
Abstract
Blood feeding red poultry mites (RPM) serve as vectors of pathogenic bacteria and viruses among vertebrate hosts including wild birds, poultry hens, mammals, and humans. The microbiome of RPM has not yet been studied by high-throughput sequencing. RPM eggs, larvae, and engorged adult/nymph samples obtained in four poultry houses in Czechia were used for microbiome analyses by Illumina amplicon sequencing of the 16S ribosomal RNA (rRNA) gene V4 region. A laboratory RPM population was used as positive control for transcriptome analysis by pyrosequencing with identification of sequences originating from bacteria. The samples of engorged adult/nymph stages had 100-fold more copies of 16S rRNA gene copies than the samples of eggs and larvae. The microbiome composition showed differences among the four poultry houses and among observed developmental stadia. In the adults’ microbiome 10 OTUs comprised 90 to 99% of all sequences. Bartonella-like bacteria covered between 30 and 70% of sequences in RPM microbiome and 25% bacterial sequences in transcriptome. The phylogenetic analyses of 16S rRNA gene sequences revealed two distinct groups of Bartonella-like bacteria forming sister groups: (i) symbionts of ants; (ii) Bartonella genus. Cardinium, Wolbachia, and Rickettsiella sp. were found in the microbiomes of all tested stadia, while Spiroplasma eriocheiris and Wolbachia were identified in the laboratory RPM transcriptome. The microbiomes from eggs, larvae, and engorged adults/nymphs differed. Bartonella-like symbionts were found in all stadia and sampling sites. Bartonella-like bacteria was the most diversified group within the RPM microbiome. The presence of identified putative pathogenic bacteria is relevant with respect to human and animal health issues while the identification of symbiontic bacteria can lead to new control methods targeting them to destabilize the arthropod host.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The red poultry mite (RPM), Dermanyssus gallinae (De Geer, 1778, Acari: Mesostigmata), is a hematophagous ectoparasite preferring bird hosts [1], but it can attack also mammals and humans [2, 3]. In humans, the RPM causes the bites, non-specific dermatitis connected with itching, and pseudo-scabies [2, 3]. In poultry production, severe RPM infestations are associated with anemia, reduced growth, and egg production losses, or even mortality increase of poultry birds [1, 4]. The RPMs transmit different bacterial and viral pathogens; however, the mode of transmissions is not yet understood [4,5,6,7]. RPMs can act either as a passive vector of the bacteria, (i.e., bacteria did not multiply inside the mites) or as a true vector. The most important pathogenic bacteria isolated from RPM include the following taxa: Pasteurella multocida, Erysipelothrix rhusiopathiae, Salmonella spp., Listeria monocytogenes, Coxiella burnetii and Spirochetes, and there is suspicion of the possible transmission of Borrelia anserine (as reviewed by Moro et al. [5], Sparagano et al. [4], Pritchard et al. [1]). Aerococcus (Aerococcus viridans and Aerococcus urinaequi) as the human pathogen [8] and P. multocida and E. rhusiopathiae sequences were identified in RPMs from poultry farms in France [7]. Furthermore, Mycobacterium sp. has been detected by reverse-transcriptome PCR (RT-PCR) in the RPMs [9]. Mycoplasma synoviae and Mycoplasma gallisepticum were in RPMs from Japanese poultry farms, but the mites used in that study were not surface cleaned [10]. In canary birds, RPMs are a reservoir of Chlamydia psittaci [11]. Apart from pathogenic bacteria, endosymbionts represent important group of bacteria associated to RPMs. Beside pathogenic bacteria, another important group of bacteria in RPMs are endosymbionts. To date, the symbionts identified in RPMs include Cardinium sp., Spiroplasma sp., Rickettsiella sp., Anistosticta sp., and Schineria sp. [5, 12]. Although RPM is of bacterial transmission importance, a study describing the D. gallinae microbiome is lacking.
Recent advance in technology, high-throughput sequencing (HTS) on Illumina platforms provide an opportunity to study microbiomes from a complex view [13, 14]. Though the previous studies showed differentiated bacterial community in RPM, their weaknesses was the lack of the quantitative or semi-quantitative advantages of real-time PCR analyses and HTS. In this study, we focused on the following goals: (i) to characterize the bacterial community associated to four farm populations of RPM using 16S ribosomal RNA (rRNA) gene metabarcoding, (ii) to identify and quantify specific pathogenic bacteria associated to RPM, (iii) and to ascertain the mite symbiotic bacterial community, and (iv) to compare the microbiome of different developmental stadia. Furthermore, we compared the data to putative 454 pyrosequencing-based bacterial transcriptome data set of the German laboratory RPM population. We provide the first comprehensive analysis of the RPM microbiome.
Materials and Methods
Origin of Mites and Collection
Four sampling sites at industrial poultry farms located in Czechia were included to the study: Ustrasice (49° 20′ 19″ N, 14° 41′ 11″ E), RPMs collected in June 2015; Bantice (48° 52′ 47″ N, 16° 10′ 40″ E), RPMs collected in July 2015; Mirovice near Pisek (49° 30′ 55.99″ N, 14° 2′ 8.95″ E), RPMs collected in February 2016 and Pohorelice (48° 58′ N, 16° 31′ E) collected in February 2016. The mites were collected manually (Ustrasice) or using Tube trap [15] (Bantice, Mirovice and Pohorelice). The traps with collected mites were moved to the laboratory and immediately processed. The mites were sampling as (i) engorged adults/nymphs, (ii) larvae, and (iii) eggs. The reason for mite separation was that neither eggs nor larvae contain any sucked blood, which is suggested as one of the source of bacteria. The samples were pooled from the traps for every site, i.e., each sample contained between 50 and 100 individuals or eggs accurately counted and placed into sterile 1.5-mL Eppendorf tubes. The list of 36 samples and sampled conditions are provided in Supplementary Table S1. The mites for analysis of 454 pyrosequencing transcriptome sequences originated from laboratory population maintained at the Institute for Parasitology of the University of Veterinary Medicine Hannover, Germany [16].
DNA Extraction
The mites or eggs in the samples were surface cleaned and homogenized following the protocol described previously [17]. DNA was extracted from homogenates using tissue Genomic DNA Mini Kit (Cat No. GT100, Geneaid, New Taipei City, Taiwan) following the manufacturer’s protocol. The extracted DNA was stored at −28 °C until analysis. The same samples were used for all analyses with the exception of transcriptome.
