
Abstract
Increasing evidence of tight links among the gut microbiota, obesity, and host health has emerged, but knowledge of the ecological processes that shape the variation in microbial assemblages across growth rates remains elusive. Moreover, inadequately control for differences in factors that profoundly affect the gut microbial community, hampers evaluation of the gut microbiota roles in regulating growth rates. To address this gap, we evaluated the composition and ecological processes of the gut bacterial community in cohabitating retarded, overgrown, and normal shrimps from identically managed ponds. Gut bacterial community structures were distinct (P = 0.0006) among the shrimp categories. Using a structural equation modeling (SEM), we found that changes in the gut bacterial community were positively related to digestive activities, which subsequently affected shrimp growth rate. This association was further supported by intensified interspecies interaction and enriched lineages with high nutrient intake efficiencies in overgrown shrimps. However, the less phylogenetic clustering of gut microbiota in overgrown and retarded subjects may offer empty niches for pathogens invasion, as evidenced by higher abundances of predicted functional pathways involved in disease infection. Given no differences in biotic and abiotic factors among the cohabitating shrimps, we speculated that the distinct gut community assembly could be attributed to random colonization in larval shrimp (e.g., priority effects) and that an altered microbiota could be a causative factor in overgrowth or retardation in shrimp. To our knowledge, this is the first study to provide an integrated overview of the direct roles of gut microbiota in shaping shrimp growth rate and the underlying ecological mechanisms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
It is now recognized that the gut microbiota serves as a virtual endocrine organ by acting as a barrier against pathogen invasion and providing complementary metabolic pathways for host nutrient acquisition [1, 2]. As a consequence, dysbiosis of the gut microbial composition is tightly associated with shrimp diseases [3] and metabolic syndrome [4], among others. Increasing evidence has shown that developmental stage [5], diet [6], health status [3], and the rearing environmental conditions [7] are the major factors in determining the composition and function of gut community. Recently, we found that shrimps reared in the same pond exhibit marked differences in body size and weight. One commonly accepted explanation is that variations in gut microbiotas among individuals lead to different growth rates [1, 2]. However, differences in gut microbiotas among identically managed shrimp cannot be attributed to the above-mentioned driving factors. It has been proposed that stochastic colonization of alien taxa in larvae gut could contribute long-term effects on the health of later life [8]. Indeed, neutral processes can explain a significant portion of the variations in fish gut community [9]. Therefore, understanding the association between shrimp gut community assembly and growth rates, and the underlying ecological processes are of essential interest from both commercial and scientific perspectives.
Recently, it has been shown that cohort pigs exhibit distinct gut bacterial assemblages, resulting in significant differences in their body weights [10]. One possible explanation is that diverse enterotypes respond differently to diet intake [10, 11]. Similarly, identically farmed sea cucumbers display distinct growth rates, whereas differences in gut bacterial composition are evident at the species level [12]. Several studies have uncovered profound changes in the composition and metabolic function of gut microbiota in obese vertebrate individuals [13–15], whereas these studies did not adequately control for differences in diet or environmental conditions. In addition, invertebrate gut microbiota is dominated by Proteobacteria [3, 12], as opposed to vertebrate gut community is dominated by Bacteroidetes and Firmicutes [13, 14, 16]. For these reasons, it is unclear whether growth rates and the gut bacterial community assembly are tightly connected in shrimp, mirroring what has been observed in vertebrates [10, 15]. The notion that a host exhibits strong selection pressures on the gut microbiota is becoming widely accepted as the evidence grows [3, 17]. However, substantial differences in the gut bacterial communities have been detected between cohabitating individuals [5, 12]. Consequently, it is unclear whether the gut community of healthy shrimp (e.g., normal growth) is more or less convergent than that of shrimp with suboptimal health (e.g., retarded or overgrown). It has been proposed that closely related lineages will coexist due to phylogenetic niche conservatism (the tendency of lineages to retain their niche-related traits) [18]. Under this premise, a healthy host can constrain the divergence among closely related species, resulting in a high level of phylogenetic relatedness in the gut bacterial community assembly. In contrast, diseased shrimp are more prone to invasion by alien bacterial species, which is concomitant with a more stochastic gut community assembly [19]. From this ecological perspective, the phylogenetic relatedness of gut community may offer a surrogate index for defining a healthy gut microbiota.
The association between the gut microbiota and growth rates appears to be more complex than the simple phylum-level Bacteroidetes/Firmicutes ratio that was initially identified [20]. Instead, it is now recognized that the microbial-mediated function (e.g., metabolic phenotype) of a given community depends on the interdependent associations among different bacterial species [21, 22] rather than the sum of the individual species’ traits [23]. The additive, synergistic, and antagonistic interactions between species are integral in microbial-mediated functions [19, 24]. Indeed, recent network studies have identified putative gut keystone taxa that are responsible for differences in host growth traits [10, 14]. Thus, network analysis affords a promising avenue for exploring these complex microbial interactions and niches [24, 25]. Evidence, albeit limited, shows that an increased shrimp gut interspecies interaction is positively associated with enhanced functional potentials [19]. We therefore hypothesized that the gut microbiota of overgrown shrimp exhibits more synergistic and complex interspecies interaction, thereby facilitating a higher efficiency in nutrient acquisition.
Identically farmed shrimps are ruled out the confounded factors, such as diet, age, and ambient conditions, in shaping gut microbiota, thus offering an ideal setting to examine the direct association among the gut bacterial community, host growth rate, and underlying ecological processes. Here, using the Illumina sequencing technique, we compared the gut bacterial community assembly among three categories of shrimp (retarded, overgrown, and normal) (Fig. S1). This design allowed us to (i) establish the connections among the shrimp gut bacterial community, digestive enzyme activity and growth rates; (ii) evaluate whether phylogenetic relatedness correlated with shrimp health states; and (iii) assess the extent to which gut bacterial interspecies interaction and function varied with shrimp growth rates.
