/aem.67.4.1775-1782.2001
Kumar PS, Brooker MR, Dowd SE, Camerlengo T (2011) Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. PLoS One 6:e20956. https://doi.org/10.1371/journal.pone.0020956
Fierer N, Lauber CL, Ramirez KS, Zaneveld J, Bradford MA, Knight R (2012) Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J 6:1007–1017. https://doi.org/10.1038/ismej.2011.159
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. https://doi.org/10.1128/aem.01541-09
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145. https://doi.org/10.1093/nar/gkn879
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peňa AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Hulsen T, de Vlieg J, Alkema W (2008) BioVenn—a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9:488. https://doi.org/10.1186/1471-2164-9-488
Suzuki S, Kaneko R, Kodama T, Hashihama F, Suwa S, Tanita I, Furuya K, Hamasaki K (2017) Comparison of community structures between particle-associated and free-living prokaryotes in tropical and subtropical Pacific Ocean surface waters. J Oceanogr 73:383–395. https://doi.org/10.1007/s10872-016-0410-0
Crespo BG, Pommier T, Fernández-Gómez B, Pedrós-Alió C (2013) Taxonomic composition of the particle-attached and free-living bacterial assemblages in the Northwest Mediterranean Sea analyzed by pyrosequencing of the 16S rRNA. Microbiologyopen 2:541–552. https://doi.org/10.1002/mbo3.92
Simon M, Grossart H-P, Schweitzer B, Ploug H (2002) Microbial ecology of organic aggregates in aquatic ecosystems. Aquat Microb Ecol 28:175–211. https://doi.org/10.3354/ame028175
Garneau MÈ, Vincent WF, Alonso-Sáez L, Gratton Y, Lovejoy C (2006) Prokaryotic community structure and heterotrophic production in a river-influenced coastal arctic ecosystem. Aquat Microb Ecol 42:27–40. https://doi.org/10.3354/ame042027
Fandino LB, Riemann L, Steward GF, Long RA, Azam F (2001) Variations in bacterial community structure during a dinoflagellate bloom analyzed by DGGE and 16S rDNA sequencing. Aquat Microb Ecol 23:119–130. https://doi.org/10.3354/ame023119
Yung CM, Ward CS, Davis KM, Johnson ZI, Hunt DE (2016) Insensitivity of diverse and temporally variable particle-associated microbial communities to bulk seawater environmental parameters. Appl Environ Microbiol 82:3431–3437. https://doi.org/10.1128/aem.00395-16
Hamdan LJ, Jonas RB (2006) Seasonal and interannual dynamics of free-living bacterioplankton and microbially labile organic carbon along the salinity gradient of the Potomac River. Estuar Coast 29:40–53. https://doi.org/10.1007/bf02784697
Parveen B, Ravet V, Djediat C, Mary I, Quiblier C, Debroas D, Humbert J-F (2013) Bacterial communities associated with Microcystis colonies differ from free-living communities living in the same ecosystem. Environ Microbiol Rep 5:716–724. https://doi.org/10.1111/1758-2229.12071
Zhang BH, Salam N, Cheng J, Li H-Q, Yang J-Y, Zha D-M, Zhang Y-Q, Ai M-J, Hozzein WN, Li W-J (2016) Modestobacter lacusdianchii sp. nov., a phosphate-solubilizing actinobacterium with ability to promote Microcystis growth. PLoS One 11:e0161069. https://doi.org/10.1371/journal.pone.0161069
Nold SC, Zwart G (1998) Patterns and governing forces in aquatic microbial communities. Aquat Ecol 32:17–35. https://doi.org/10.1023/a:1009991918036
Tamura T (2014) The family Sporichthyaceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes: Actinobacteria. Springer, Berlin, pp 883–888. https://doi.org/10.1007/978-3-642-30138-4_182
DeLong EF, Franks DG, Alldredge AL (1993) Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol Oceanogr 38:924–934. https://doi.org/10.4319/lo.1993.38.5.0924
Kirchman DL (2002) The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol Ecol 39:91–100. https://doi.org/10.1111/j.1574-6941.2002.tb00910.x
Gomez-Pereira PR, Fuchs BM, Alonso C, Oliver MJ, van Beusekom JEE, Amann R (2010) Distinct flavobacterial communities in contrasting water masses of the North Atlantic Ocean. ISME J 4:472–487. https://doi.org/10.1038/ismej.2009.142
Teixeira LM, Merquior VLC (2014) The family Moraxellaceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes: Gammaproteobacteria. Springer, Berlin, pp 443–476. https://doi.org/10.1007/978-3-642-38922-1_245
Baldrian P, Kolařík M, Štursová M, Kopecký J, Valášková V, Větrovský T, Žifčáková L, Šnajdr J, Rídl J, Vlček Č, Voříšková J (2011) Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J 6:248–258. https://doi.org/10.1038/ismej.2011.95
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 31528001) and Natural Science Foundation of Guangdong Province, China (No. 2016A030312003). W-J Li was also supported by Guangdong Province Higher Vocational Colleges & Schools Pearl River Scholar Funded Scheme (2014).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary materials
ESM 1
(DOCX 332 kb)
Rights and permissions
About this article

Cite this article
Li, JL., Salam, N., Wang, PD. et al. Discordance Between Resident and Active Bacterioplankton in Free-Living and Particle-Associated Communities in Estuary Ecosystem. Microb Ecol 76, 637–647 (2018). https://doi.org/10.1007/s00248-018-1174-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-018-1174-4