PCR
To validate the prepared templates, PCR amplification of the 16S rRNA gene was performed with universal bacterial primers [18] on all the samples used for analyses. To avoid contamination, the master mix was made with ddH2O instead of template DNA, which was used as negative control; the positive control included DNA of Escherichia coli. The amplification and cloning were done according to protocol described previously [17]. Further, the PCR amplicons were pooled per stadium and sites and cloned according to the previously described protocol [19]. The selected clones were sequenced (Sanger sequencing) by Macrogen (http://www.macrogen.com, Seoul, Korea). Sequences were assembled in CodonCode Aligner 5.1.5 (CodonCode Corporation, Victoria Centerville, MA, USA). Chimeric sequences were identified using Mallard and Pintail software [20, 21]. After this step, we obtained 131 sequences that were further processed in UPARSE-USEARCH pipeline to operational taxonomic units (OTUs) at 97% of similarity [22]. The representative sequences were assigned to bacterial taxa using the Ribosomal database project (RDP) naive Bayesian rRNA classifier [23] and compared with GenBank sequences using nucleotide BLAST [24]. The taxon-specific primers were used to verify the presence of selected taxa in the samples (Supplementary Table S2), the PCR products of correct size were randomly cloned and sequenced to verify primers specificity. The obtained Bartonella-like and Rickettsiella-like sequnces of 16S rRNA gene were deposited in GenBank - Access Numbers: MF086616-MF086655.
Phylogenetic Analyses
Partial 16S rRNA gene sequences were assembled with CodonCode Aligner, version 1.5.2 (CodonCode Corporation, Dedham, MA, USA) and assigned to bacterial taxonomy using the Ribosomal database project naive Bayesian classifier [23]. The sequences were aligned using SILVA Incremental Aligner v.1.2.11 [25]. The best-fit model of nucleotide substitution was selected using jModelTest v.2.1.7 [26, 27] based on the AIC criterion. General time-reversible models were suggested for Bartonella-like (GTR + I + G, proportion of invariable sites = 0.337, gamma shape = 0.393), and for Rickettsiella sequence analysis (GTR + G, gamma shape = 0.342). The phylogenies were inferred through Bayesian analysis with PhyloBayes-MPI v.1.4e [28,29,30] and maximum-likelihood analysis in PhyML v.3.0 [31]. The phylograms were finalized using Figtree v.1.4.2 (http://tree.bio.ed.ac.uk/; [32]).
Illumina Amplicon Analyses
The identification of bacteria by Illumina amplicon sequencing was based on 16S rRNA using CS1_515F and CS2_806R primers targeting the V4 of 16S rRNA gene [33] and previously described conditions [34]. The master mix with ddH2O was used as negative control. The products were sequenced at the DNA Services Facility, Research Resources Center, University of Illinois (Chicago, IL, USA) on MiSeq platform (Illumina, San Diego, CA, USA) [35]. The forward and reverse sequences (2 × 250 bp) were aligned and processes using UPARSE – USEARCH Pipeline according the standard operation procedure [22]. The operational taxonomic units OTUs were described according to the Ribosomal database project, training set no. 15 [36]. Then, the representative sequences for each OTU were compared to those in GenBank using the BLASTn [24]. Raw sequences of 16S rRNA gene are accessible through SRA study accession number SRP095486.
The shared file and standardization was done in MOTHUR v.1.36.1 software [37] according to the MiSeq standard operation procedure (MiSeq SOP [38]. The data were standardized by subsampling to 22,615 sequences and all further analyses were done with standardized data set in MOTHUR and PAST 3.06 software [39]. The taxonomical features of the samples were visualized by KRONA projection [40]. The inverse Simpson diversity index was calculated in MOTHUR. The tested variables described the effect of site and stadium population on OTUs distribution. For the analyses of microbiome, we applied protocol for analyzes of Tyrophagus putrescentiae microbiome [41]. The tested variables included the stadium of RPM and sampled sites. At first, the effect of the variables and their interaction was tested by two-way permutational multivariate analyses of variance (PERMANOVA) using the Bray–Curtis and Jaccard distance matrices in 1000 permutations in PAST, followed by testing the factors individually in one-way PERMANOVA. Redundancy Analysis (RDA) models based on the same matrices as described above were constructed using the “vegan” R package [42]. To visualize the coordinates resulting from the best dbRDA models, triplots were created. A heatmap depicting OTU abundance and clustering in dendrograms was produced using the “gplots” R package [43]. The OTU abundance data were logarithmically transformed as suggested by Anderson et al. [44]. We prefer the LOG2 transformation, which permits better visualization of less abundant species. Venn diagrams were used to highlight shared OTUs among eggs/larvae/engorged nymphs and adults or among the poultry houses. We included only those OTUs to the analyses, which were present in all three replicates per treatment. The METASTATS analyze [45] was applied to describe the differences in the microbiome of adults, eggs, and juveniles and among the adults from different samples sites. It was calculated in MOTHUR using 100,000 permutations.
Quantitative PCR
The universal primers were used to quantify the numbers of 16S rRNA copies of bacteria in the samples. To avoid the influence of chloroplast DNA to analyzes, we selected Com1 and 769R primers for quantification of total bacteria copies [46]. The specific primers for Cardinium and Wolbachia were used (Supplementary Table S2). Amplifications were conducted using a StepOnePlus™ Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). SYBR green (Bio-Rad Laboratories, Hercules, CA, USA) was used as a double-stranded DNA (dsDNA) binding dye. Baseline and threshold calculations were performed with the StepOnePlus software version 2.3. (Thermo Fisher Scientific). The amplification consisted of 40 cycles, including a denaturation of 30 s at 95 °C, annealing for 35 s at 54–60 °C (Supplementary Table S2), and elongation for 45 s at 72 °C. Melting curves were recorded to ensure qPCR specificity. The design included three samples per stadium and poultry house and two technical replicates. The qPCR standard was prepared as was described previously [47]. DNA samples were diluted by 1/10. The resulting data were standardized by recalculation per one mite or egg. The data were log10-transformed and analyzed by the Kruskal–Wallis nonparametric test, using Dunn potshot comparison and a Bonferroni correction using XLSTAT 2015.
Analysis of D. gallinae Transcriptome Sequences-Based 454 Pyrosequencing
Dermanyssus gallinae transcriptome sequences [16] obtained from egg, juvenile as well as adult mite stages comprised 232,097 singleton reads, 35,788 isotigs, and 56 contigs. As analyses in Schicht et al. [16] were conducted in 2013, all sequences were reanalyzed and blasted again by Blast2GO software suite [48] using the nr-data base of the NCBI to discover bacteria sequences in the transcriptome data set. BLASTn top hits species were verified for their bacterial origin by using uniprot (http://www.uniprot.org/) taxonomy search.
Results
Identification of Bacteria by Conventional PCR and Sanger Sequencing
Altogether 131 cloned sequences of 16S rRNA gene by universal primers formed 26 OTUs (see the Supplementary Table S3) in the DNA samples of eggs, larvae, or adults/nymphs of RPM. The following OTUs were identified: The Actinobacteria sequences (S10) showing 99% of similarity Tsukamurella paurometabola. Rhizobiales sequences formed five OTUs with 94–98% similarity to Bartonella (S5, S13, S15, S31, and S32). The Ricketsiella-like sequences formed OTU of 94% similarity to Diplorickettsia massiliensis (S24). Further, two OTUs (2 and 3) contained sequences of 93% similarity to Wolbachia and two Sphingobacteriales sequences formed OTU (16) with 97% similarity to Candidatus Cardinium GenBank Access. No. KC677579 from RPM [49]. Although Cardinium (OTU16); Wolbachia (OTUs 2 and 3); and Bartonella sequences (OTUs 5, 13, and 15) were detected among the clones from larvae, but not in the eggs, their presence was confirmed by taxa specific primers (Table S1).