Materials and Methods
Experimental Design and Sample Collection
The shrimp ponds investigated in this study are located in Xiangshan, Ningbo, the Eastern China. The ponds are approximately uniform in size (3000 m2) and depth (1.2 m). For subsequent standardization managements, the farmers had analyzed and adjusted the water parameters before inoculation, to make sure that the rearing conditions are similar across the ponds. On 8 April 2016, congeneric larval shrimps (Litopenaeus vannamei) were inoculated with a stocking density of 480,000 individuals into each pond. The ponds were identically managed, including seawater inputs, daily water exchange rate (5%), feed type, and schedule. After 70 days of cultivation (17 June), we found that shrimps in the same pond exhibited substantial differences in body size and weight. The size that accounted for the most proportion (approximately 90%) was artificially categorized into normal shrimps (Fig. S1). The expected size at this growth stage was further confirmed by the farmers. Subsequently, we sorted the sizes that markedly smaller or larger than that of normal shrimps into retarded or overgrown individuals, respectively (Fig. S1). We collected shrimp and water samples from each pond. Shrimps were separately stored and aerated in tanks with water from the corresponding pond during transportation. To minimize the spatial variability within the ponds, water samples were taken from six representative points in each pond (in similar locations across the six ponds) and were combined to form a composite sample representing each pond. Water samples were stored in an icebox.
DNA Extraction
On the sampling day, shrimps from each pond were sorted into three categories according to their body size and weight (Fig. S1). Because an adequate amount of DNA could not be obtained from the intestine of a single retarded shrimp in the initial trial runs, for each pond, the intestines of three shrimp from each size category were dissected on ice and pooled to compose one sample. To collect the planktonic microbial biomass, a 0.5-L water sample for each pond was pre-filtered through nylon mesh (100 μm pore size) and subsequently filtered onto a 0.22-μm membrane (Millipore, Boston, MA, USA). Genomic DNA (gDNA) was extracted using the FAST DNA Spin kit (MO BIO Laboratories, Carlsbad, CA, USA) according to the manufacturer’s protocols. The gDNA extracts were quantified using a NanoDrop ND-2000 spectrophotometer (NanoDrop Technologies, Wilmington, USA) and then stored at −80 °C prior to amplification.
Digestive Enzyme Activity
To measure the digestive enzyme activity for a given gut community, the stomach and hepatopancreas from the same three shrimp were extracted and pooled. For the measurement, ice-cold distilled water with a volume (ml) four times the weight (g) of the tissues was added. The tissues were then homogenized at 4 °C and centrifuged at 6000 rpm for 10 min, and the supernatant was collected to measure the digestive enzyme activity. The activities of amylase (kit no. A016-1), pepsin (kit no. A080-1), and lipase (kit no. A054) were measured using commercial assay kits (Nanjing Jiancheng Institute, Nanjing, China) in accordance with the manufacturer’s instructions. Digestive enzyme activity was expressed as a relative unit per milligram of soluble protein (U mg−1).
Bacterial 16S rRNA Gene Amplification and MiSeq Sequencing
The bacteria-specific primers 338F (5′-GTACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) were used to amplify the V3–V4 regions of bacterial 16S rRNA gene [26]. Polymerase chain reaction (PCR) condition was performed as previously described [25]. PCR products were visualized in 1.5% agarose gel to verify product band size. Each sample was amplified in triplicate and pooled to minimize the reaction-level PCR bias. Amplicons for each sample were combined and purified using a PCR fragment purification kit.
After purification, DNA concentration was measured using a PicoGreen-iT dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA). Identical amounts of amplicons from each sample were pooled in a single tube, and the amplicons were sequenced using MiSeq platform (Illumina, San Diego, CA, USA), producing 2 × 300 bp paired-end reads.
Processing of Illumina Sequencing Data
The paired-end reads were joined with FLASH [27]. The assembled sequences were processed following the Quantitative Insights Into Microbial Ecology pipeline (QIIME v1.9.0) [28]. Briefly, the sequences with ambiguous bases or truncated at any site of more than three consecutive bases receiving a Phred quality score (Q) < 20 were deleted, as were truncated reads that had <75% of their original length. Chimeric sequences were identified using the UCHIME algorithm [29] and removed, as were singletons. Bacteria phylotypes were identified using UCLUST [30] and classified into the operational taxonomic units (OTUs) at a 97% cutoff. The most abundant sequence of each OTU was selected as the representative sequence and then taxonomically assigned in the Greengenes database (release 13.8) [31] using PyNAST [32]. After taxonomies had been assigned, OTUs that were affiliated with Archaea, chloroplasts, Eukaryota, and those unassigned at the Bacteria domain level were excluded from the dataset. The filtered alignments were then used to generate a maximum-likelihood tree using FastTree [33] for phylogenetic analysis. To correct for unequal sequencing depth, we used a 20× randomly rarefied subset of 15,600 sequences per sample to calculate the diversity and distance between samples. Sequence data are deposited in Sequence Read Archive at DDBJ under the accession no. DRA005153.