Phylogenetic Analyses of Bartonella-like and Rickettsiella-like Sequences
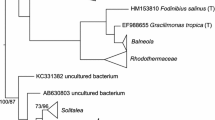
The phylogenetic analysis of 31 Bartonella-like sequences obtained from RPM using universal primers revealed two separate clusters (Fig. 1). The both clusters were located between Bartonella tamiae and the rest of Bartonella altogether with the cloned sequences from stinkbugs [50], ants [51,52,53,54,55], bees [56] including Bartonella apis [57], Varroa mites [17], astigmatid stored product mites [58,59,60], and the sequences obtained from human skin [61]. The first group (A) of 14 clones from RPM formed sister groups to Bartonella symbiont from the ants [51,52,53,54,55]. The second group (B, 17 clones) formed sister cluster to the whole Bartonella genus with the exception of B. tamiae.

Phylogenetic analysis of Bartonella spp. cloned from D. gallinae. Phylogeny was inferred by Bayesian analysis employing the GTR model with a proportion of invariable sites (+I) and a gamma distribution with four rate categories (+G). The initial alignment consisted of 31 nearly full-length sequences of the 16 rRNA gene from this study, and 507 reference sequences available in the RDP database. These sequences came from 55 type strains representing the families Bartonellaceae and Brucellaceae, and 452 related uncultured bacteria. Branch lengths correspond to mean posterior estimates of evolutionary distances (scale bar, 0.05). Branch labels indicate the Bayesian posterior probability and supporting bootstrap value for selected branches from maximum-likelihood analysis. The phylogram was rooted with Caulobacter mirabilis sequence AJ227774 as an outgroup
The analyses of cloned sequences of Rickettsiella sp. from universal primers indicated that the sequences were related to Rickettsiella and Diploricketssia genera (Fig. S1). Nine cloned sequences from RPM formed sister group to Candidatus Rickettsiella viridis symbiont of pea aphids (Acyrthosiphon pisum) [62, 63]. The sequences were closely related to those obtained from oribatid mite Nanorchestes [64] and bacterium from human skin [61]. For further description, we are using term Rickettsiella-like and Bartonella-like symbionts.
Microbiome Description Based on Illumina Amplicon Sequencing
Altogether 1,493,621 16S rRNA sequences of V4 region were obtained from Illumina for four tested sites in Czechia (Bantice, Mirovice, Pohorelice, and Ustrasice). The sequences formed 225 OTUs at 3% of dissimilarity level. The Krona projections were constructed for developmental stadia (Fig. S3) and sampled sites for adults/nymphs (Fig. S4). The list of identified OTUs is given in the Supplementary Table S4.
Both parameters, numbers of OTUs and inverse Simpson diversity index differed in the developmental stadia, K 2,32 = 11.488, P = 0.002 and K 2,32 = 12.182, P = 0.001, respectively. Both parameters were two times higher in the eggs and larvae microbiome samples than in adults/nymphs samples (Fig. S5).
The developmental stadium and site of sampling and their interaction significantly influenced microbiome composition as showed the Bray–Curtis and Jaccard matrices: two-way PERMANOVA (Table 1). When the factors were tested separately, both were significant (P < 0.001) in both matrices. The Bofferoni-corrected P values indicated differences among the sampling sites. The larvae microbiome did not differ from the eggs, but eggs and larvae microbiomes were significantly different from engorged nymphs/adults. The dbRDA using Bray–Curtis and Jaccard matrix (Fig. 2a, b) confirmed that both site and developmental stadia had significant effect to OTUs composition, i.e., site F = 9.55; P = 0.001 and F = 6.53; P = 0.001; stadium F = 11.04; P = 0.001 and F = 6.75; P = 0.001 for Bray–Curtis and Jaccard matrix, respectively. Based on dbRDA, the samples of engorged nymphs and adults were separated from samples of larvae and juveniles. The dbRDA analysis separated OTUs to the following groups: (i) adults associated: Bartonella-like (OTUs 2, 72, 79); Tsukamurella strandjordii (OTU 6); and Kocuria rhizophila (OTU 9); (ii) associated to Ustrasice site: Rickettsiella-like (OTU 3) and Pantoea vagans (OTU 12) and (iii) eggs and larvae associated, e.g., Wolbachia (OTUs 5, 20), Cardinium (OTU 1), Psychrobacter pulmonis (OTU 15) and Romboutsia sedimentorum (OTU 13). When we compared the situation to the Krona projections (Figs. S3 and S4) it is apparent that Wolbachia (OTUs 5, 20) and P. vagans (OTU 12) had low proportions in adults, while Cardinium (OTU 1) consisted 15% of sequences in the adult microbiome.

dbRDA of sampled site and OTUs based on Bray–Curtis (a) and Jaccard (b) dissimilarity matrix in D. gallinae microbiome. The comparison of developmental stadia and sites is based on ellipses (95%). In Bray–Curtis dbRDA, the CAP1 and CAP2 explained 43 and 37% of variance in data set. In Jaccard dbRDA, the CAP1 and CAP2 explained 39 24% of variability
When the presence/absence of OTUs was compared among developmental stadia, i.e., eggs, larvae and engorged nymphs/adults, all of them shared between 8 and 38 OTUs depending on the sampling site (Fig. S6). The highest unique OTUs numbers were in the samples of larvae, i.e., from 8 to 44 OTUs, followed by eggs, i.e., 9–13 OTUs and no unique OTU was in adult samples in all four studied sites. When the presence/absence of OTU was compared per sites in developmental stadia separately (Fig. 3), six OTUs was shared in the samples of engorged adults/nymphs in all sites, 27 and 26 OTUs in larvae and eggs, respectively. The OTUs that were shared between adults from observed sites, which should be considered as core species: Cardinium spp. (OTU 1); Bartonella-like. (OTUs 2, 4, 72, 79, 142); K. rhizophila (OTU 9); P. vagans (OTU 12); Staphylococcus saprophyticus (OTU 14); and Bacillus toyonensis (OTU 142). On the top of the core species, the following species/genera were also detected in all the larva and egg samples: Cardinium (OTU 1), Bartonella-like (OTU 2, 4, 72), K. rhizophila (OTU 9), and P. vagans (OTU 12).

Venn diagrams constructed form the samples of red poultry mite D. gallinae microbiome to compare the distribution of OTUs among sampled poultry houses (Bantice, Mirovice, Pohorelice, Ustrasice); a eggs; b larvae; c engorged adults/nymphs
The heatmap (Fig. S2a, b) showed separation of the samples engorged adults/nymphs from larvae and juveniles, but no specific OTU clusters for eggs and larvae is visible on untransformed data (Fig. S2a). There is a specific cluster of OTUs from the samples of eggs and larvae on LOG2 transformed data (Fig. S2b). However, there is also the OTUs cluster, which is not specific for developmental stadia and sampled sites, composed from S. saprophyticus (OTU14), Cardinium (OTU1), K. rhizophila (OTU9), and Bartonella-like bacteria (OTUs2, 4, and 72).
The METASTATS analysis was used to compare microbiome among adults, larvae, and eggs of the RPM (Table 2). The following OTUs had relative abundance significantly higher in the sample of larvae or eggs than in adults: Cardinium (OTU 1), Wolbachia (OTUs 5, 20), R. sedimentorum (OTU 13), P. vagans (OTU 12), Alcaligenes aquatilis (OTU 7), S. saprophyticus (OTU 14), and P. pulmonis (OTU 15). Opposite situation was found for the following species: Bartonella-like (OTUs 2, 4, 72, 79), T. strandjordii (OTU 6) and K. rhizophila (OTU 9). These bacteria had significantly higher relative abundance in microbiome of engorged adults/nymps than in larvae or eggs. No statistically significant difference in relative abundance among developmental stages was observed in Ricketsiella-like (OTU 3). However, the adults microbiome contained OTUs with relative abundance significantly influenced by samples sites (Table 3).
qPCR Quantification
The numbers of copies of 16S rRNA obtained from universal primers (Table S2) significantly differed between stadia (K 2,22 = 47.49, P < 0.001) of RPM (Fig. 4). The numbers of copies in engorged nymphs and adults were 100-fold higher than in eggs and larvae (Fig. 4a). The same trend was also found for Cardinium and Wolbachia copies (K 2,22 = 32.13, P < 0.001 and K 2,22 = 46.70, P < 0.001). The numbers of Cardinium in engorged nymphs and adults were 10-fold higher than in eggs and 100-fold higher than in larvae (Fig. 4b) while Wolbachia had 10-fold higher numbers in engorged nymphs and adults than in the eggs and larvae.

The quantification of numbers of copies of 16S rRNA gene in developmental stadia in D. gallinae microbiome. The data are LOG transformed and presented as the box and whiskers plots. a Numbers of copies obtained by universal primers. b Numbers of copies obtained by Cardinium specific primers. c Numbers of copies obtained from Wolbachia. The significant differences in Kruskal–Wallis test are indicated by different letters
Predicted Microbiome Derived from NGS 454 Pyrosequencing Data
BLASTn analysis (performed in December 2016) revealed 25,525 sequences of the RPM transcriptome sequences. Following taxonomy search of top hit species, 811 sequences were allocated to bacterial taxa (cf. Supplementary Table S5). Overall, 299 different bacterial taxa were obtained and the top 20 hit species/genera are shown in Table 4. More than one fourth of the predicted bacteria sequences showed similarity to Bartonella species and represented 11 of the 20 top hit species. Among the Bartonella taxa, the sequences showed similarity to Bartonella vinsonii (12 sequences, mean similarity 68.3–96.0%) and Bartonella birtlesii (1 sequence, mean similarity 81.3%) sequences. Additionally, the next sequences showed similarity to Bartonella spp. (40 sequences, mean similarity 67.4–97.0%), B. grahamii (23 sequences, mean similarity 71.3–97.2%), Bartonella quintana (17 sequences, mean similarity 68.5–90.6%), Candidatus Bartonella ancashi (15 sequences, mean similarity 66–95%) and Bartonella clarridgeiae (5 sequences, mean similarity 67.3–93.2%).
Referring to already identified endosymbionts of RPM, top hits were obtained with two sequences of similarity to S. eriocheiris (mean similarity 87.0%), but no sequences for Cardinium, Rickettsiella, Anistosticta, and Schineria. The sequences similar to different Wolbachia records available in database were detected as top hits (Supplementary Table S5). Except to putative symbiotic bacteria, we found sequences similar to S. saprophyticus (mean similarity 72.2–100%), Kocuria spp. (mean similarity 93.4% and 100%) and one Alcaligenes aquatilis (mean similarity 97.3%).
Top hit bacterial taxa of potential pathogenicity with mean similarity values >90% were, e.g., four sequences of Salmonella enterica (mean similarity 72.1–99%), two sequences of Francisella tularensis (mean similarity 82.1 and 90%), one sequence of A. viridans (mean similarity 97.1%) and three sequences of Mycobacterium abscessus (mean similarity 74.9 and 97.2%). Two sequences of the laboratory population showed top hits for P. multocida (range of mean similarity 71.8–76.5%).
Discussion
The Comparison of RPM Microbiomes
The RPM microbiome has been characterized according to 16S rRNA gene as well as the whole transcriptome data of a laboratory RPM population from Germany [16]. The microbiome of the field adult RPM consisted of between 15 and 64 OTUs, but 10 OTUs covered from 90 to 99.9% of all sequences analyzed. The heterogeneity in samples among the four poultry houses confirmed findings of previous reports that the bacterial community varied between locations [65]. Thus, the difference between RPM populations seems to be linked to the geographical sites and, moreover, the difference likely influence the poultry farming practices [65]. Based on our findings we hypothesize that the next factor influencing the microbiome structure would be the different developmental stage of these mites.
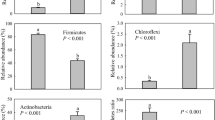
Moro et al. [7] determined phylotypes in RPM and suggested that the bacteria are saprophytes, symbionts, opportunistic pathogens, or pathogenic agents. Our results obtained of the RPM microbiome analyses from the four poultry houses in Czechia and the German laboratory population confirmed this classification. However, the function of different bacterial taxa might be distinct than previously suggested [4, 1] due to the more accurate identifications in this study or new information available in the literature. Among the putative saprophytes, Kocuria consisted from 4 to 7% of sequences in the adult RPM microbiomes of Czechia. We detected Kocuria also to be low abundance in the transcriptome of the German laboratory RPM population. In this context, Kocuria have been recently identified in stored product mites (Acari: Astigmata) and are suggested as the bacterial species present in the environment [66, 67].