Statistical Analysis
Shrimp body weight, digestive enzyme activity, and bacterial diversity among categories were compared using a one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. Non-metric multidimensional scaling (NMDS) and analysis of similarity (ANOSIM) were performed to evaluate the overall differences in bacterial community structure [34]. The correlations among the shrimp gut bacterial community, digestive enzyme activity and growth rates were examined by the Mantel test using the ‘vegan’ package in R v3.1.0 [35]. Then, structural equation modeling (SEM) was used to evaluate the interrelationships among these variables in Amos 18.0 (IBM, Chicago, IL, USA). The a priori and theoretical assumptions made to establish the SEM were as follows: (i) the gut bacterial community can directly influence shrimp growth rate, and (ii) the gut bacterial community can lead to changes in enzyme activity, which subsequently affects shrimp growth rate. The data matrix was fitted to the model using the maximum-likelihood estimation method. Favorable model fits were suggested by non-significant χ 2 test (P > 0.05), high goodness-of-fit index (GFI >0.90), and low root square mean errors of approximation (RMSEA <0.05). The indicator values (IndVal) method was used to identify indicative bacterial families associated with each investigated group [36]. Only the IndVal values that were both significant (P < 0.05) and >0.4 were considered, using the package ‘labdsv’ in R [35, 37].
A null model analysis of multivariate dispersion (PERMDISP) was performed to test whether the dispersion among biological replicates for each group is indistinguishable from the null expectation [38]. In quantifying the phylogenetic diversities (i.e., phylobetadiversity) among the three shrimp groups, we calculated the mean nearest taxon distance (MNTD) separating OTUs into three communities [39]. MNTD is the mean phylogenetic distance separating each species in the community from its closest relative, which is sensitive to changes in lineage close to the phylogenetic tips [40]. To evaluate the degree of non-random phylogenetic relatedness, the ‘standardized effect size’ of the phylogenetic community structure (ses.MNTD) was calculated for MNTD based on the difference between phylogenetic distances in the observed communities versus null communities generated with 999 randomizations divided by the standard deviation of phylogenetic distances in the distribution [40, 41]. Negative ses.MNTD values with low quantiles (P < 0.05) indicate that co-occurring species are more closely related than expected by chance (i.e., convergence), whereas positive ses.MNTD values with high quantiles (P > 0.95) suggest an overdispersion (i.e., divergence) of co-occurring species [39, 41]. Further, a larger absolute magnitude of ses.MNTD value reflects the stronger effects of deterministic processes [5]. These analyses were implemented in the R environment with the package ‘Picante’ [35, 40].
Interspecies interaction was evaluated using an open-accessible pipeline (http://ieg2.ou.edu/MENA) [42]. To quantitatively compare the differences in gut interspecies interaction among shrimp categories, a set of topology properties was calculated, including average path length, clustering coefficient, modularity, and positive co-occurrences [42, 43]. The network was visualized in Cytoscape 3.3.0 [44].
To obtain bacterial metabolic functional traits, the sequencing data were re-analyzed as described above, with the exceptions for taxonomical classification, against a closed reference (Greengenes database 13.8) and then rarified to 10,870 reads per sample. Subsequently, the OTUs table was normalized by dividing the abundance of each OTU by its predicted 16S copy number to produce the KEGG (Kyoto Encyclopaedia of Genes and Genomes) classified functions of the community by Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) [45]. The significances in the KEGG pathways between normal and retarded/overgrown shrimp were tested using a response ratio analysis at a 95% confidence interval [46].
Results
Shrimp Categories and Digestive Enzyme Activity
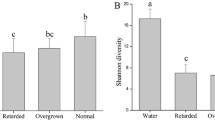
We sorted shrimp into three categories according to size: overgrown (8.27 ± 0.39 cm, mean ± standard deviation), normal (6.47 ± 0.09 cm), and retarded (4.50 ± 0.19 cm) subjects (Fig. S1). The average body weights of these three categories were 24.07 ± 3.07 g, 12.15 ± 1.29 g and 3.62 ± 0.48 g, respectively, which were significantly different among categories (F ANOVA = 167.5, P < 0.001). In addition, the measured digestive enzyme activity, including amylase, pepsin, and lipase activities, exhibited consistent differences across the three categories, with the highest activity in overgrown shrimp and the lowest in retarded shrimp (Fig. 1). Notably, amylase, pepsin and lipase activities were positively and significantly (Pearson r > 0.81, P < 0.001 in all cases) correlated with shrimp body size and body weight (Table S1).

Comparison of digestive amylase (a), pepsin (b), and lipase (c) activities among the three shrimp categories. Different lowercase letters indicate significant differences among groups. Means were compared using one-way analysis of variance (ANOVA), with P < 0.05 considered significant
Distribution of Taxa and Phylotypes
After quality control, we obtained a total of 577,456 high-quality sequences and 15,697–32,578 sequences per sample (mean = 24,060 ± 3952, standard deviation). Then, the unequal sequencing depths were rarified to 15,600 sequences per sample, resulting in 8373 OTUs across the samples. The dominant phyla were Gammaproteobacteria (mean relative abundance, 41.1%) and Alphaproteobacteria (30.3%), followed by Bacteriodes (16.3%) and Actinobacteria (4.5%) (Fig. S2). Deltaproteobacteria (0.82%), Betaproteobacteria (0.47%), and Firmicutes (0.32%) were found at low abundances (data not shown). The relative abundances of the dominant phyla/classes in water samples and in shrimps from the three size categories were distinct and significantly different among the groups (Fig. S2). For example, the relative abundance of Gammaproteobacteria was low in water samples (5.3 ± 0.2%) but was dramatically enriched in retarded shrimps (73.7 ± 20.4%) and significantly decreased to 33.3 ± 9.5% in normal ones. In contrast, the relative abundances of Alphaproteobacteria and Actinobacteria exhibited an opposite pattern (Fig. S2).
Variation in Bacterial Communities across Habitats and Shrimp Categories
The evenness and α-diversity of bacterial communities were compared among categories that were defined a priori, including the number of observed species, Shannon index and phylogenetic diversity. The evenness was significantly lower in the retarded shrimp but was comparable in the other groups (Fig. S3). However, there were significant differences in α-diversity (P < 0.001 for the three diversity indices) among groups. In general, the α-diversity was higher in water samples than in shrimps. Furthermore, normal shrimps harbored a higher bacterial α-diversity than did retarded shrimps, with an intermediate α-diversity level in overgrown shrimps (Fig. S3).
Based on the UniFrac distance of OTUs detected across the samples, an NMDS ordination biplot revealed a clear clustering in the bacterial communities (Fig. 2a). This pattern was further corroborated by an ANOSIM, which demonstrated that both the habitats (R = 0.498, P = 0.0006) and shrimp categories (Global r = 0.549, P < 0.0001) were important determinants of bacterial community composition. In addition, the bacterial community compositions were significantly distinct between each pair of groups (Table S2). A permutational multivariate analysis of variance showed that shrimp category explained 20.1% (P = 0.014) of the variation in gut bacterial communities.

Non-metric multidimensional scaling (NMDS) ordination of the dissimilarity (UniFrac distance) in bacterial community composition (a). The structural equation model shows the effects of the shrimp gut bacterial community on enzyme activity and body weight/size (b). Numbers on arrows are standardized path coefficients. Arrow widths show the strength of the causal relationship. Percentages (R 2) indicate the variance explained by the gut bacterial community and enzyme activity. *P < 0.05; **P < 0.01
Notably, the shrimp gut bacterial communities were significantly (Mantel test, P < 0.05 in all cases) correlated with shrimp weight/size and enzyme activities (Table S3). For this reason, we constructed SEMs to explore the direct and indirect effects of the shrimp gut microbiota on digestive activity and body weight/size. The model fit the data well: χ 2 = 1.52, P = 0.45, GFI = 0.96, RMSEA = 0.001. Our SEMs explained 83.7% of variance in shrimp body weight/size (Fig. 2b). Gut bacterial community was found to have significantly positive (λ = 0.56, P = 0.015) effects on enzyme activities, which in turn substantially promoted (λ = 0.68, P = 0.002) shrimp body weight/size. In contrast, the direct effects of gut bacterial community on shrimp body weight/size were relatively weak (λ = 0.19, P = 0.045) (Fig. 2b). Overall, the shrimp growth rate was directly determined by the gut microbiota-mediated digestive activities (Fig. 2b).
Identification of Key Gut Bacterial Families for Indicating Shrimp Categories
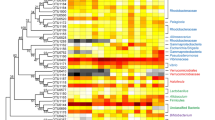
Given that the gut bacterial communities were distinct among the shrimp categories, we next investigated which assemblages characterized such differences. It has been shown that ecological cohesion is highest at the bacterial family level, which can accurately indicate shrimp health status [7]. Thus, we screened the indicators at the bacterial family level using IndVal values >0.4. Twenty-six bacterial families were identified, with significant (P < 0.05 in all cases) changes in their relative abundances among the three categories (Fig. 3). A majority (16 of 26) of these indictor families was most abundant in normal shrimps. In contrast, the relative abundances of Rhodospirillaceae, Saprospiraceae, Bdellovibrionaceae, and Marinicellaceae were highest in overgrown shrimps. Interestingly, two potential pathogens, Pseudoalteromonadaceae and Vibrionaceae, had enriched relative abundances in retarded shrimps (Fig. 3).

The 26 indicative bacterial families among the shrimp size categories, with their indicator values and significance. The diameters of the circles are proportional to the relative abundances of the family, with red, blue, and green circles indicating the peak relative abundances in retarded, overgrown, and normal shrimp, respectively
Ecological Processes Governing Shrimp Gut Bacterial Community Assembly
Using the MNTD, we compared the phylogenetic community composition of shrimp gut microbiotas among the three categories. The mean MNTD value was highest in normal shrimp and was significantly lower in retarded shrimps (Fig. 4), suggesting that phylogenetic clustering is stronger in normal shrimps than that in retarded and overgrown subjects. In addition, the ses.MNTD values obtained using the null model were significantly less than zero (Fig. 4); thus, the shrimp gut bacterial communities tended to be more phylogenetically clustered than would be expected by chance, regardless of the shrimp category. Similarly, the Bray-Curtis distance-based PERMDISP results also significantly (P < 0.001 in all cases) varied from the null random expectation for each category (Table 1). However, it should be noted that the absolute magnitude of ses.MNTD value in normal shrimps was the highest (Fig. 4), indicating a greater importance of deterministic processes in shaping the gut bacterial community in normal shrimps.

Variation of mean nearest taxon distance (MNTD) and the standardized effect sizes of MNTD (ses.MNTD) of gut bacterial communities in retarded, overgrown, and normal shrimp
To further test the possible effects of phylogenetic patterns on shrimp growth rate, we evaluated interspecies interaction in each shrimp category. The OTU table was split into three datasets: retarded, overgrown, and normal shrimp gut microbiotas. The OTUs detected in five or more replicates were selected for correlation calculation, resulting in 299, 548, and 734 OTUs obtained from retarded, overgrown, and normal shrimp samples, respectively (Table 2). In all cases, the network connectivity distribution curves fitted well with the power law model (R 2 > 0.80, Table 2), which suggested that the constructed networks were scale-free. However, the network plots revealed that interspecies interaction was more complex and better connected in retarded and overgrown shrimps than in normal shrimp (Fig. 5), although more OTUs in normal shrimps were retained in the analysis (Table 2). This pattern is evidenced by the lower average path, higher average clustering coefficient and average degree (Table 2), revealing that the nodes in the network are closer [47], are better connected with their neighbors [48], and have more strength connections in overgrown and retarded shrimps [42]. In contrast, the network is better to be separated into modules (as evidenced by higher modularity; Table 2) in normal shrimps, which suggests that the niches are occupied by different patches of co-located functional units [47]. Interestingly, the percentage of positive co-occurrence was highest in overgrown shrimps (Table 2), indicating a higher degree of cooperative activities in the gut microbiota of overgrown shrimps.