Undetermined Rhizobiales taxa identified by Moro et al. [7] were suggested as saprophagous species. Here, we identified Bartonella-like sequences among the cloned 16S rRNA sequences from universal eubacterial primers and, moreover, confirmed Bartonella classification by the taxa specific primers. In addition, the obtained sequences from universal bacterial primers clustered with Illumina amplicon obtained Bartonella-like sequences. OTU2 is similar to cluster B, while OTUs 4 and 72 to cluster A (Fig. 1). The clones’ sequences of 16S rRNA gene showed 97% similarity to previously deposited RPM sequences of Rhizobiales (EF674511 and EF674514) [7]. We ascertained that the OTUs of Bartonella species hold between 30 and 65% of sequences in the adult RPM microbiome. This proportion is even higher than was observed for Alphaproteobacteria (lower than 5%) of RPM, previously [65]. Analysis of the German laboratory RPM population transcriptome confirmed high-abundance Bartonella-like sequences, being more than a quarter of the predicted bacterial sequences similar to Bartonella species.
Bartonella are Gram-negative, slow-growing, and facultative intracellular bacterial pathogens that infect mainly mammalian hosts causing them intraerythrocytic bacteremia. Bartonella transmission is possible via blood-sucking arthropod vectors [68], including ticks and mites [69, 70]. Following a family outbreak of B. quintana infection, the suspected vectors were RPMs, which migrated into the apartment from a hole in the roof [71]. The phylogenetic analyses of cloned Sanger sequences from RPM, however, showed that the sequences belonged to Bartonella suggested as symbionts of honeybees [57], bumble bees [56], ants [51,52,53,54,55], [72], stinkbugs [50], Varroa mites [17] and astigmatid mites [60, 67, 73]. The sequences formed two groups indicating that additional Bartonella-like bacteria taxa are associated to RPM. This fact supports high diversity of Bartonella-like OTUs from Illumina amplicons in RPM. Unfortunately, the weakness of this study is that Bartonella was identified based on 16S rRNA gene, which is, however, not suitable marker for the identification this genus [74, 75]. Future studies are necessary to demonstrate pathogeny or symbiotic-related genes and transmission models of Bartonella-like bacteria in RPM.
The reported symbiotic taxa in RPM include Cardinium [7, 9, 49] and Wolbachia [7]. In this study, Cardinium sequences had a low percentage (<1%) in the adult microbiome in Pohorelice and Mirovice, but 25 and 32% in Bantice and Mirovice areas, respectively. Among the bacterial sequences of the German laboratory RPM, Cardinium was not detected differently from the case Wolbachia and S. eriocheiris, which were absent in that sample. The Wolbachia sequences of Czech RPM populations covered only low percentage (<1%) in the adult microbiomes in poultry houses from Bantice, Mirovice, and Ustrasice; however, ca. 4% was observed in Pohorelice. In the RPM isolate from Germany, top hits for Wolbachia resulted in 1.2% of predicted bacterial data. These results indicated that the observed populations varied in proportion of these intracellular symbionts. Previously, Rickettsia sequences were detected and suggested as RPMs symbionts [7, 12].
In this study, Rickettsiella-like sequences formed 56% of the microbiome in adults from Ustrasice, while in the samples from other poultry houses of Czechia, it was a very low percentage (<1%) in their microbiomes. Moreover, Rickettsiella-like bacteria has not been detected among data of the laboratory RPM population from Germany [16]. In this study, the cloned Sanger sequences from RPM showed lower similarity (94%) to D. massiliensis, which has been described as an obligate intracellular gamma-proteobacterium in the tick Ixodes ricinus [76]. Regarding our phylogenic analyzes were the sequences closely similar to Candidatus Rickettsiella viridis symbiont of pea aphids (A. pisum) [62, 63] and sequences of bacteria in oribatid mite Nanorchestes [64], thereby, we indicated that the sequences belong to novel RPM symbiotic bacterium. This bacterium should be described to more detail in the future.
One opportunistic pathogen Tsukamurella was suggested in RPM [7]. It was confirmed in our study and the cloned Sanger sequences of 16S rRNAs showed 99% similarity to T. paurometabola and T. strandjordii [77]. In the present study, Tsukamurella represented from 3 to 19% of sequences of the adult RPM microbiome. Tsukamurellae are members of the mycolic acid-containing aerobic actinomycetes and T. paurometabola originally isolated from the mycetomes and ovaries of bed bugs [77]. Human infections with Tsukamurella are very rare and connected to immunosuppression and postoperative wounds [78]. The occurrence of T. paurometabola in bodies of the bugs suggests endosymbiotic relationships in RPM. The sequences of 98% identity to Spiroplasma have been reported from mites in France [7]. No Spiroplasma was found in microbiome neither was detected by specific primers in the RPMs populations of Czechia. Differently, in the transcriptome of laboratory RPM, two sequences were found, the sequences showed 87% similarity to S. eriocheiris. His result suggests that the Spiroplasma can occur in some RMP populations.
Microbiome Differences between Developmental Stadia and Symbionts
The life cycle of RPM includes eggs from which after 2 days the larval stage would hatch from. The larvae are non-parasitic, but then they change into protonymph, deuteronymph, and finally adults, which are all blood-sucking stages found on the host [1]. It means that eggs and larvae should be considered free of bacteria joined with feeding of blood on the host. However, these non-hematophagous stages might contain those bacteria transmitted maternally (transovarial transmission). In this study, the eggs and blood unfed larvae were surface cleaned by routinely used protocols [12], the high bacterial diversity in the eggs and larvae should be the result of surface contamination. The surface of the eggs contains micro-structures [79] suitable for bacterial attachment and it is possible that not all bacteria are remove by the cleaning method. We observed from 86 to 208 those OTUs among the samples. It means that the surface cleaning of eggs or larvae is not absolute or might be linked to transovarial transmission. The relatively high bacterial diversity might be explained by the presence of low numbers of various bacteria, which occurs in the “dirty” RPM environment. These bacteria formed the Illumina amplicon profiles from the samples. It is supported by the differences in the numbers of 16S rRNA gene copies observed by qPCR, i.e., the engorged adult/nymph stages had 100-fold more copies of 16S rRNA than eggs and larvae. The bacteria might just have been in the blood meal of engorged mites without any colonization of the mites, but the low number of OTUs and presence in the eggs/larvae did not support this suggestion.
We found Cardinium sequences most prevailing in eggs and larvae microbiome, i.e., 22 and 35% of obtained sequences were detected. Symbiont Wolbachia covered from 4 to 7% of the sequences in egg and larva microbiomes; however, in adults, it was lower than 1%. It, itself, indicates the transovarial and transstadial transmission of Cardinium and Wolbachia. This is not surprising due to the cases described in mites [80, 81].