Network interaction graph for retarded (a), overgrown (b), and normal (c) shrimp gut bacterial communities. Each node represents a bacterial OTU. The colors of the nodes indicate the OTUs affiliated to different major classes. A blue edge indicates a positive interaction, whereas a red edge indicates a negative interaction between two individual nodes
Changes in Functional Composition among the Shrimp Categories
The mean nearest sequenced taxon index (an index for evaluating the accuracy of PICRUSt [45]) in the community to its nearest relative with a sequenced reference genome was 0.16 ± 0.03 (mean ± standard deviation) across the gut samples. A comparable mean NSTI was detected for the human gut microbiota, for which PICRUSt produced an accurate metagenome prediction [45]. Thus, the predicted functional traits in this study are reliable. The carbohydrate and protein digestion and absorption pathways were all significantly decreased in retarded shrimps compared with normal shrimps, whereas those in overgrown shrimps exhibited an opposite pattern (Fig. 6). Compared with normal shrimps, divergent responses of environmental information process and carbohydrate metabolism were also observed between retarded and overgrown shrimps (Fig. 6). In contrast, pathways of infectious disease were consistently enriched, whereas the immune system (with the exception of the RIG-I-like receptor signaling pathway) was decreased in retarded and overgrown shrimps (Fig. 6). Thus, it is likely that retarded and overgrown shrimps are more prone to pathogen invasion.

Comparisons of gut bacterial KEGG pathways between retarded and normal and between overgrown and normal shrimp using the response ratio methods. Circles in the right column indicate a significant increase in the abundance of the corresponding pathway, and those in the left column indicate a decrease compared with normal shrimp
Discussion
Accumulating evidence has disclosed a tight connection among the gut microbiota, obesity, and host health [49, 50]. Thus, unraveling the ecological mechanisms that govern gut microbiota assembly is expected to guide microbial modifications for sustaining host health [49, 51]. However, previous studies have only focused on the biogeochemical factors that determine the differences in gut bacterial communities between lean and obese animals and humans [10, 16]. This study is thus the first attempt to jointly define the underlying ecological processes of the gut microbiota in contributing shrimp with distinct growth rate.
It is a challenge to identify the causal relationship between the gut microbiota and obesity due to inadequately control for differences in host age, diet, environmental conditions, and other factors that have profound impacts on the gut microbial community [3, 5]. However, the gut bacterial communities were significantly distinct among the shrimp categories in the present study (Fig. 2), which was also evident at the coarse phylum/class level (Fig. S2). This pattern raises the question of which factors drive such variations. A plausible explanation is that stochastic processes contribute to the divergence of gut bacterial communities [9]. However, this would not be the case here because the gut bacterial communities significantly deviated from the null random expectation for each category (Table 1). Alternatively, it is likely that priority effects cause a contingency in the structure of communities, resulting in alternative stable states [52]. This assertion is supported by recent data showing that the early colonizers of the larval gut exert a long-term effect on the risk of chronic disease and health later in life [8]. Consistently, substantial inter-individual variations in the gut bacterial composition have been observed in cohabiting larval Atlantic cod [53] and juvenile sea cucumbers [12]. However, it should be noted that the priority effects do not rule out host selection on the gut microbiota from the local species pool. For example, Alphaproteobacteria and Actinobacteria lineages were strongly selected against by shrimp, whereas Gammaproteobacteria species appear to be adaptive to shrimp gut conditions (Fig. S2). In agreement with our previous report showing that the relative abundances of Alphaproteobacteria and Actinobacteria were higher in healthy shrimps than in diseased ones (here, retarded and overgrown shrimp), while Gammaproteobacteria species exhibit an opposite pattern [3]. Notably, the variations in gut bacterial community could directly translate into changes in digestive enzyme activities, which in turn shape shrimp growth rate (Fig. 2). Similarly, close associations have been described between the gut bacterial community and host body weight in cohabiting pigs [10] and sea cucumbers [12]. These findings corroborate the notion that dysbiosis of the gut microbiota is linked to host growth traits, mirroring what has been observed in vertebrates [10, 13, 14], although shrimp gut assemblages are apparently distinct from those of vertebrates.
Then, we examined which assemblages contribute to the differences in shrimp growth rates. Opportunistic pathogens of the Pseudoalteromonadaceae and Vibrionaceae lineages, their relative abundances were highest in retarded shrimps and were lowest in normal shrimps (Fig. 3). Our previous work has shown that the relative abundance of Vibrionaceae is closely associated with shrimp disease severity [3]. Under this premise, retarded and overgrown shrimps are at an increased risk for developing disease. Prevotellaceae and Enterobacteriaceae are known to be positively associated with host obesity in vertebrates [10, 54], whereas no difference was detected between large and small sea cucumber cohorts [12]. In contrast, we found that overgrown shrimp were characterized by maximum relative abundances of Rhodospirillaceae, Saprospiraceae, Bdellovibrionaceae, and Marinicellaceae (Fig. 3). Therefore, it is likely that the connection between obesity and gut bacterial family profile depends on the host trophic level. Nevertheless, we can make educated guesses regarding functionality based on the current knowledge of the biology and ecology of these indicative bacterial families. For example, Saprospiraceae and Marinicellaceae species harbor demonstrated abilities for hydrolysis and utilization of complex carbon sources [55, 56], thereby aiding host in the acquisition of additional nutrients. Consistent with this idea, in the present study, the relative abundances of Saprospiraceae and Marinicellaceae were positively (Pearson r > 0.60, P < 0.01 in all cases, data not shown) correlated with the measured digestive enzyme activities. A loss of microbial diversity appears as the most constant finding of intestinal dysbiosis [4]. However, we found no significant difference in gut bacterial diversity (Fig. S3), although the community compositions changed significantly between overgrown and normal shrimps (Fig. 2; Table S2). A possible explanation for this discrepancy is that a high abundance of Bdellovibrio species (Fig. 3) restores the gut bacterial diversity and maintaining host health [57]. Indeed, the reintroduction of key predators in digestive ecosystem has been proposed as a therapeutic approach for restoring the disease-associated dysbiosis of gut microbiota [4]. To conclude, the enrichment of predators might limit the population of dominant species, which is a probable mechanism to maintain gut species diversity in obese shrimps. However, it is worthwhile to note that an altered gut community might be a stable but harmful balance [58].
The ses.MNTD distribution mean deviated significantly from zero (Fig. 4); thus, the gut bacterial assemblages exhibited a significantly higher phylogenetic clustering than would be expected by chance (Table 1), which suggests that the gut bacterial communities were structured by host filtering (akin to environmental filtering) [41]. Notably, we found that phylogenetic clustering was stronger in normal shrimps than in retarded and overgrown subjects (Fig. 4), revealing that the importance of host filtering decreases in sub-healthy (retarded and overgrown, if not termed as diseased) shrimps. In other words, sub-healthy shrimps exert a weaker selection on the gut microbiota, thereby resulting in less phylogenetically convergent communities. This view arises from the fact that, the occurrence of shrimp disease reduces the strength of selection on the gut microbiota [19]. How can phylogenetically related species coexist in the shrimp gut? In theory, closely related species occupy a similar niche and should thus compete more intensively than species of distinct lineages [41]. However, in praxis, biological invasion studies have demonstrated that the presence of phylogenetically related species in a resident community can increase the success of alien colonizers [59]. One possible explanation for this discrepancy is that bacterial strains can switch to the utilization of a different available nutrient source, thereby facilitating the closely related species exhibit niche differences. This assertion is supported by the evolutionary plasticity and redundancy theory, which infers that close relatives have differently routed pathways, thus allowing the coexistence of phylogenetically related species [60]. According to frequency-dependent regulation, niche differences promote coexistence by favoring species when they drop to low density and are threatened with extinction [61]. As a consequence, bacterial evenness and diversity were higher in normal shrimp (Fig. S3). Alternatively, gut species are strongly selected by similar selective pressures in normal shrimp, thereby eliminating more different and less related taxa [62]. Consistent with this notion, it has been shown that gut bacterial communities tend to be less physically inhibited (e.g., host filtering) in diseased shrimp, which leads to a stochastic gut community assembly [19]. We therefore infer that the convergence of gut bacterial phylogenetic clustering depends on the host health state but warrant further study to validate this pattern.
It has become clear that interspecies interaction determines the microbial-mediated functions [10, 14, 19]. We found that the interspecies interaction was more complex, better connected, and more cooperative in retarded and overgrown shrimps, whereas normal shrimps have a greater modularity, corresponding to functional units [47] (Table 2). Given the key functionalities encoded and conferred by the gut microbiota [2, 15], it is tempting to speculate that the increased interspecies interaction in overgrown shrimps would promote digestive capacity. Consistently, we found that the measured digestive activities were the highest in overgrown shrimps (Fig. S2). This enhanced digestive activity could be due to the bloom of lineages with efficient nutrient intake, such as Marinicellaceae and Saprospiraceae species (Fig. 3). Similarly, a recent network analysis showed that the gut microbiota of obese children had a higher correlation density than that of normal-weight children, which is coincident with an increased fermentation capacity [14]. In contrast, the gut microbiota of normal shrimps exhibits a lower percentage of positive co-occurrence (Table 2). This result adds to the increasing evidence that strong competition, as occurs in a diverse community, favors allelopathic species [63, 64]. It has been proposed that increased allelopathic interactions could facilitate the resident community barrier against pathogen invasion [64]. Under this scenario, retarded and overgrown shrimps are more prone to pathogen invasion. Consistently, the abundances of functional pathways involved in diseases infection increased significantly in overgrown and retarded shrimps (Fig. 6). In addition, compared with normal shrimps, the metabolic pathways divergently changed between overgrown and retarded shrimps, whereas other pathways were less altered in overgrown shrimps (Fig. 6). It is known that organisms are capable of reallocating energy from anabolism to immunological activity in response to infection [65]. Thus, these differences may explain why the growth rates are divergent (retarded and overgrown), although both states exhibit higher percentages of cooperative syntrophy in relation to normal shrimps. Apparently, obesity is not a serious concern in aquaculture. However, it is worth noting that overgrown shrimps that have been threatened by pathogen invasion (Fig. 6) may spread these pathogens and impact their cohorts [66]. Indeed, there was a disease outbreak in the later stage (data not shown). For these reasons, additional studies are required to characterize the temporal dynamic of the gut microbiota in retarded and overgrown shrimps.
To the best of our knowledge, this is the first attempt to explore the direct connection between the gut microbiota and the shrimp growth rate. We found that marked changes in the gut bacterial community contributed to variations in digestive activities, subsequently affecting shrimp growth rates. The high efficiency of nutrient intake in overgrown shrimps was evidenced by an intensified interspecies interaction in the gut bacterial community and enriched lineages with high nutrient intake efficiencies. However, the less phylogenetic clustering of gut microbiota in retarded and overgrown shrimps may offer empty niches for pathogens invasion. Given the adequate control for differences in factors that have profound impacts on the gut microbiota, we speculate that the differences in gut community assembly can be attributed to the legacy of random colonization in larval shrimps (priority effects) and that an altered microbiota may be a causative factor for retardation or overgrown. The results further validate our previous idea that the larval stage is likely the best opportunity to establish the desired gut microbiota via the preemptive colonization of probiotics [51]. Together, these findings greatly add our understanding of the causative role of the gut microbiota in retarded and overgrown shrimp as well as the underlying ecological processes, instead of well-studied vertebrate microbiota.