RPM as the Reservoir of Human Pathogenic Bacteria
Previous studies provided the findings that RPMs are reservoirs or passive vectors for some human pathogens, because the multiplication of pathogenic bacteria in RPM mite bodies is unknown (Moro et al. [5], Sparagano et al. [4], and Pritchard et al. [1]. Moro et al. [7] observed sequences of similarity to E. rhusiopathiae and suggested importance of pathogen associations to these mites [65]. Here, we found 192 out of 1,493,621 analyzed sequences of 97% identity to this pathogen. E. rhusiopathiae bacteria were reported causing erysipelas in poultry, which is characterized as an acute septicemic infection that may result in sudden high mortality [82]. The bacteriological analyses of laying hens and mites during erysipelas disease outbreaks caused by E. rhusiopathiae in poultry flocks, revealed that RPM collected from the same house at the end of the production period of the following flock were negative for the presence of E. rhusiopathiae [83]. It should be noted, that the observed hens in this study were of good health and no signs of disease were recorded. In the field samples, we did not find sequences similar to P. multocida, which occurs in RPM and is suggested as an important pathogen [7]. However, two sequences of the laboratory population showed top hits for P. multocida (range of mean similarity 71.8–76.5%). The next analyses are necessary to identify if these bacterial taxa are of human pathogenic importance.
Conclusions
At population level, the mites are infested by various intracellular symbionts and putative symbiotic bacteria simultaneously. The differences also influence the way of bacterial transmission depending on the distribution of the bacteria inside the mites. The association of the RPM to Bartonella-like putative symbiotic bacteria was indicated. This study suggests a potential transovarial transmission and transstadial transmission of Cardinium and Wolbachia. There is also hint of the transovarial transmission of Bartonella-like bacteria, because the relative proportion of Bartonella-like sequences in the eggs and larvae microbiomes is similar as in the case of Cardinium. A different situation to Bartonella-like was observed for Tsukamurella, which represents only a small proportion of the sequences found in the eggs and larvae.
References
Pritchard J, Kuster T, Sparagano O, Tomley F (2015) Understanding the biology and control of the poultry red mite Dermanyssus gallinae: a review. Avian Pathol 44:143–153. doi:10.1080/03079457.2015.1030589
Lucky AW, Sayers CP, Argus JD, Lucky A (2001) Avian mite bites acquired from a new source—pet gerbils: report of 2 cases and review of the literature. Arch Dermatol 137:167–170. doi:10.1001/pubs.Arch Dermatol.-ISSN-0003-987x-137-2-dob00013
Cafiero MA, Camarda A, Circella E, Santagada G, Schino G, Lomuto M (2008) Pseudoscabies caused by Dermanyssus gallinae in Italian city dwellers: a new setting for an old dermatitis. J Eur Acad Dermatol Venereol 22:1382–1383. doi:10.1111/j.1468-3083.2008.02645.x
Sparagano OAE, George DR, Harrington DWJ, Giangaspero A (2014) Significance and control of the poultry red mite, Dermanyssus gallinae. Annu Rev Entomol 59:447–466. doi:10.1146/annurev-ento-011613-162101
Moro CV, De Luna CJ, Tod A, Guy JH, Sparagano OAE, Zenner L (2009) The poultry red mite (Dermanyssus gallinae): a potential vector of pathogenic agents. Exp Appl Acarol 48:93–104. doi:10.1007/s10493-009-9248-0
Moro CV, Fravalo P, Amelot M, Chauve C, Zenner L, Salvat G (2007) Colonization and organ invasion in chicks experimentally infected with Dermanyssus gallinae contaminated by Salmonella enteritidis. Avian Pathol 36:307–311. doi:10.1080/03079450701460484
Moro CV, Thioulouse J, Chauve C, Normand P, Zenner L (2009) Bacterial taxa associated with the hematophagous mite Dermanyssus gallinae detected by 16S rRNA PCR amplification and TTGE fingerprinting. Res Microbiol 160:63–70. doi:10.1016/j.resmic.2008.10.006
Rasmussen M (2016) Aerococcus: an increasingly acknowledged human pathogen. Clin Microbiol Infect 22:22–27. doi:10.1016/j.cmi.2015.09.026
De Luna CJ, Arkle S, Harrington D, George DR, Guy JH, Sparagano OAE (2008) The poultry red mite Dermanyssus gallinae as a potential carrier of vector-borne diseases. Ann N Y Acad Sci 1149:255–258. doi:10.1196/annals.1428.085
Huong CTT, Murano T, Uno Y, Usui T, Yamaguchi T (2014) Molecular detection of avian pathogens in poultry red mite (Dermanyssus gallinae) collected in chicken farms. J Vet Med Sci 76:1583–1587. doi:10.1292/jvms.14-0253
Circella E, Pugliese N, Todisco G, Cafiero MA, Sparagano OAE, Camarda A (2011) Chlamydia psittaci infection in canaries heavily infested by Dermanyssus gallinae. Exp Appl Acarol 55:329–338. doi:10.1007/s10493-011-9478-9
De Luna CJ, Moro CV, Guy JH, Zenner L, Sparagano OAE (2009) Endosymbiotic bacteria living inside the poultry red mite (Dermanyssus gallinae). Exp Appl Acarol 48:105–113. doi:10.1007/s10493-008-9230-2
Hodkinson BP, Grice EA (2015) Next-generation sequencing: a review of technologies and tools for wound microbiome research. Adv Wound Care (New Rochelle) 4:50–58. doi:10.1089/wound.2014.0542
Shendure J, Ji H (2008) Next-generation DNA sequencing. Nat Biotechnol 26:1135–1145. doi:10.1038/nbt1486
Mul M, van Niekerk T, Chirico J, Maurer V, Kilpinen O, Sparagano O, Thind B, Zoons J, Moore D, Bell B, Gjevre A-G, Chauve C (2009) Control methods for Dermanyssus gallinae in systems for laying hens: results of an international seminar. World Poultry Sci J 65:589–599. doi:10.1017/S0043933909000403
Schicht S, Qi W, Poveda L, Strube C (2014) Whole transcriptome analysis of the poultry red mite Dermanyssus gallinae (de Geer, 1778). Parasitology 141:336–346. doi:10.1017/S0031182013001467
Hubert J, Erban T, Kamler M, Kopecky J, Nesvorna M, Hejdankova S, Titera D, Tyl J, Zurek L (2015) Bacteria detected in the honeybee parasitic mite Varroa destructor collected from beehive winter debris. J Appl Microbiol 119:640–654. doi:10.1111/jam.12899
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. John Wiley and Sons, New York, pp. 115–175
Kopecky J, Perotti MA, Nesvorna M, Erban T, Hubert J (2013) Cardinium endosymbionts are widespread in synanthropic mite species (Acari: Astigmata). J Invertebr Pathol 112:20–23. doi:10.1016/j.jip.2012.11.001
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ (2005) At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl Environ Microbiol 71:7724–7736. doi:10.1128/AEM.71.12.7724-7736.2005
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ (2006) New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl Environ Microbiol 72:5734–5741. doi:10.1128/AEM.00556-06
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. doi:10.1038/nmeth.2604
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi:10.1128/AEM.00062-07
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi:10.1093/nar/25.17.3389
Pruesse E, Peplies J, Glockner FO (2012) SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829. doi:10.1093/bioinformatics/bts252
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi:10.1080/10635150390235520
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772–772. doi:10.1038/nmeth.2109
Rodrigue N, Lartillot N (2014) Site-heterogeneous mutation-selection models within the PhyloBayes-MPI package. Bioinformatics 30:1020–1021. doi:10.1093/bioinformatics/btt729
Jow H, Hudelot C, Rattray M, Higgs PG (2002) Bayesian phylogenetics using an RNA substitution model applied to early mammalian evolution. Mol Biol Evol 19:1591–1601. doi:10.1093/oxfordjournals.molbev.a004221
Lartillot N, Lepage T, Blanquart S (2009) PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25:2286–2288. doi:10.1093/bioinformatics/btp368
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi:10.1093/sysbio/syq010
Rambaut A (2007) FigTree, a graphical viewer of phylogenetic trees. Molecular evolution, phylogenetics and epidemiology: research, software and publications of Andrew Rambaut and members of his research group. http://tree.bio.ed.ac.uk/software/figtree/. Accessed 27 July 2015
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi:10.1038/ismej.2012.8
Aizenberg-Gershtein Y, Izhaki I, Santhanam R, Kumar P, Baldwin IT, Halpern M (2015) Pyridine-type alkaloid composition affects bacterial community composition of floral nectar. Sci Rep 5:11536. doi:10.1038/srep11536
Earley ZM, Akhtar S, Green SJ, Naqib A, Khan O, Cannon AR, Hammer AM, Morris NL, Li X, Eberhardt JM, Gamelli RL, Kennedy RH, Choudhry MA (2015) Burn injury alters the intestinal microbiome and increases gut permeability and bacterial translocation. PLoS One 10:e0129996. doi:10.1371/journal.pone.0129996
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM (2014) Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42:D633–D642. doi:10.1093/nar/gkt1244
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi:10.1128/AEM.01541-09
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi:10.1128/AEM.01043-13
Hammer O, Harper DAT, Ryan PD (2001) PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron 4:4 http://palaeo-electronica.org/2001_1/past/issue1_01.htm. Accessed 6 August 2016
Ondov BD, Bergman NH, Phillippy AM (2011) Interactive metagenomic visualization in a web browser. BMC Bioinformatics 12:385. doi:10.1186/1471-2105-12-385
Erban T, Ledvinka O, Nesvorna M, Hubert J (2017) Experimental manipulation shows a greater influence of population than dietary perturbation on the microbiome of Tyrophagus putrescentiae. Appl Environ Microbiol 83. doi:10.1128/AEM.00128-17
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2016. vegan: Community Ecology Package. CRAN - The Comprehensive R Archive Network. http://CRAN.R-project.org/package=vegan. Accessed 6 August 2016
Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B (2016) gplots: Various R programming tools for plotting data. CRAN - The Comprehensive R Archive Network. https://CRAN.R-project.org/package=gplots. Accessed 6 August 2016
Anderson MJ, Ellingsen KE, McArdle BH (2006) Multivariate dispersion as a measure of beta diversity. Ecol Lett 9:683–693. doi:10.1111/j.1461-0248.2006.00926.x
White JR, Nagarajan N, Pop M (2009) Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol 5:e1000352. doi:10.1371/journal.pcbi.1000352
Dorn-In S, Bassitta R, Schwaiger K, Bauer J, Holzel CS (2015) Specific amplification of bacterial DNA by optimized so-called universal bacterial primers in samples rich of plant DNA. J Microbiol Methods 113:50–56. doi:10.1016/j.mimet.2015.04.001
Hubert J, Kopecky J, Nesvorna M, Perotti MA, Erban T (2016) Detection and localization of Solitalea-like and Cardinium bacteria in three Acarus siro populations (Astigmata: Acaridae). Exp Appl Acarol 70:309–327. doi:10.1007/s10493-016-0080-z
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. doi:10.1093/bioinformatics/bti610
Duron O (2013) Lateral transfers of insertion sequences between Wolbachia, Cardinium and Rickettsia bacterial endosymbionts. Heredity 111:330–337. doi:10.1038/hdy.2013.56
Matsuura Y, Kikuchi Y, Meng XY, Koga R, Fukatsu T (2012) Novel clade of alphaproteobacterial endosymbionts associated with stinkbugs and other arthropods. Appl Environ Microbiol 78:4149–4156. doi:10.1128/AEM.00673-12
Russell JA, Moreau CS, Goldman-Huertas B, Fujiwara M, Lohman DJ, Pierce NE (2009) Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc Natl Acad Sci U S A 106:21236–21241. doi:10.1073/pnas.0907926106
Anderson KE, Russell JA, Moreau CS, Kautz S, Sullam KE, Hu Y, Basinger U, Mott BM, Buck N, Wheeler DE (2012) Highly similar microbial communities are shared among related and trophically similar ant species. Mol Ecol 21:2282–2296. doi:10.1111/j.1365-294X.2011.05464.x
Hu Y, Lukasik P, Moreau CS, Russell JA (2014) Correlates of gut community composition across an ant species (Cephalotes varians) elucidate causes and consequences of symbiotic variability. Mol Ecol 23:1284–1300. doi:10.1111/mec.12607
Bonasio R, Zhang G, Ye C, Mutti NS, Fang X, Qin N, Donahue G, Yang P, Li Q, Li C, Zhang P, Huang Z, Berger SL, Reinberg D, Wang J, Liebig J (2010) Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science 329:1068–1071. doi:10.1126/science.1192428
Stoll S, Gadau J, Gross R, Feldhaar H (2007) Bacterial microbiota associated with ants of the genus Tetraponera. Biol J Linn Soc 90:399–412. doi:10.1111/j.1095-8312.2006.00730.x
Martinson VG, Danforth BN, Minckley RL, Rueppell O, Tingek S, Moran NA (2011) A simple and distinctive microbiota associated with honey bees and bumble bees. Mol Ecol 20:619–628. doi:10.1111/j.1365-294X.2010.04959.x
Kesnerova L, Moritz R, Engel P (2016) Bartonella apis sp. nov., a honey bee gut symbiont of the class Alphaproteobacteria. Int J Syst Evol Microbiol 66:414–421. doi:10.1099/ijsem.0.000736
Hubert J, Kopecky J, Perotti MA, Nesvorna M, Braig HR, Sagova-Mareckova M, Macovei L, Zurek L (2012) Detection and identification of species-specific bacteria associated with synanthropic mites. Microb Ecol 63:919–928. doi:10.1007/s00248-011-9969-6