References
Sommer F, Bäckhed F (2013) The gut microbiota—masters of host development and physiology. Nat Rev Microbiol 11:227–238
Tap J, Furet JP, Bensaada M, Philippe C, Roth H, Rabot S, Lakhdari O, Lombard V, Henrissat B, Corthier G (2015) Gut microbiota richness promotes its stability upon increased dietary fibre intake in healthy adults. Environ. Microbiol. 17:4954–4964
Xiong J, Wang K, Wu J, Qiuqian L, Yang K, Qian Y, Zhang D (2015) Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Appl. Microbiol. Biotechnol. 99:6911–6919
Mosca A, Leclerc M, Hugot JP (2016) Gut microbiota diversity and human diseases: should we reintroduce key predators in our ecosystem? Front. Microbiol. 7:455
Yan Q, Li J, Yu Y, Wang J, He Z, Van Nostrand JD, Kempher ML, Wu L, Wang Y, Liao L, Li X, Wu S, Ni J, Wang C, Zhou J (2016) Environmental filtering decreases with fish development for the assembly of gut microbiota. Environ. Microbiol. doi:10.1111/1462-2920.13365
Ng'ambi J, Li R, Mu C, Song W, Liu L, Wang C (2016) Dietary administration of saponin stimulates growth of the swimming crab Portunus Trituberculatus and enhances its resistance against vibrio alginolyticus infection. Fish Shellfish Immunol 59:1050–4648
Xiong J, Zhu J, Zhang D (2014) The application of bacterial indicator phylotypes to predict shrimp health status. Appl. Microbiol. Biotechnol. 98:8291–8299
Sabes-Figuera R, Knapp M, Bendeck M, Mompart-Penina A, Salvador-Carulla L (2015) The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 26:24–29
Burns AR, Stephens WZ, Stagaman K, Wong S, Rawls JF, Guillemin K, Bohannan BJM (2016) Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J 10:655–664
Ramayo-Caldas Y, Mach N, Lepage P, Levenez F, Denis C, Lemonnier G, Leplat JJ, Billon Y, Berri M, Doré J, Rogel-Gaillard C, Estellé J (2016) Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J 10:2973–2977
Miyanaga A, Shimizu K, Noro R, Seike M, Kitamura K, Kosaihira S, Minegishi Y, Shukuya T, Yoshimura A, Kawamoto M (2011) Enterotypes of the human gut microbiome. Nature 473:174–180
Sha Y, Liu M, Wang B, Jiang K, Sun G, Wang L (2016) Gut bacterial diversity of farmed sea cucumbers Apostichopus japonicus with different growth rates. Microbiology 85:109–115
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031
Riva A, Borgo F, Lassandro C, Verduci E, Morace G, Borghi E, Berry D (2016) Pediatric obesity is associated with an altered gut microbiota and discordant shifts in Firmicutes populations. Environ. Microbiol. doi:10.1111/1462-2920.13463
Holmes E, Li JV, Marchesi JR, Nicholson JK (2012) Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. 16:559–564
Ferrer M, Ruiz A, Lanza F, Haange SB, Oberbach A, Till H, Bargiela R, Campoy C, Segura MT, Richter M (2013) Microbiota from the distal guts of lean and obese adolescents exhibit partial functional redundancy besides clear differences in community structure. Environ. Microbiol. 15:211–226
Sullam KE, Essinger SD, Lozupone CA, O’Connor MP, Rosen GL, Knight R, Kilham SS, Russell JA (2012) Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol. Ecol. 21:3363–3378
Wiens JJ, Ackerly DD, Allen AP, Anacker BL, Buckley LB, Cornell HV, Damschen EI, Davies TJ, Grytnes JA, Harrison SP (2010) Niche conservatism as an emerging principle in ecology and conservation biology. Ecol. Lett. 13:1310–1324
Zhu J, Dai W, Qiu Q, Dong C, Zhang J, Xiong J (2016) Contrasting ecological processes and functional compositions between intestinal bacterial community in healthy and diseased shrimp. Microb. Ecol. 72:975–985
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI (2009) A core gut microbiome in obese and lean twins. Nature 457:480–484
Duran-Pinedo AE, Paster B, Teles R, Friaslopez J (2011) Correlation network analysis applied to complex biofilm communities. PLoS One 6:e28438
Hallam SJ, McCutcheon JP (2015) Microbes don’t play solitaire: how cooperation trumps isolation in the microbial world. Environ. Microbiol. Rep. 7:26–28
Egan S, Gardiner M (2016) Microbial dysbiosis: rethinking disease in marine ecosystems. Front. Microbiol. 7:991
Faust K, Raes J (2012) Microbial interactions: from networks to models. Nat Rev Microbiol 10:538–550
Xiong J, Chen H, Hu C, Ye X, Kong D, Zhang D (2015) Evidence of bacterioplankton community adaptation in response to long-term mariculture disturbance. Sci Rep 5:15274
Herlemann DP, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ, Andersson AF (2011) Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5:1571–1579
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963
Caporaso JG, Kuczynski J, Stombaugh J (2010) QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
De Santis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied Environ Microbiol 72:5069–5072
De Santis TZ, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, Phan R, Andersen GL (2006) NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 34:394–399
Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26:1641–1650
Clarke KR (1993) Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18:117–143
R Development Core Team (2013) R: A language and environment for statistical computing
Dufrêne M, Legendre P (1997) Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol. Monogr. 67:345–366
Roberts DW (2007) labdsv: Ordination and multivariate analysis for ecology. R package version
Chase JM, Kraft NJ, Smith KG, Vellend M, Inouye BD (2011) Using null models to disentangle variation in community dissimilarity from variation in α-diversity. Ecosphere 2:art24
Stegen JC, Lin X, Fredrickson JK, Chen X, Kennedy DW, Murray CJ, Rockhold ML, Konopka A (2013) Quantifying community assembly processes and identifying features that impose them. ISME J 7:2069–2079
Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, Blomberg SP, Webb CO (2010) Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26:1463–1464
Webb CO, Ackerly DD, McPeek MA, Donoghue MJ (2002) Phylogenies and community ecology. Ann Rev Ecol Syst 33:475–505
Deng Y, Jiang Y, Yang Y, He Z, Luo F, Zhou J (2012) Molecular ecological network analyses. BMC Bioinformatics 13:113
Newman MEJ (2003) The structure and function of complex networks. SIAM Rev. 45:167–256
Shannon P, Markiel A, Ozier O, Baliga NS, Wang J, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13:2498–2504
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Thurber RLV, Knight R (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotech 31:814–821
Wu J, Xiong J, Hu C, Shi Y, Wang K, Zhang D (2015) Temperature sensitivity of soil bacterial community along contrasting warming gradient. Appl. Soil Ecol. 94:40–48
Newman ME (2006) Modularity and community structure in networks. Proc Nat Acad Sci USA 103:8577–8582
Guimera R, Sales-Pardo M, Amaral LA (2007) Classes of complex networks defined by role-to-role connectivity profiles. Nat Physcis 3:63–69
Bäckhed F, Fraser C, Ringel Y, Sanders ME, Sartor RB, Sherman P, Versalovic J, Young V, Finlay BB (2012) Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe 12:611–622
Gilbert JA, Quinn RA, Debelius J, Xu ZZ, Morton J, Garg N, Jansson JK, Dorrestein PC, Knight R (2016) Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 535:94–103
Xiong J, Dai W, Li C (2016) Advances, challenges, and directions in shrimp disease control: the guidelines from an ecological perspective. Appl Microbiol Biotech 100:6947–6954
Fukami T (2015) Historical contingency in community assembly: integrating niches, species pools, and priority fffects. Ann Rev Ecol Evol Syst 46:1–23
Fjellheim AJ, Playfoot KJ, Skjermo J, Vadstein O (2012) Inter-individual variation in the dominant intestinal microbiota of reared Atlantic cod (Gadus morhua L.) larvae. Aquac. Res. 43:1499–1508
Harakeh SM, Khan I, Kumosani T, Barbour E, Almasaudi SB, Bahijri SM, Alfadul SM, Ajabnoor GMA, Azhar EI (2016) Gut microbiota: a contributing factor to obesity. Front Cell Infet Microbiol 6:95
Mcilroy SJ, Nielsen PH (2014) The family Saprospiraceae. pp 863–889
Galitskaya P, Akhmetzyanova L, Selivanovskaya S (2016) Biochar carrying hydrocarbon decomposers promotes degradation during the early stage of bioremediation. Biogeosci Discuss 13:5739–5752
Lebba V, Santangelo F, Totino V, Nicoletti M, Gagliardi A, De Biase RV, Cucchiara S, Nencioni L, Conte MP, Schippa S (2013) Higher prevalence and abundance of Bdellovibrio bacteriovorus in the human gut of healthy subjects. PLoS One 8:e61608
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R (2012) Diversity, stability and resilience of the human gut microbiota. Nature 489:220–230
Stecher B, Chaffron S, Käppeli R, Hapfelmeier S, Freedrich S, Weber TC, Kirundi J, Suar M, Mccoy KD, Von MC (2010) Like will to like: abundances of closely related species can predict susceptibility to intestinal colonization by pathogenic and commensal bacteria. PLoS Path 6:e1000711
Wagner A (2011) Genotype networks shed light on evolutionary constraints. Trends Ecol. Evol. 26:577–584
Adler PB, Hillerislambers J, Levine JM (2007) A niche for neutrality. Ecol. Lett. 10:95–104
Mayfield MM, Levine JM (2010) Opposing effects of competitive exclusion on the phylogenetic structure of communities. Ecol. Lett. 13:1085–1093
Inglis RF, Gardner A, Cornelis P, Buckling A (2009) Spite and virulence in the bacterium Pseudomonas Aeruginosa. Proc Nat Acad Sci USA 106:5703–5707
Jousset A, Schulz W, Scheu S, Eisenhauer N (2011) Intraspecific genotypic richness and relatedness predict the invasibility of microbial communities. ISME J 5:1108–1114
Yiu JHC, Dorweiler B, Woo CW (2016) Interaction between gut microbiota and toll-like receptor: from immunity to metabolism. J Mol Med. doi:10.1007/s00109-016-1474-4
Mallon CA, Elsas JDV, Salles JF (2015) Microbial invasions: the process, patterns, and mechanisms. Trends Microbiol. 23:719–729
Acknowledgements
This work was supported by the Zhejiang Province Public Welfare Technology Application Research Project (2016C32063), and the Open Fund of Key Laboratory of Marine Biogenetic Resources, Third Institute of Oceanography (HY201601); the Project of Science and Technology Department of Ningbo (2015C10062); and the KC Wong Magna Fund in Ningbo University.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
ESM 1
(PDF 284 kb)
Rights and permissions
About this article

Cite this article
Xiong, J., Dai, W., Zhu, J. et al. The Underlying Ecological Processes of Gut Microbiota Among Cohabitating Retarded, Overgrown and Normal Shrimp. Microb Ecol 73, 988–999 (2017). https://doi.org/10.1007/s00248-016-0910-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-016-0910-x