Hubert J, Nesvorna M, Kopecky J, Sagova-Mareckova M, Poltronieri P (2015) Carpoglyphus lactis (Acari: Astigmata) from various dried fruits differed in associated micro-organisms. J Appl Microbiol 118:470–484. doi:10.1111/jam.12714
Kopecky J, Nesvorna M, Hubert J (2014) Bartonella-like bacteria carried by domestic mite species. Exp Appl Acarol 64:21–32. doi:10.1007/s10493-014-9811-1
Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Comparative Sequence Program NISC, Murray PR, Turner ML, Segre JA (2012) Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 22:850–859. doi:10.1101/gr.131029.111
Tsuchida T, Koga R, Fujiwara A, Fukatsu T (2014) Phenotypic effect of “Candidatus Rickettsiella viridis,” a facultative symbiont of the pea aphid (Acyrthosiphon pisum), and its interaction with a coexisting symbiont. Appl Environ Microbiol 80:525–533. doi:10.1128/AEM.03049-13
Lukasik P, van Asch M, Guo H, Ferrari J, Godfray HCJ (2013) Unrelated facultative endosymbionts protect aphids against a fungal pathogen. Ecol Lett 16:214–218. doi:10.1111/ele.12031
Moquin SA, Garcia JR, Brantley SL, Takacs-Vesbach CD, Shepherd UL (2012) Bacterial diversity of bryophyte-dominant biological soil crusts and associated mites. J Arid Environ 87:110–117. doi:10.1016/j.jaridenv.2012.05.004
Moro CV, Thioulouse J, Chauve C, Zenner L (2011) Diversity, geographic distribution, and habitat-specific variations of microbiota in natural populations of the chicken mite, Dermanyssus gallinae. J Med Entomol 48:788–796. doi:10.1603/ME10113
Erban T, Klimov PB, Smrz J, Phillips TW, Nesvorna M, Kopecky J, Hubert J (2016) Populations of stored product mite Tyrophagus putrescentiae differ in their bacterial communities. Front Microbiol 7:1046. doi:10.3389/fmicb.2016.01046
Hubert J, Kopecky J, Sagova-Mareckova M, Nesvorna M, Zurek L, Erban T (2016) Assessment of bacterial communities in thirteen species of laboratory-cultured domestic mites (Acari: Acaridida). J Econ Entomol 109:1887–1896. doi:10.1093/jee/tow089
Regier Y, O’Rourke F, Kempf VA (2016) Bartonella spp.—a chance to establish one health concepts in veterinary and human medicine. Parasit Vectors 9:261. doi:10.1186/s13071-016-1546-x
Reeves WK, Dowling APG, Dasch GA (2006) Rickettsial agents from parasitic Dermanyssoidea (Acari: Mesostigmata). Exp Appl Acarol 38:181–188. doi:10.1007/s10493-006-0007-1
Klangthong K, Promsthaporn S, Leepitakrat S, Schuster AL, McCardle PW, Kosoy M, Takhampunya R (2015) The distribution and diversity of Bartonella species in rodents and their ectoparasites across Thailand. PLoS One 10:e0140856. doi:10.1371/journal.pone.0140856
Melter O, Arvand M, Votypka J, Hulinska D (2012) Bartonella quintana transmission from mite to family with high socioeconomic status. Emerg Infect Dis 18:163–165. doi:10.3201/eid1801.110186
Larson HK, Goffredi SK, Parra EL, Vargas O, Pinto-Tomas AA, McGlynn TP (2014) Distribution and dietary regulation of an associated facultative Rhizobiales-related bacterium in the omnivorous giant tropical ant, Paraponera clavata. Naturwissenschaften 101:397–406. doi:10.1007/s00114-014-1168-0
Valerio CR, Murray P, Arlian LG, Slater JE (2005) Bacterial 16S ribosomal DNA in house dust mite cultures. J Allergy Clin Immunol 116:1296–1300. doi:10.1016/j.jaci.2005.09.046
Kosoy M, Hayman DTS, Chan K-S (2012) Bartonella bacteria in nature: where does population variability end and a species start? Infect Genet Evol 12:894–904. doi:10.1016/j.meegid.2012.03.005
Tsai Y-L, Chang C-C, Chuang S-T, Chomel BB (2011) Bartonella species and their ectoparasites: selective host adaptation or strain selection between the vector and the mammalian host? Comp Immunol Microbiol Infect Dis 34:299–314. doi:10.1016/j.cimid.2011.04.005
Mediannikov O, Sekeyova Z, Birg M-L, Raoult D (2010) A novel obligate intracellular gamma-proteobacterium associated with ixodid ticks, Diplorickettsia massiliensis, gen. nov., sp. nov. PLoS One 5:e11478. doi:10.1371/journal.pone.0011478
Kattar MM, Cookson BT, Carlson LDC, Stiglich SK, Schwartz MA, Nguyen TT, Daza R, Wallis CK, Yarfitz SL, Coyle MB (2001) Tsukamurella strandjordae sp. nov., a proposed new species causing sepsis. J Clin Microbiol 39:1467–1476. doi:10.1128/JCM.39.4.1467-1476.2001
Almehmi A, Pfister AK, McCowan R, Matulis S (2004) Implantable cardioverter-defibrillator infection caused by Tsukamurella. W V Med J 100:185–186
Kucerova Z, Stejskal V (2009) Morphological diagnosis of the eggs of stored-products mites. Exp Appl Acarol 49:173–183. doi:10.1007/s10493-009-9256-0
Ma W-J, Vavre F, Beukeboom LW (2014) Manipulation of arthropod sex determination by endosymbionts: diversity and molecular mechanisms. Sex Dev 8:59–73. doi:10.1159/000357024
Pietri JE, DeBruhl H, Sullivan W (2016) The rich somatic life of Wolbachia. MicrobiologyOpen 5:923–936. doi:10.1002/mbo3.390
Chirico J, Eriksson H, Fossum O, Jansson D (2003) The poultry red mite, Dermanyssus gallinae, a potential vector of Erysipelothrix rhusiopathiae causing erysipelas in hens. Med Vet Entomol 17:232–234. doi:10.1046/j.1365-2915.2003.00428.x
Eriksson H, Brannstrom S, Skarin H, Chirico J (2010) Characterization of Erysipelothrix rhusiopathiae isolates from laying hens and poultry red mites (Dermanyssus gallinae) from an outbreak of erysipelas. Avian Pathol 39:505–509. doi:10.1080/03079457.2010.518313
Acknowledgements
This study was supported by the project of the Ministry of Agriculture of the Czech Republic RO0417 and by COST Action FA1404 (COREMI) (http://www.coremi.eu/home.html). Computational resources were supplied by the Ministry of Education, Youth and Sports of the Czech Republic under the Projects CESNET (Project No. LM2015042) and CERIT-Scientific Cloud (Project No. LM2015085) provided within the program Projects of Large Research, Development and Innovations Infrastructures. We thank Vlastislav Machandr for his kind help with sample collection and Martin Markovic for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 2144 kb).
Rights and permissions
About this article

Cite this article
Hubert, J., Erban, T., Kopecky, J. et al. Comparison of Microbiomes between Red Poultry Mite Populations (Dermanyssus gallinae): Predominance of Bartonella-like Bacteria. Microb Ecol 74, 947–960 (2017). https://doi.org/10.1007/s00248-017-0993-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-017-0993-z